
Abstract
Campylobacter jejuni is an important zoonotic pathogen known to be resistant to a wide range of antibiotics worldwide. Campylobacter jejuni may be intrinsically resistant to antibiotics or can acquire antibiotic resistance determinants through gene transfer. However, the knowledge of molecular mechanisms of antimicrobial resistance among Campylobacter isolates from wild birds, especially in Lithuania, is limited. Whole genome sequencing (WGS) is a tool for better understanding the evolutionary and epidemiologic dynamics of C jejuni. This study describes a draft whole genome sequence of C jejuni MM26-781 isolated from a common pigeon (Columba livia) in Lithuania in 2011 and assigned to ST-6424 (CC179) sequence type. The draft genome sequence contained 1.68 Mb, comprising 1651 coding genes, 40 transfer RNAs, 1 ribosomal RNA, and 69 pseudogenes with an average G + C content of 30.4%. The RAST (Rapid Annotation using Subsystem Technology) pipeline annotated (NCTC11168) a total of 305 subsystems in the genome of C jejuni MM26-781 strain, with most of the genes associated with amino acids and derivatives related to metabolism (18.93%) and protein metabolism (14.43%). The genes and mutations related to antibiotic resistance, including gyrA and gyrB genes associated with quinolone resistance, blaOXA-448 gene (locus tag C9371_07715) associated with resistance to β-lactams, rpoB gene associated with resistance to rifamycin, vgaE gene associated with resistance to streptogramin and efflux system CmeABC (cmeA, cmeB, cmeC), efflux pump PmrA, and transcriptional regulator CmeR responsible for multidrug resistance in C jejuni MM26-781 chromosome, were identified. Also, the virulence factors, including ciaB, cadF, ceuE, pldA, motB, and bd1A genes, were identified by WGS data analysis.
Introduction
Campylobacteriosis is the most commonly reported foodborne zoonosis in Lithuania and is of high concern to the food safety worldwide. 1 The causative agents of this infection are primarily associated with poultry, followed by cattle, and have been identified in different species of wild animals and birds.2,3 Antimicrobial resistance in Campylobacter is common and an increase in resistance has been observed in the past decade. 4 Application of modern bacterial identification and characterization methods such as high-throughput whole genome sequencing (WGS) allows better understanding of molecular mechanisms of antimicrobial resistance. This method is a revolutionary tool in public health microbiology and is gradually replacing classical typing methods in surveillance of infectious diseases.5,6 The aim of this study was to characterize the whole genome sequence of Campylobacter jejuni strain MM26-781 obtained from common pigeon (Columba livia) in Lithuania and to identify antimicrobial resistance determinants of this isolate.
Materials and Methods
C jejuni used in the study
Campylobacter jejuni MM26-781 strain isolated from common pigeon in 2011 and assigned to ST-6424 (CC179) sequence type (the Campylobacter collection of the Department of Food Safety and Quality, Lithuanian University of Health Sciences) was selected for WGS. Previously, this C jejuni strain sequence type and clonal complex were assigned by submitting the DNA sequences to the Campylobacter PubMLST database (strain ID number 23759) 7 Antimicrobial susceptibility testing was performed with 5 antimicrobials, including ciprofloxacin, ceftriaxone, gentamicin, tetracycline, and erythromycin, by the agar dilution method described in our previous study. 8 The tested C jejuni MM26-781 strain revealed exceptionally high-level resistance to ciprofloxacin and ceftriaxone (MIC [minimum inhibitory concentration] >256 μg/mL).
Genomic DNA preparation
The C jejuni MM26-781 strain isolated from common pigeon was stored at −80°C in brain heart infusion broth (Oxoid Ltd., Basingstoke, UK) with 30% glycerol (Stanlab, Lublin, Poland). The frozen bacterial culture was recovered on Blood Agar Base No. 2 (Oxoid) supplemented with 5% defibrinated horse blood (E&O Laboratories, Bonnybridge, Scotland) and incubated under microaerophilic conditions (5% oxygen, 10% carbon dioxide, and 85% nitrogen) at 37°C for 48 hours. DNA was extracted using the PureLink Genomic DNA Mini Kit (Invitrogen, Carlsbad, CA, USA) based on manufacturer’s instruction and finally eluted in 50 μL of sterile water. The amount and integrity of gDNA (genomic DNA) was quantified using Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and 0.8% to 1% agarose gel, respectively.
WGS, assembly, and annotation
A sequencing library was prepared with the Nextera XT Sample Preparation Kit (Illumina, San Diego, CA, USA) following manufacturer’s guidelines. Campylobacter jejuni genome was sequenced at the NGS-MiSeq core facility of University of Copenhagen using MiSeq instrument (Illumina) with 250-bp (base pairs) paired-end reading cycles. CLC Genomics Workbench v. 6.5.1 (CLC Denmark) was used for the adapter and quality trimming of the raw reads. De novo assembly was performed using the SPAdes v3.9.0 genome assembler 9 using assembly parameters: k automatic selection based on read length, repeat resolution, mismatch careful mode, mismatch corrector, and a wide range of k-mer sizes: 21, 33, 55, 77, 99, 127. The assembled sequences were annotated using the NCBI (National Center for Biotechnology Information) GenBank annotation pipeline (PGAP). 10 The subsystems annotation was obtained using the SEED-based automated annotation system after the data were uploaded to RAST (Rapid Annotation using Subsystem Technology) genome server. 11 ResFinder 3.0 was used for identification of intrinsic genes associated with the phenotypic antimicrobial resistance of the strain using thresholds of 90% identity and 60% gene coverage.12,13 Also, the coding sequences (CDSs) of the genome were subjected to Resistance Gene Identifier (RGI 4.2.2; CARD 3.0.0) analysis information in the Antibiotic Resistance Database (ARDB). 14 The MM26-781 strain was compared with the reference genome of C jejuni NCTC11168 (NCBI GenBank AL111168.1). Along with detection of intrinsic antibiotic resistance genes, PathoFinder 1.1 was used for the potential prediction of a bacteria’s pathogenicity. 15 The Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession PYWF00000000. The version described in this article is version PYWF01000000 (Table 1).
Project information for the sequencing of Campylobacter jejuni strain MM26-781 isolated from common pigeon in Lithuania.

Results and Discussion
Comparative genome analysis
The draft genome of C jejuni MM26-781 consisted of 32 contigs with a 30.4% G + C content and total length of 1 683 133 bp (N50, 122 854 bp). The RAST annotations of assemblies identified a total of 305 subsystems, 40 transfer RNAs, 1 ribosomal RNA, 1651 CDSs, and 69 pseudogenes (Figure 1). The number of antimicrobial resistance genes including CmeABC and PmrA multidrug efflux pumps, CmeR transcriptional repressor, 16 fluoroquinolone resistance genes (gyrA, gyrB) and β-lactamase resistance gene (blaOXA-448) were identified within the subsystem of virulence, disease, and defense. Both efflux pumps belong to the resistance nodulation division family of transporters and contribute to multidrug resistance of antimicrobials.17,18 BASys was used to create genome map with the COG (clusters of orthologous genes) functional categories (Figure 1). 19 The ResFinder 3.0 tool allowed us to identify the resistant blaOXA-448 gene (encoding β-lactamase), 5 amino acid changes in gyrA gene, and 2 amino acid changes in cmeR gene (for nucleotide identity cutoff—99.46%, query length 747/747). Along with these detected antimicrobial factors, 6 mutations with amino acid changes including deletion Lys123→del in the L22 ribosomal protein (rpIV) were observed (Table 2). Mutations in the L22 ribosomal protein confer macrolide resistance in a variety of pathogenic and nonpathogenic bacteria. 20 However, C jejuni MM26-781 strain was sensitive to erythromycin. Nevertheless, the mutations might also affect the level of other proteins and confer resistance to other different antimicrobial agents. 21 The CARD data analysis of C jejuni MM26-781 identified genes related to antibiotic resistance including rifamycin resistance mediated by rpoB, streptogramin resistance mediated by vgaE, and antibiotic efflux pumps responsible for multidrug resistance. Using a CRISPR-finder 22 and PathogenFinder, clustered regularly interspaced palindromic repeat systems (CRISPR) between 86 215 and 86 580 bp and CRISPR between 307 644 and 307 731 bp were detected. CRISPR systems may increase expression of genes which enhance the virulence of C jejuni. The PathogenFinder analysis revealed pathogenic potential for C jejuni MM26-781 as a human pathogen. This strain matched 121 pathogenic families (92.5%) indicating a high risk for human infections. Among this, C jejuni MM26-781 strain harbored cytolethal distending toxin (CDT) composed of cdtA, cdtB, and cdtC genes. The CDT is a virulence factor causing damage in the host’s DNA chromosome and cell death.23-25 In addition, the sequence analysis identified 6 virulence genes, ciaB, ceuE, cadF, pldA, motB, and bd1A, responsible for invasion, cell adhesion, flagellar motility, and biofilm formation, respectively. The presence of putative virulence factors in C jejuni MM26-781 strain isolated from common pigeon, including invasion with better colonization, may pose a risk for zoonotic transmission to a human host.
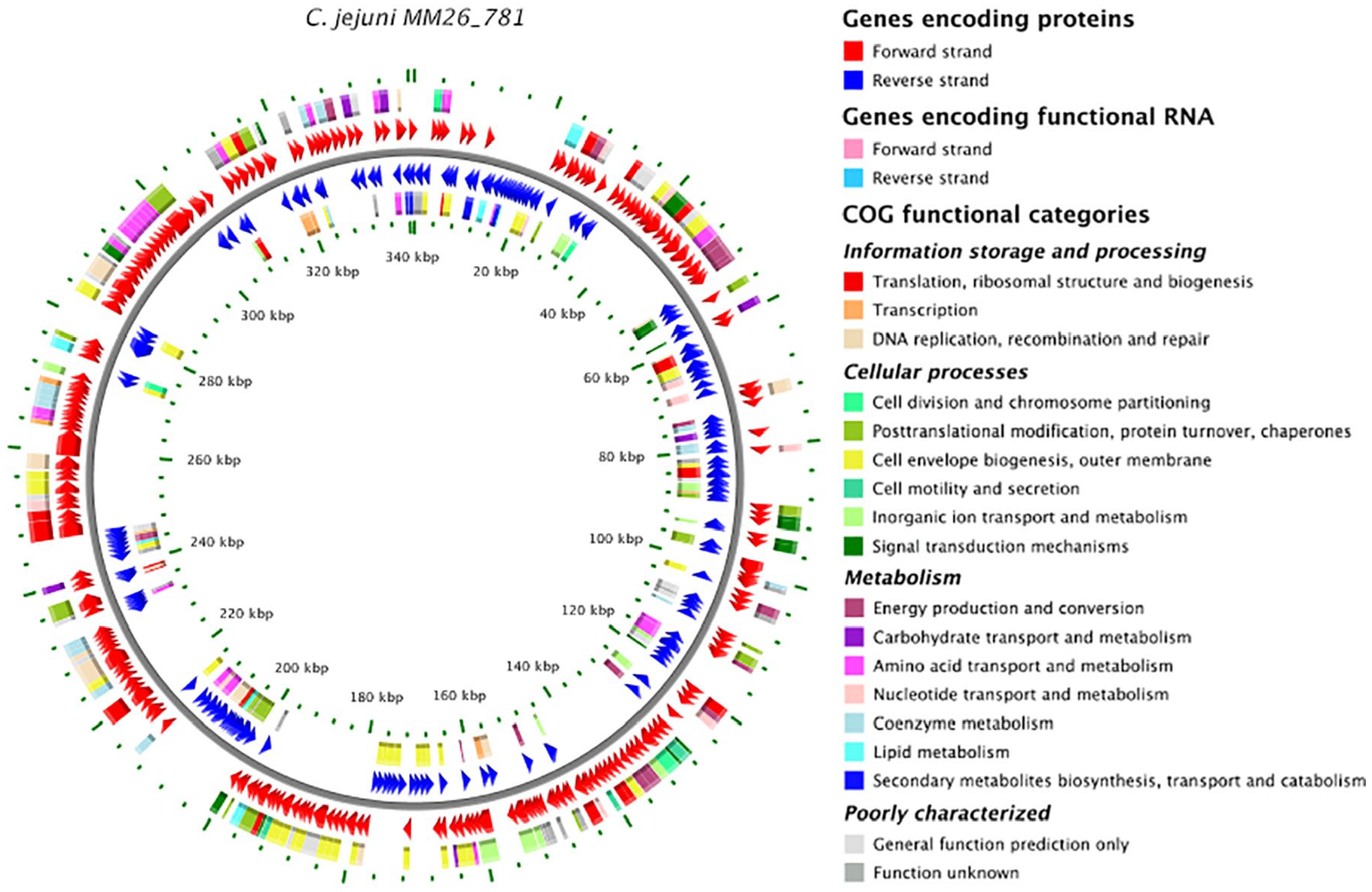
Schematic representation of the draft genome sequence of Campylobacter jejuni MM26-781 with the clusters of orthologous genes functional categories. BASys was used to create this genome map.
Nucleotide and amino acid changes of L22 ribosomal protein, cmeR, and gyrA genes of Campylobacter jejuni..

Conclusions
The draft whole genome sequence of C jejuni strain MM26-781 isolated from common pigeon was characterized in this study. This is the first report of such type from Lithuania and we believe that the genomic data of C jejuni MM26-781 strain with information on CRISPR, fluoroquinolones, β-lactamase, and multidrug-resistant determinants including virulence factors will facilitate further understanding of this important zoonotic pathogen. The findings of this study demonstrate the pathogenic potential of C jejuni isolated from common pigeon and suggest that WGS can be used to identify resistance determinants and virulence factor prediction.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MM and EK devised the study and the main conceptual ideas; JA and EK performed bioinformatic analysis; JA wrote the manuscript with input from all authors; MM, EK, SR and AN contributed to the interpretation of the results. All authors contributed to manuscript revision, read and approved the submitted version.