
Abstract
Dual analyses of the interactions between Ostreid herpesvirus 1 (OsHV-1) and the bivalve Crassostrea gigas during infection can unveil events critical to the onset and progression of this viral disease and can provide novel strategies for mitigating and preventing oyster mortality. Among the currently used “omics” technologies, dual transcriptomics (dual RNA-seq) coupled with the analysis of viral DNA in the host tissues has greatly advanced the knowledge of genes and pathways mostly contributing to host defense responses, expression profiles of annotated and unknown OsHV-1 open reading frames (ORFs), and viral genome variability. In addition to dual RNA-seq, proteomics and metabolomics analyses have the potential to add complementary information, needed to understand how a malacoherpesvirus can redirect and exploit the vital processes of its host. This review explores our current knowledge of “omics” technologies in the study of host-pathogen interactions and highlights relevant applications of these fields of expertise to the complex case of C gigas infections by OsHV-1, which currently threaten the mollusk production sector worldwide.
Introduction
An in-depth understanding of molecular host-pathogen interactions is needed to develop effective measures of prevention, mitigation, and control of diseases in aquatic and terrestrial plant and animal stocks in wild and cultured settings. Recent advances in “omics” approaches have greatly expanded the potential for integrated analyses of large sets of molecular data to reveal markers of desirable traits, such as the resistance to adverse environmental conditions and diseases, and to develop targets for therapy and strategies for selection programs.1–4
Because whole genome sequencing projects can be limited by the complexity and size of the genetic material, the increased feasibility of high-throughput (HT) sequencing technologies has boosted the application of transcriptome sequencing (RNA-seq), including the dual sequencing of host and pathogen. 5 Dual sequencing (dual-seq) is the term used to describe sequencing approaches aiming to simultaneously capture and analyze pathogen and host nucleic acids, while being able to distinguish each of their contributions. From a technical point of view, dual-seq leverages on the increased readout of HT sequencers and on the relatively reduced error rates associated with these data.6,7 Dual-seq exploits the fraction of HT-readouts called “sequencing dark matter,” 8 which basically consist of the off-target reads usually discharged during reference (-genome or -transcriptome) mapping. 9 Initially, these approaches were limited to RNA and, therefore, they were described as “dual RNA-seq.”10,11 Indeed, dual RNA-seq provides a significant advancement in the under-standing of molecular interactions ranging from pathogenic to commensal and mutualistic relationships in a cost-effective manner.12,13 Along with the novelty of the simultaneous sequencing of host and pathogen transcriptomes, dual RNA-seq allows for a time-resolved analysis of molecular interactions occurring during infection of organisms, including non-model host species, with significant ecological and evolutionary implications.14,15
Proteome-centered studies have advanced at a similar pace, often as part of multi “omics” studies, with bacterial proteomes,16–18 protein-protein interactions (PPIs) in host-pathogen systems,19–21 and small open reading frame (sORF) micropeptides22,23 representing some of the current frontier topics. Proteins are the functional cores to multitudes of processes, including viral penetration, propagation, antiviral host responses, and viral subversion of host defenses.19,21,24–26 Thus, the characterization of protein/peptide profiles during the host-virus interaction is crucial to elucidate the complex events underlying infections and diseases. Viral genomes encode a small, although variable, number of proteins, the timely interactions of which with the molecular host’s machinery can ensure the virus’ replication success, often as a result of PPIs. 27 Recent developments in method workflows and technological improvements in mass spectrometry platforms enable a detailed characterization of host-virus and virus-virus PPIs. Advanced dual proteomic methods in modern virology include antibody-based immunoaffinity purification-mass spectrometry (AP-MS) and the yeast 2-hybrid (Y2H) method, among others. 28 Furthermore, metabolomics-based approaches can be used to identify changes in the profile of small molecules participating in the metabolism of proteins, nucleic acids, lipids, and sugars. Accordingly, metabolomic studies can provide additional information about how viruses re-wire the host metabolism to promote infection and pathogenesis.29–31 Therefore, incorporating the metabolomic data into the frame of knowledge resulting from other “omics” is expected to accelerate a holistic interpretation of host-virus interactions.
Irrespective of the chosen “omics,” the need to manage large amounts of molecular data to correctly infer and validate biological hypotheses imposes a rigorous step-by-step evaluation of the work, including experimental design, sampling protocols, and data processing (eg, statistics, bioinformatics pipelines, visualization, and database matching).32,33
At the cutting edge of the “omics” integration and multiplatform research, the analysis of host-associated microbiota by metabarcoding of marker genes, shotgun metagenomics, and metabolomics has provided diagnostics and predictive capabilities that can now help elucidate complex interspecies relationships and perturbations, such as symbiotic, commensalistic, opportunistic, and pathogenic.34,35
Despite the steadily increasing number of transcriptomics- and proteomics-based studies, not many papers refer to the in vivo analysis of host-pathogen interactions. This review aims to report and discuss a dual analysis approach and technical research aspects related to Ostreid herpesvirus 1 (OsHV-1) and its preferred host Crassostrea gigas. This model represents a unique case of study because, in the absence of bivalve cell lines for in vitro testing, dual analyses require a successful infection in vivo, in field, or laboratory conditions.
Dual RNA Sequencing
The first dual RNA-seq studies were focused on viruses and fungi affecting humans and mouse-infecting protists.36–41 Subsequent protocol improvements allowed for both the qualitative and quantitative recovery of bacterial RNA and expanded the application of the field of dual RNA-seq to bacterial infections. 11 However, the differences in RNA structure and quantity between bacteria and eukaryotic hosts make the enrichment of infected cells or bacterial RNAs necessary. 5 Rarely, in vivo dual-seq approaches have included viruses as pathogens, although both RNA viruses (those viruses whose genome and transcriptional products are made by RNA) and a part of DNA viruses (those viruses whose replication relies on RNA) are suitable dual RNA-seq targets.
Regarding bivalve hosts, dual RNA-seq approaches have been performed to study Quahog Parasite Unknown (QPX) infections in the hard clam Mercenaria mercenaria, 42 Mytilicola infections in the mussel Mytilus edulis, 43 and malacoherpesvirus infections in the oyster C gigas,44–47 in the ark shell Scapharca broughtonii, 48 and in the gastropod Haliotis diversicolor supertexta. 49 However, dual RNA-seq can take advantage of the availability of both reference genomes only for OsHV-1/C gigas samples, whereas in all the other dual studies a de novo assembly of the host transcriptome is necessary. The absence of genome sequences, which are used to accurately discriminate between host and pathogen reads, can bias dual RNA-seq results of phylogenetically similar organisms (eukaryotes such as crustacean Mytilicola spp. and lophotrochozoan Mytilus spp.).
For the first time in 2015, 2 research groups performed a dual RNA-seq analysis to investigate the OsHV-1 infection of C gigas by sequencing samples collected up to 120 hours after experimental infection 44 and the other based on sequencing highly infected oysters collected from the Goro lagoon, Italy. 45 Both studies contributed to the identification of key antiviral pathways and candidate markers of viral infections in oysters. At the same time, these first dual RNA-seq data have contributed to an improved understanding of the viral transcriptome, demonstrating that the relative ratios between OsHV-1 expression values are highly comparable over heterogeneous samples, 50 as well as on different hosts. 48 These experiments have shown that the sequencing coverage plays an essential role in obtaining productive dual-seq data, because an abundant number of viral reads could be obtained only with more than 4 billion total sequenced bases (Figure 1). Considering the genome size of malacoherpesviruses (around 200 kb), tens of thousands of viral reads are needed to attain a considerable coverage of the viral open reading frames (ORFs).

Read coverage in dual RNA-seq experiments performed on malacoherpesvirus-infected mollusks. The diagram shows the distribution of total host-pathogen sequenced bases (in billions) versus the malacoherpesvirus sequenced bases (in millions) for a selection of RNA-seq samples obtained using polyA-selected libraries (only the square-framed sample Italy_2018 refers to a ribo-depleted library). High-quality reads were mapped on malacoherpesvirus genomes (either OsHV-1 or HaHV-1) using the CLC mapper tool by setting the values 0.8 of similarity fraction over 0.5 of the read length. Sequence Read Archive (SRA) sample IDs: Zhang et al, 2012 (SRR334248, SRR334249, SRR334250, SRR334251, SRR334252, SRR334253, SRR334254, SRR334255, SRR334256, SRR334257, SRR334258, SRR334259), Rosani et al 45 (E-MTAB-2552), He et al 44 (SRR2002940, SRR2002941, SRR2002942, SRR2002947, SRR2002948, SRR2002949), and Bai et al 48 (PRJNA471241); samples labeled as Italy_2018 represent unpublished data.
To avoid unsuccessful sequencing runs, it is important to estimate the viral RNA fraction over the total RNA before sequencing. Usually, RNA samples are selected on the basis of the OsHV-1 DNA loads in the oyster tissues (a proxy to estimate the virus infectivity). However, OsHV-1 DNA and OsHV-1 RNA did not correlate well, 44 as the abundance of viral DNA often does not imply a similar abundance of sequenced viral RNA reads (Figure 2). To overcome this problem, it is necessary to use a viral transcription marker, namely, a viral gene, the expression value of which correlates well with the expression of the whole viral transcriptome. The OsHV-1 DNA polymerase (ORF100) has been used to measure the copy number of viral DNA in a sample, 51 but the ORF100 expression level is an unreliable predictor of the total amount of viral RNA. 50 Recently, Mushegian et al 52 annotated the OsHV-1 ORF104 as a possible major capsid protein, and we proposed this ORF as a reliable estimator of the total viral RNA in a sample. To validate this hypothesis, we tested OsHV-1 ORF104 and its Haliotid herpesvirus 1 (HaHV-1) homolog ORF68 in oyster and haliotid RNA-seq samples subjected to HT-RNA sequencing (Figure 3). Our data demonstrated a good correlation between the ORF104 expression levels and the number of sequenced OsHV-1 reads. Likewise, the ORF68 expression level is a promising marker for the HaHV-1 transcription, since the measured values were consistent with the abundance of viral reads in an infected sample compared with its paired (uninfected) control.

Relative amounts of OsHV-1 DNA and OsHV-1 RNA in variously infected Crassostrea gigas. The diagram illustrates the number of OsHV-1 DNA copies per microliter compared with the number of OsHV-1 viral reads obtained by RNA-seq for 6 unpublished oyster samples subjected to HT RNA-seq (S1-S6).

Candidate markers for malacoherpesvirus transcription. The diagram illustrates the distribution of OsHV-1 ORF104 and HaHV-1 ORF68 expression values versus the total number of sequenced viral reads. Virus expression values are reported as delta of the Ct of the viral reads compared with the corresponding value of a host housekeeping gene (Elongation factor-1α for C gigas and Y-box binding protein 1 for H diversicolor supertexta).
Perspectives on Dual Non-coding RNA Sequencing
Most dual RNA-seq experiments in bivalves have been performed on polyA-selected RNAs, since this procedural approach allows a high specificity in the selection of coding RNAs. Conversely, random priming enables the capture of the complete RNAome, and size selection procedures can be subsequently applied to focus on the desired RNA fraction of long or short RNAs. 53 To reduce the noise of ribosomal RNAs, ribo-depletion commercial kits are commonly used, whereas a precise size fractionation on gel can allow the selection of specific short RNAs (eg, microRNAs [miRNAs] and PIWI-interacting RNAs [piRNAs]) from physiological RNA degradation products. The use of ribo-depleted libraries in bivalves has been limited, probably because of the possibly reduced performance of commercial kits on invertebrate ribosomal RNA. Regarding recovery of viral genomes, Shi et al 54 demonstrated the good performance of ribo-depleted libraries on invertebrate samples, although a real comparison between polyA and ribo-depleted libraries is still lacking. We showed that the number of OsHV-1 reads obtained from 2 paired libraries produced using either polyA selection or ribo-depletion is very similar (Figure 1—the 2 violet samples named Italy_2018). The main difference was in the number of reads mappable on the C gigas genome, since only 45% of the reads of the ribo-depleted library positively matched the current oyster genome release compared with 80% of the reads from the paired polyA-selected library.
Similarly, studies on small non-coding RNA (sncRNA) of bivalves have begun to appear but are still rare. In 2014, Chen et al 55 sequenced small RNAs of scallops infected by the acute viral necrobiotic virus (AVNV), an OsHV-1 variant. The authors demonstrated a different sncRNA size distribution between the control sample and the viral-infected sample, since control sncRNAs are characterized by a 2-peak distribution denoting the presence of both 22-nt sncRNAs (miRNAs) and 31-nt sncRNA (piRNAs), whereas the 22-nt peak was the only recognizable peak in the infected sample. Unfortunately, the reads have not been submitted to public repositories and it is not clear if any sncRNA read matched the viral genome, a situation symptomatic of the presence of viral-encoded sncRNAs or of an active antiviral RNA interference pathway, as widely reported in arthropods 56 and in few other mollusk species. 57
The analysis of the sncRNA reads obtained from the OsHV-1-infected oyster sample Italy_2018 (see Figure 1) resulted in an sncRNA size distribution similar to the one reported for the AVNV-infected sample, with a main 21-nt peak including most of the reads (Figure 4—blue bars). However, the size distribution of the sncRNA reads mapping on the OsHV-1 genome did not present a well-defined size distribution, suggesting that these sncRNA reads originated mainly from physiological RNA degradation processes (Figure 4—orange bars). Because malacoherpesviruses belong to Herpesvirales, an order which includes several viruses encoding sncRNAs, 58 they are interesting models to study host-pathogen interactions mediated by sncRNAs. Moreover, malacoherpesviruses shared some features with the White Spot Syndrome Virus (WSSV) which also encodes 1 sncRNA. 59 As a matter of fact, the existence of viral-encoded small RNAs is not yet confirmed in the published studies and additional experiments are needed to further investigate this point.

Size distribution of sncRNA reads in 1 oyster sample infected by OsHV-1. Blue bars represent the size distribution (14-41 nucleotides) of all the high-quality sncRNA reads obtained from the Italy_2018 samples (primary x-axis), whereas the orange bars are the sncRNA reads mapped on the OsHV-1 genome (secondary x-axis).
Dual Proteomics and Metabolomics
Proteomics and metabolomics provide complementary information to RNA-seq data during host-pathogen interactions. They can provide cost-effective strategies to substantiate inferences made from differential gene expression studies, support the functional characterization of gene products and post-translational modifications, and supply metabolic information in line with the biochemical phenotype. This is clearly reflected by their progressive development and rise in popularity within the fields of human and plant virology during the past decade.60–63 These techniques are also increasingly being used to investigate different host-pathogen interactions in bivalves.64–69 However, in vivo applications of proteomics and/or metabolomics specifically with molluskan virus models are rare, and dual approaches to distinguish virus- from host-encoded proteins and products have not yet been explored.
Regarding C gigas and OsHV-1, Corporeau et al 70 used a two-dimensional gel electrophoresis (2-DE) proteomic approach followed by liquid chromatography with tandem mass spectrometry (LC-MS/MS) to identify 25 differentially expressed proteins between juvenile oysters infected with a low load and a high load of OsHV-1 at 24 hpi. Precisely, the oysters were injected in the adductor muscle with 100 µL inoculate, considered either virulent (high load) or weakly virulent (low load) and corresponding to 2 × 109 or 4 × 106 copies of injected viral genome, respectively, as quantified by real-time polymerase chain reaction (PCR). 70 Key findings included an increase in the abundance of voltage-dependent anion channels (VDACs) and a switch from aerobic respiration toward aerobic glycolysis, or the Warburg effect. Such features of the infection process appear to be shared with other viruses, such as human papillomavirus (HPV), human cytomegalovirus (HCMV; β-herpesvirus), Kaposi’s sarcoma herpesvirus (KSHV), and hepatitis C virus.71–74 These findings were recently substantiated by Delisle et al, 75 who developed a specific C gigas VDAC antibody and identified associations between VDAC abundance and virus susceptibility during field exposures.
Although Corporeau et al 70 still remain the only proteomic-based analysis to date using an OsHV-1 virus challenge, Green et al 76 employed a similar 2-DE approach to identify the differential expression of proteins during an experimental challenge simulated with viral-like double-strand RNA (dsRNA) Poly I:C. In another study, Masood et al 77 also investigated the oyster response to Poly I:C, using a quantitative shotgun proteomic approach to improve the coverage of the oyster proteome, and detected significant changes in the abundance of 720 proteins post injection. Interestingly, poor correlations were found between the abundances of coding RNAs and related proteins, evidence which advocates the importance of including multiple levels of analysis (eg, RNA and proteins, time points).
Metabolomic insights during OsHV-1 infection were also recently obtained, 78 supporting the involvement of VDAC and the Warburg effect. Upregulation of Irg-1 and immune-responsive gene 1 protein/cis-aconitic acid decarboxylase (IRG1/CAD) was also implicated, with the detection of itaconate as an inducible trigger at the crossroads of numerous pathways/mechanisms. With proteomics and metabolomics thus far being separately applied to the oyster-OsHV-1 model to scan single time points and providing relatively low coverage of the intertwined host-virus proteome or metabolome, there is considerable scope to expand their functionalities to assess spatiotemporal molecular dynamics and thus enrich the dual analysis approach (Figure 5).
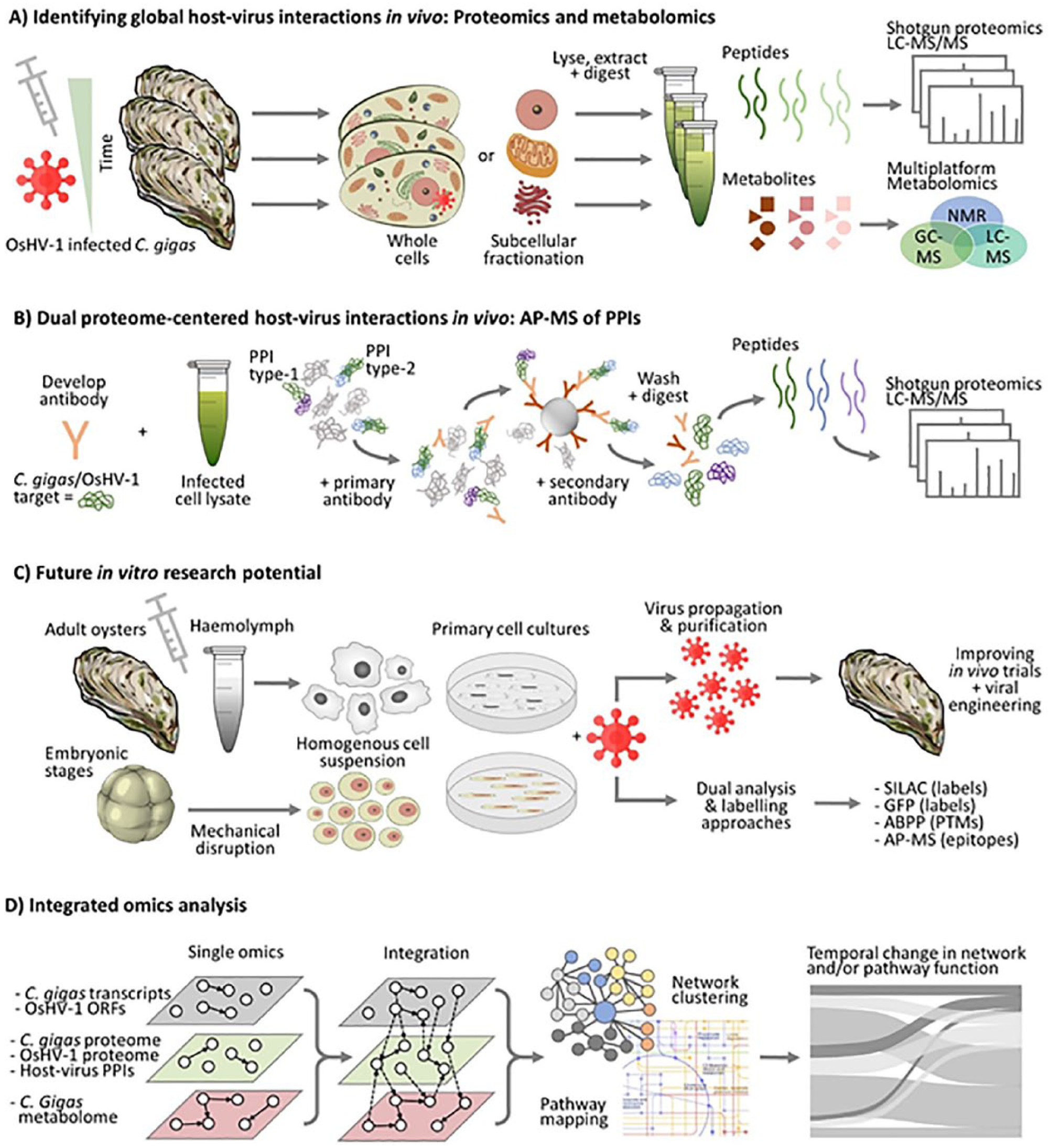
Rational and perspective use of proteomic and metabolomic analyses in the study of OsHV-1 and Crassostrea gigas interactions. (A) Detailed temporal, and spatial, analyses of the global proteome and metabolome during the progression of OsHV-1 infection to reveal dynamic changes in metabolism. Broad shotgun proteomics is one appropriate method of choice, being discovery focused and with access to complete genomic information. Multiplatform metabolomics is required for comprehensive metabolome coverage. (B) General workflow for investigating host-host or host-virus PPIs during infection via affinity purification (AP) using antibodies and shotgun proteomics (requires a priori knowledge of target). (C) Development of a reliable primary cell culture infection model (in lieu of an immortalized cell line) will support the use of analytical methods currently suited to in vitro systems, and with the potential to produce quantities of purified OshV-1 virions. (D) Workflows are being developed to integrate multiple omics data at different temporal and spatial scales, but meaningful integration remains a considerable challenge, requiring careful experimental design and multidisciplinary collaboration.
With access to genomic and/or transcriptomic information, modern LC-MS/MS platforms and data workflows can typically yield identification of 1000 to 3000 proteins in a sample; anticipated improvements in chromatography are likely set to enhance this by increasing the peptide separation quality and sampling depth. 79 Perhaps, more challenging for host-virus interaction studies involving proteomics is being able to separate those features representing anti- and pro-pathogenic cellular responses, for instance, to identify which pathogen-encoded proteins interact with which host proteins to suppress or hijack normal host protein function, and characterize these networks in both space and time. 80
Owing to the importance of PPIs during infection, their analysis in numerous host-virus models (eg, hepatitis C virus, influenza A virus, Zeka virus, potato leafroll virus [PLRV])81,82 currently represents a major goal. To capture these PPIs, protein complexes can be isolated using affinity purification (AP), either with a tagged “bait” protein, or if an antibody is available via co-immunoprecipitation (co-IP). AP followed by digestion and LC-MS/MS of the peptides is a particular method which has gained considerable traction in virology. 83 A major advantage of AP-MS is that it does not require prior knowledge of the bait protein’s interaction partners (thus providing novel insight into a particular protein of interest). It also allows unbiased detection of PPIs under physiological conditions, and it can be used for in vivo studies.28,84 Recent analysis of malacoherpesvirus proteins50,52 additionally provides unique information to facilitate development and scope for AP-MS methods to be used for this particular host-virus model. Undoubtedly, the application of dual proteomics techniques could be expanded greatly by advancing the feasibility of primary cell cultures of bivalve embryos and haemocytes.85–87 In fact, the identification of oyster-OsHV-1 PPIs is essential to understand the biology of the infection and could lead to novel targets for monitoring disease progression, manipulation of mechanistic components for functional studies, and to the development of innovative strategies enabling a reduced oyster susceptibility to the virus.
Metabolomic methods are already well established, and analytical platforms (such as proton nuclear magnetic resonance [ 1 H NMR], gas chromatography-mass spectrometry [GC-MS], liquid chromatography-mass spectrometry [LC-MS], capillary electrophoresis-mass spectrometry [CE-MS]), bioinformatics pipelines, and spectral databases are improving rapidly, with a range of sensitive HT technologies currently being developed.88,89 However, unfortunately, there is not yet a single platform that can measure all metabolites due to their different physicochemical properties (as opposed to transcriptomic and proteomic approaches based on the detection of only nucleic and amino acids, respectively). Thus, multiplatform studies and various metabolite extraction techniques are required to obtain an extensive coverage of the metabolome. Such methods are readily available, 31 and different frameworks for the dual metabolomics analysis of host-pathogen interactions have been developed.90,91 With such approaches starting to be incorporated, the primary challenge for comprehensive host-virus interaction studies which combine multiple ‘omics technologies will with no doubt revolve around methods for data integration and interpretation.
Conclusions and Perspectives
At present, one of the best models for studying viral host-pathogen interactions in mollusks is represented by Malacoherpesviridae, which affect oysters (OsHV-1) or abalones (HaHV-1). Whereas future dual analyses can enrich the molecular data already available for this model, it is important to explore the functional roles of the viral proteins predicted from the sequenced ORFs, comparatively for OsHV-1 and HaHV-1. At the same time, the development of molluskan cell lines could provide a valuable resource to dissect and analyze each step of the viral infection, with the final aim to find inhibitors or inhibition strategies effective at the earliest phases of the malacoherpesvirus infection.
Footnotes
Acknowledgements
We are grateful to the Aquaculture Biotechnology Research Group at the Auckland University of Technology for ongoing discussions that facilitated the formulation of this work.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was partially funded by the H2020 project VIVALDI (Scientific basis and tools for preventing and mitigating farmed mollusk diseases; grant agreement 679589 of the European Commission). We are grateful to the University of Padova/Department of Biology for the post-doc fellowship to U.R. (BIRD 2016-168432).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
UR and PV outlined the review. UR and TY wrote the first draft, with CMB providing experimental data on HaHV-1-infected abalones. All authors contributed to and critically revised the manuscript.