
Abstract
Sigma factors are bacterial transcription factors that bind the core RNA polymerase and direct transcription initiation at a specific promoter site. These specialized sigma factors bind the promoters of genes appropriate to the environmental conditions and selectively increase the transcription of those genes. Here, we attempt to identify sigma factors in 5 genomes belonging to the Enterobacter cloacae complex (Ecc), a group of gram-negative bacteria that are important nosocomial pathogens. This process includes the identification of orthologous sequences, conserved motifs, domains, families, phylogenetic profiles, and protein-protein associations of these components. Based on the reference genome, genome-wide comparison revealed that the genomes of Enterobacter asburiae JCM6051, Enterobacter nimipressuralis CIP 104980, Enterobacter hormaechei ATCC49162, Enterobacter kobei JCM 8580, and Enterobacter ludwigii EN-119 encode 10 sigma factors that exist in the reference strain Enterobacter cloacae subsp cloacae ATCC13047. Moreover, the sequence similarity, protein domains and families of the sigma factors, protein-protein association, and phylogenetic profile indicate that the sigma factor proteins of these 5 strains may have evolutionary relatedness and functional characteristics important to their various environmental niches. Interestingly, the absence of RpoS in E kobei, which contributes to bacterial survival under environmental stress conditions, indicates that RpoS might have been independently acquired and may play different roles relating to pathogenicity, host range determination, and/or niche adaptation. Future work such as RNA sequencing will be directed towards investigating the roles that these sigma factors play in the biology of the Ecc.
Introduction
Enterobacter spp (genus Enterobacter), a group of rod-shaped bacteria of the family Enterobacteriaceae are gram-negative bacteria that are classified as facultative anaerobes. The Enterobacter genus is ubiquitous in nature; the presence of these bacteria in the intestinal tracts of animals results in their wide distribution in soil and water. They are also found in plants. In humans, multiple Enterobacter species, including Enterobacter asburiae JCM6051, Enterobacter nimipressuralis CIP 104980, Enterobacter hormaechei ATCC49162, Enterobacter kobei JCM 8580, and Enterobacter ludwigii EN-119, are known to act as opportunistic pathogens (disease-causing organisms) responsible for nosocomial infections, such as urinary tract infections, cholecystitis, osteomyelitis, and neonatal meningitis. 1
Regulation of gene expression enables the cell to control the production of proteins needed for its life cycle or for adaptation to extracellular changes. Various steps during transcription and translation are therefore subject to different regulatory mechanisms. The most important step in gene regulation is the initiation of transcription in which DNA-dependent RNA polymerase (RNAP) is the key enzyme. 2 However, RNAP alone cannot initiate transcription. Initiation of transcription requires an additional polypeptide known as a sigma factor (σ). Sigma factors are bacterial transcription factors that bind core RNAP and direct transcription initiation at a specific promoter site.3–5 Recognition of a promoter sequence is directed by the σ-subunit of the RNAP holoenzyme. 6
Multiple sigma factors are identified in different species of genus Enterobacter. Strains of the Ecc are widely encountered in nature but they can act as pathogens. Biochemical and molecular studies of E cloacae have shown genomic heterogeneity, and this classification is composed of 6 species: E asburiae JCM6051, E nimipressuralis CIP 104980, E hormaechei ATCC49162, E kobei JCM 8580, and E ludwigii EN-119 are the species most frequently isolated from human clinical specimens. 7 Various sigma factors are involved in different functions, such as sigma factor σ70 RpoD (growth related/housekeeping), σ54 RpoN (nitrogen utilization), σ32 RpoH (heat shock response), σ28 RpoF (flagellar synthesis, chemotaxis), 8 and sigma factors (RpoS) contributing to drug resistance and the regulation of important stress-related transcription factors. 9 The Ecc is involved in extraintestinal infections and is known to possess virulence-associated characteristics, including the ability to adhere to and invade eukaryotic cells and spread within the host. 10 Comparative genomics analyses of several genomes to find similarities and differences between various sigma factors have proven to be extremely useful for gaining insight into the epidemiology and evolution of the pathogenicity of bacterial species. 10 Our analysis indicates that the sigma factors appear to have been acquired same way and may play the same roles relating to pathogenicity, host range determination, and/or niche adaptation. Identification of these sigma factors will further improve identification of the redefined species based on novel sigma factor markers. Future work will be directed towards investigating the roles that these sigma factors play in the biology of the Ecc.
Materials and Methods
E cloacae complex data acquisition
The assembled genome sequence data of the 5 Ecc strains were retrieved from National Center for Biotechnology Information (NCBI). The accession numbers of these genomes are as follows: E asburiae JCM6051 (CP011863-CP011867), E nimipressuralis CIP104980 (MKER00000000), E hormaechei ATCC49162 (MKEQ00000000), E kobei JCM 85580 (MKXD00000000)T, and E Ludwigii EN-119 (CP017279). All 5 sequenced Ecc strains were obtained from different sources and cause various infectious diseases that are listed in Table 1.
List of genomes and their source of isolation.

Preprocessing of genome sequence data and annotation
The genome sequences of E asburiae JCM6051, E nimipressuralis CIP 104980, E hormaechei ATCC49162, E kobei JCM 8580, and E ludwigii EN-119 were refined and used for annotation analysis. The annotation was conducted using the RAST annotation server 11 with default parameters. The annotated genomes of E asburiae JCM6051, E nimipressuralis CIP 104980, E hormaechei ATCC49162, E kobei JCM 8580, and E ludwigii EN-119 obtained using the SEED Viewer of the RAST server were used for analysis, including the coding sequences, RNAs, GC contents, and general feature categories.
Gene prediction and analysis using the reference genome
The sigma factor proteins from E cloacae subsp cloacae ATCC13047T were obtained from UniProt, NCBI, and the literature and were used as bait sequences in a local BLAST search (BLASTN, BLASTX) of the RAST server using E cloacae subsp cloacae as the reference strain to examine the sigma factors in the genomes of E asburiae JCM6051, E nimipressuralis CIP 104980, E hormaechei ATCC49162, E kobei JCM 8580, and E ludwigii EN-119. The following parameters against the genome sequence of each strain were used with the RAST server: gap penalty, 5; gap opening, 100; and gap extension penalty, 6.66.
In silico analysis of protein domains and families
The identification of domains can provide insight into their function and association. NCBI’s Conserved Domain Database (CDD), UniProt, and InterPro are resources for the annotation of protein sequences with the location of conserved domains and functional sites inferred from domain footprints.12–13 The amino acid sequences of E asburiae JCM6051, E nimipressuralis CIP 104980, E hormaechei ATCC49162, E kobei JCM 8580, and E ludwigii EN-119 sigma factor proteins were uploaded in FASTA format in the CD search tool of the NCBI CDD, InterPro, and UniProt databases. A search was performed against position-specific score matrices for the fast identification of conserved domains in protein sequences, with a maximum of 500 hits, composition-based statistics adjustment, and concise result mode. The results were given in a list of domain hits based on their specific accession numbers, sequence interval, and e-value. In this way, proteins with similar domain architecture were identified. Protein sequences of all sigma factors from each strain were further analysed in Pfam using the HMMER search algorithm to identify significant Pfam matches with family, description, alignment, bit score, and e-value.
Functional characterization of genes
The AmiGO 1.8 tool was used to analyse Gene Ontology (GO) data and to identify gene products and all associated functions among all the sigma factors retrieved from the 5 genomes. One-by-one amino acid sequences of all genomes were submitted to BLAST AmiGO with an expected threshold of 0.1. BLAST results were filtered against all data sources, all species, all gene product types, and all gene product associations in the form of a high-scoring gene matrix, which identifies their cellular components, molecular functions, and biological processes.
Alignment and tree building
A powerful and comprehensive suite of molecular biology and next-generation sequencing (NGS) analysis tools, MUSCLE, 14 was used to align the genome sequences to check the extent of similarity between them. Geneious performed pairwise and multiple alignments of the FASTA files using ClustalW. Heatmaps in linear topology were also obtained to check the extent of percentage similarity. Evolutionary relationships among various biological species or other entities were drawn in the form of a phylogenetic/evolutionary tree using the MEGA 7 (Molecular Evolutionary Genetics Analysis) tool. 15 The settings in MEGA 7 to build a tree were as follows: the neighbour-joining method, minimum evolution method, and UPGMA, with standard analysis preferences, such as phylogeny reconstruction, statistical method, and Poisson model.
Results and Discussion
The sigma factor is operational in the Ecc
Without a comprehensive survey of a genome, it is essentially impossible to differentiate orthologues from various species; orthologues are gene products whose similarity to one another arose from the duplication of an ancestral gene. At the subsystem level in RAST, we noted that most genes associated with environmental process, virulence, and metabolism were the same among various species, which indicates that either the Ecc belongs in various environments or these strains may have similar features within various environments that are closely related in their genomes.
A list of the 11 genes known as sigma factors demonstrated to be functional in virulence or niche adaptation were retrieved from various databases such as UniProt and NCBI by BLASTn and BLASTp analyses for E cloacae ATCC 13047 as a reference strain (Table 2). The 10 genes are RpoA, RpoB, RpoC, RpoD, RpoE, RpoF, RpoH, RpoS, RpoN, and RpoZ. Gene sequences of these selected sigma factors were used in BLAST against E asburiae JCM6051, E nimipressuralis CIP 104980, E hormaechei ATCC49162, E kobei JCM 8580, and E ludwigii EN-119 on the RAST server to identify these genes in the 5 genomes. It was noted that all sigma factor genes were present among all the genomes of E asburiae, E nimipressuralis, E hormaechei, E kobei, and E ludwigii, except RpoS, which was absent in E kobei (Table 2). RpoS is a conserved stress regulator that plays a critical role in survival under stress conditions in Escherichia coli and other γ-proteobacteria. 16 RpoS is also involved in the virulence of many pathogens, including Salmonella and Vibrio species. The lack of RpoS in the E kobei genome difference might result in substantial changes in RpoS-regulated gene expression. 16 The absence of the RpoS sigma factor in E kobei may also lead to missing functions, as RpoS is critical in general stress responses and can not only function in promoting survival during environmental stresses but also prepare the cell for stresses, so its absence might have a negative impact on bacterial survival.
Genome-wide prediction of sigma factors in Enterobacter cloacae complex.

Protein domains and family identification
Proteins can be composed of single or multiple domains, and similar domains can be found in proteins with different functions. Information retrieved from the domain annotation indicated that a number of sigma factor proteins contain the same domains. For example, sigma factor RpoA contains the RNAP_alpha_NTD domain and the RNA pol L protein family in the 6 strains compared in this study. The N-terminal domain of the α-subunit of bacterial RNAP (Table S1) is essential for RNAP assembly and basal transcription in vivo and in vitro. 17 Similarly, many other sigma factors contain similar domains and belong to the same protein families. The conservation in protein domains and protein families determines the function and evolutionary relationships of the Ecc. Almost all sigma factors from E asburiae JCM6051, E nimipressuralis CIP 104980, E hormaechei ATCC49162, E kobei JCM 8580, and E ludwigii EN-119 have similar domains, which perform similar and important functions.
Functional characterization of genes
Bacteria respond to changing environmental conditions by switching the global pattern of expressed genes. A key mechanism for global transcriptional switches depends on sigma factors that bind the RNAP core enzyme and direct it towards the appropriate stress response genes. 11 Functional annotation of sigma factors retrieved from 6 strains in the Ecc was conducted in various ways, including the AmiGO 1.8 tool, which was used for the analysis of GO data and the literature. BLAST results were filtered against all data sources, all species, all gene product types, and all gene product associations in the form of a high-scoring gene product list, which identifies their cellular component, molecular function, and biological processes (Table S2). Some of the sigma factors, eg, RpoA, RpoB, RpoC, RpoD, RpoE, and RpoZ, were identified to play molecular functions such as protein binding, whereas RpoS and RpoN were exceptional and identified to function as sigma factors (Table S2). A housekeeping sigma factor (σ70 in Escherichia coli, σ A in Bacillus subtilis) is required for most transcription during growth, whereas other sigma factors act as master regulators for stress responses, such as heat shock or entry in to stationary phase (σ H and σ S , respectively, in E coli) or for developmental programmes such as growth of flagella (σ F in E coli) and sporulation (σ H , σ F , σ E , σ G , and σ K in B subtilis). 18 In this study, which is consistent with studies in the literature, it seems that all important sigma factors are found in these 6 Ecc strains, indicating the relatedness of their functions. RpoS is exceptionally absent from E kobei. It was reported that sigma factor RpoS is the master regulator of the general stress response and involved in multiple signal integration in the Enterobacteriaceae family and other related bacteria. 19 It seems that absence of RpoS may lead to the failure of stress regulation and activation of other RpoS-dependent genes in E kobei.
Evolutionary association of sigma factor proteins by sequence alignment
Multiple sequence alignment and visualization was performed using MUSCLE and Heatmap to identify regions of similarity and score the sigma factors of E asburiae JCM6051, E nimipressuralis CIP 104980, E hormaechei ATCC49162, E kobei JCM 8580, E cloacae ATCC 13047, and E ludwigii EN-119 that may be a consequence of structural, functional, or evolutionary relationships between the sequences. Figure 1 shows the graphical representation of the sequence alignment via heatmap. Those sequences which showed 100% similarity are represented in same colour, eg, RpoA showed 100% similarity among all strains, except RpoA encoded by E kobei, which had 99% similarity. Similarly, all sigma factors displayed 100% to 99% similarity among all strains. RpoE is exceptional, with minimum similarity, as shown in Figure 1. Distance values range from 0.0000 to 0.0089, which are depicted by the gradient of colours ranging from dark red (lowest distance value indicating high similarity between genes) to green (highest distance value indicating low similarity between genomes) (Figure 1). Members of the same family are expected to share a common evolutionary history and thus at least some functional aspects. 20 Comparison of the proteins encoded in the 6 different genomes form 3 main clusters, as reported by Hoffmann and Roggenkamp, 21 and multiple clades, which illustrates the evolutionary lineages between these species (Figure 2).
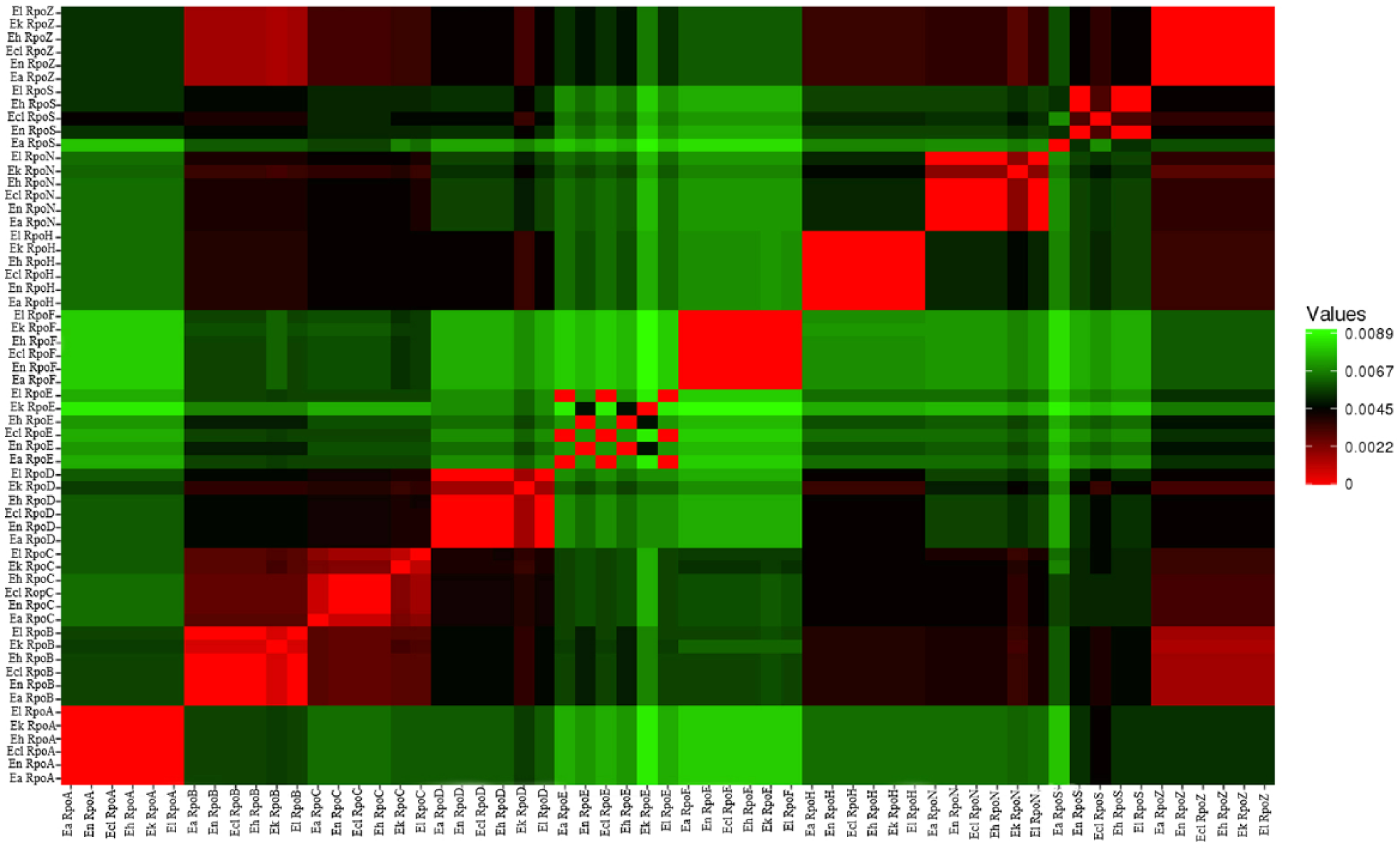
Heatmaps of sigma factors showing percentage similarities between each other and with reference genome. The heatmap shows the pairwise gene conservation distances of these strains. Dendrograms across the top and left of the heatmap show the relationship of genes based on genes conservation. The strain names are indicated to the right and bottom of the heatmap.

Evolutionary tree of Enterobacter cloacae complex showing similarities and differences between each other.
Evolutionary and epidemiological studies have predicted that Enterobacteriaceae involved in extraintestinal infections possess virulence-associated characteristics. 22 The Ecc can be considered an emerging group of pathogens, of which E hormaechei is the most commonly isolated nosocomial pathogen.23,24 Previous studies have demonstrated that most E hormaechei carried genes that encoded resistance to aminoglycosides and third-generation cephalosporins and reduced susceptibility to fluoroquinolones. 24 Similarly, E cloacae have an intrinsic resistance to ampicillin and cephalosporins. 22 Thus, similar to E hormaechei and E cloacae, it might be possible that sigma factors from E asburiae, E nimipressuralis, E kobei, and E ludwigii may have virulence-associated properties and are resistant to antibiotics. Variation in antibiotic resistance is also responsible for wide variations in protein structure. 25
Conclusions
In conclusion, advancements in NGS technologies have provided a wealth of genomics data with the availability of many new bacterial genome sequences. The computational prediction of sigma factors in 5 Ecc genomes will be useful for identifying new pathogenicity-associated genes and their mechanism within the genomes of bacteria that contain sigma factors. Our results show that the identification of sigma factors and their sequence similarity may indicate similar roles of these proteins and their evolutionary association. However, the absence of RpoS in E Kobei indicates that RpoS may have been independently acquired and play different roles relating to pathogenicity, host range determination, and/or niche adaptation. Thus, comparing the results from a few different genomes and constructing a short list of common hits may be the most effective way to compare bacterial genome sequences.
Footnotes
Author Contributions
FN and MI performed experiments for sequencing, annotation and drafting the manuscript. GZ, AH and AMY helped in drawing Phylogenetic tree as well as protein domain analysis. ZB and MI designed the experiment.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from Zhejiang National Natural Science Foundation of China (LY17C010006) and Opening foundation of the State Key Laboratory for Diagnosis and Treatment of Infectious Diseases and Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital Medical College, Zhejiang University (2016KF10).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.