
Abstract
Background:
Leber hereditary optic neuropathy (LHON) is an inherited progressive optic neuropathy usually caused by mitochondrial DNA mutations. Recently, autosomal recessive (arLHON), which is caused by biallelic mutations in the DNAJC30 gene (usually c.152A > G), has been described. The onset of LHON before the age of 12 is uncommon and it is typically associated with a more variable clinical course and a more favorable visual prognosis than adult-onset LHON.
Materials and Methods:
Detailed clinical findings of a female child with vision loss due to arLHON together with choroideremia (CHM) carrier state are presented.
Results:
Genetic testing for the three most common mitochondrial LHON pathogenic variants was negative. On suspicion of arLHON, genetic testing was continued with the next-generation sequencing (NGS) of the nuclear DNA, identifying a homozygous pathogenic variant in DNAJC3°c.152A > G, p.(Tyr51Cys), but no alterations in the CHM gene. Idebenone treatment was started 4.5 months after the first evaluation. Clinical diagnosis of the CHM carrier state was confirmed by multiplex ligation-dependent probe amplification (MLPA) assay, which revealed a heterozygous deletion of all exons of the CHM.
Conclusions:
In children with acute or subacute, simultaneous, or sequential vision loss that is unresponsive to immunomodulatory treatment, LHON should be considered as a possible diagnosis. Our case emphasizes the diagnostic advantage of sequencing DNAJC30 in parallel with the mitochondrial DNA, especially in Eastern European descent patients. Genomic rearrangement testing should be considered for patients with a CHM carrier phenotype who have negative results on sequencing tests.
Keywords
Introduction
Leber hereditary optic neuropathy (LHON) is one of the most frequent inherited optic neuropathies and one of the most common inherited mitochondrial diseases, usually caused by one of the three pathogenic variants of mitochondrial DNA (mtDNA), m.3460G > A, m.11778G > A, or m.14484T > C.1,2 Recently, an autosomal recessive form of LHON (arLHON) has been described, caused by biallelic mutations in the DNAJC30 gene (typically a missense mutation NM_032317.2 c.152A > G, NP_115693.2 p.Tyr51Cys), dividing LHON into arLHON and mitochondrial LHON (mtLHON). 3 The homozygous p.Tyr51Cys DNAJC30 variant has been estimated to account for up to 27% of genetically diagnosed LHON families in the founder population of Eastern Europe and up to 5% of families in non-founder populations. 4 Although data are limited, recent reports suggest that the arLHON patient subgroup has a higher male prevalence, earlier age of disease onset, and a higher rate of idebenone-treated and spontaneous recovery than mtLHON patients.3,4 The phenotypic presentation of arLHON does not differ from mtLHON, with the subacute phase followed by an atrophic chronic stage about one year after onset. In the subacute stage, pseudopapilledema with circumpapillary telangiectatic microangiopathy and swelling of the retinal nerve fiber layer (RNFL) without leakage at fluorescein angiography occur. As the disease progresses, retinal ganglion cell layer (RGCL) loss leads to temporal optic atrophy, which eventually becomes diffuse. 3
Disease onset before the age of 12 is estimated to affect around 8–10% of the total mtLHON population. It tends to have a better visual prognosis and a different natural history of visual loss progression. 5 Although most children with mtLHON present with classic acute visual loss, an unexpectedly large number of children have an insidious, subclinical, and slowly progressive presentation, with diagnostic delays of 3–15 years. 6
Choroideremia (CHM; Online Mendelian Inheritance in Man identifier [OMIM], #303100) is a rare X-linked recessive chorioretinal dystrophy that causes progressive degeneration of the choriocapillaris, retinal pigment epithelium (RPE), and photoreceptors of the retina in the affected males. The first symptom is typically nyctalopia in childhood, followed by visual field constriction in early adulthood, and eventually legal blindness by the fifth decade of life. The prevalence of CHM is estimated to be between 1 in 50 000 and 1 in 100 000 individuals, with the highest in Finland. 7 Carrier females of CHM are typically asymptomatic but may be recognized by characteristic patchy areas of pigmentary mottling of varying severity, which can be reliably observed with fundus autofluorescence (FAF) imaging. This phenotype is consistent with random X-chromosome inactivation. 8
The CHM gene, implicated in choroideremia, contains 15 exons spanning approximately 150 kb of genomic DNA and is ubiquitously expressed. 9 CHM exhibits high genetic heterogeneity, with the major pathogenic variants being small sequence variants (small intragenic deletions/insertions and missense, nonsense, and splice site variants). Up to 20% of patients have single-exon, multiexon, or whole-gene deletions, which may not be detected by exon sequencing methods. 10
To the best of our knowledge, this is the first report of LHON in a choroideremia carrier.
Case report
An 11-year-old Eastern European descent girl was admitted to our hospital due to subacute vision loss, noted shortly after the summer holidays when the child was unable to read. The child's medical history revealed no prior vision problems or eye disease. Family history revealed that the child's father (unavailable for investigation) was blind due to presumed retinal dystrophy. There was no ocular pain with eye movement, headache, or other neurological symptoms. At presentation, her best-corrected visual acuity (BCVA) was 20/400 in the left eye and 20/400 in the right eye. Using Ishihara pseudoisochromatic color plates she could only identify 1 of 12 test plates with both eyes. The cover test revealed a mild alternating exotropia. Fundus examination showed moderate bilateral optic disc pseudo-edema with hyperemia and circumpapillary telangiectatic microangiopathy, more evident in the left eye. Additionally, patchy peripheral pigmentary mottling was found in both eyes. FAF imaging (Zeiss Clarus 500, Carl Zeiss Meditec AG, Jena, Germany) showed corresponding hypo-autofluorescent speckles symmetrically in both eyes. A horizontal spectral-domain optical coherence tomography (SD-OCT) scan (Spectralis, Heidelberg Engineering, Heidelberg, Germany) through the macula demonstrated subtle abnormalities in the interdigitation and ellipsoid zone in both eyes. At the same time, the SD-OCT of the optic discs showed RNFL swelling (Figure 1) in both eyes.
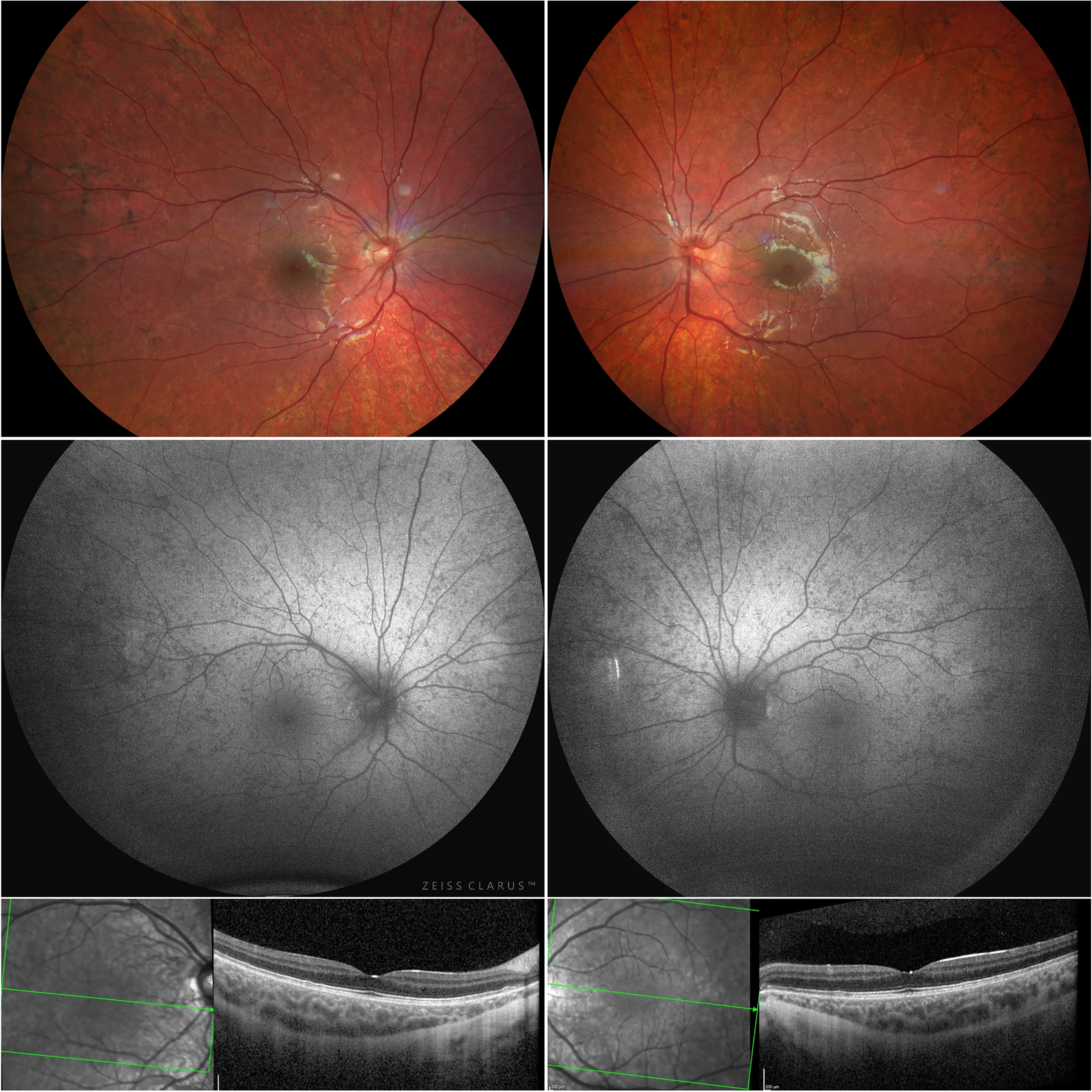
Wide-angle fundus images showed pseudopapilledema and granular, patchy hyperpigmentation (upper panel) at presentation. The corresponding FAF images demonstrated speckled pattern hypo-autofluorescent lesions throughout the fundus (middle panel), while the SD-OCT B-scans through the fovea showed subtle changes in the IZ and EZ (lower panel). Abbreviations: FAF, fundus auto-fluorescence; SD-OCT, spectral-domain optical coherence tomography; IZ, interdigitation zone; EZ, ellipsoid zone.
Brain magnetic resonance imaging (MRI) revealed hyperintensity in the posterior portion of both optic nerves and optic chiasm, with enlargement of the chiasm on T2-weighted images (Figure 2). The remainder of the brain was normal. Based on these results, a diagnosis of chiasmal optic neuritis was made. The results of all other clinical investigations, including aquaporin-4 antibody testing, anti-myelin oligodendrocyte protein antibody testing, cerebrospinal fluid (CSF) composition analysis, and CSF cytology for viral or bacterial etiology, were negative. Despite receiving 1 g of intravenous methylprednisolone daily for five days, her visual acuity did not improve. Treatment was then continued with intravenous immunoglobulin (IVIG), which was also ineffective.
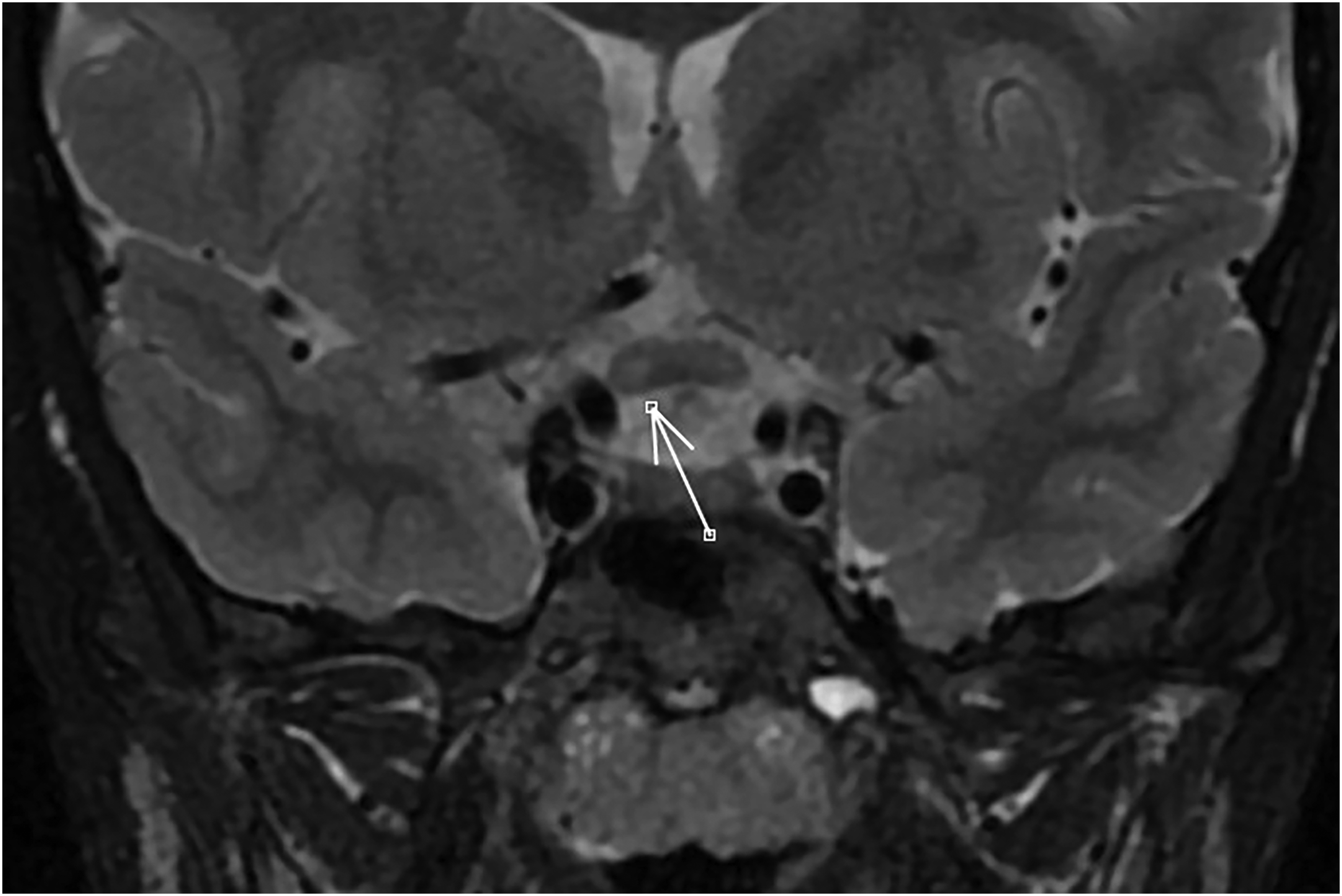
Magnetic resonance imaging T2-weighted brain scans demonstrated central hyperintensity in the posterior portion of both optic nerves and the optic chiasm, and enlargement of the chiasm (more accentuated on the right side (arrow)).
The electrophysiological testing showed delayed latencies with low amplitudes of the visual evoked potentials (VEPs), while the full-field electroretinogram (ERG) was normal. The finding of optic pathway dysfunction, combined with the patient's clinical presentation and lack of response to treatment, raised the suspicion of LHON.
On follow-up examination three weeks later, BCVA was 20/200 in both eyes. Color vision had slightly improved. There was no afferent pupillary defect. On fundoscopic examination, the optic discs were slightly less hyperemic.
Genetic testing results were negative for typical LHON pathogenic variants and the whole mtDNA sequencing. By then, 3 months after the first evaluation, the visual acuity had recovered to 20/70 in the right eye and 20/40 in the left eye. The pseudopapilledema had evolved to mild temporal paleness of the discs. Color vision was normal in the left eye and slightly reduced in the right. On suspicion of autosomal recessive LHON, genetic testing was continued with the next-generation sequencing (NGS) panel testing of the nuclear DNA for known optic neuropathy and retinal dystrophy causal genes. This identified a homozygous pathogenic DNAJC30 variant c.152A > G, p.(Tyr51Cys) and confirmed the diagnosis four months after the first referral. Subsequently, idebenone treatment was initiated at a dosage of 300 mg three times daily (TID), as recommended by the manufacturer. At the time of initiation of the treatment (4.5 months after first referral), the BCVA was 20/50 in the right eye and 20/32 in the left eye. Despite progressive RNFL and RGCL thinning and optic nerve pallor, BCVA and visual field defects continued to improve during the follow-up period of 11 months (Figure 3).
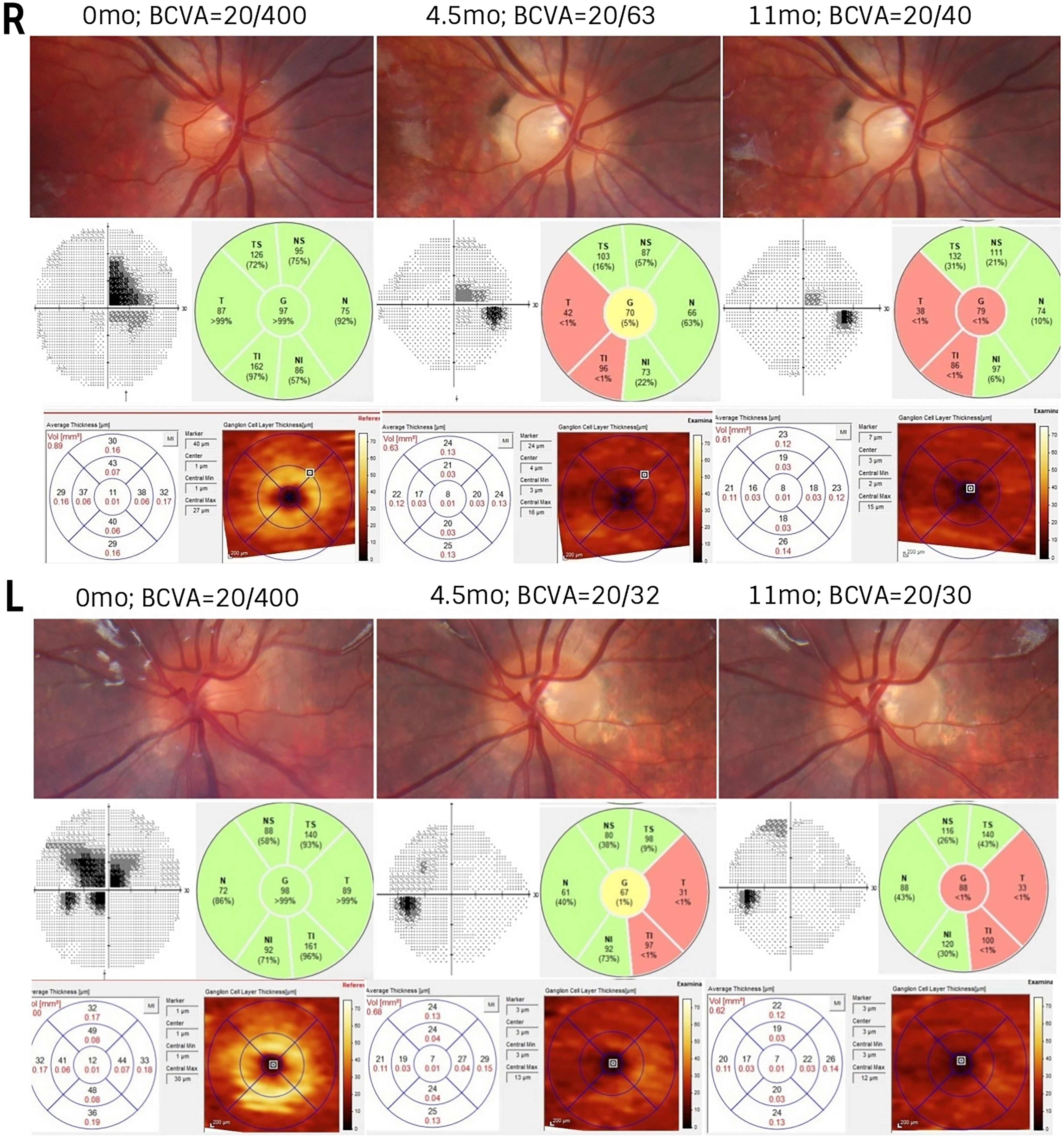
Optic nerve photographs at presentation, 4.5 months, and 11 months (top panels). Corresponding visual fields showed central scotoma in both eyes at presentation with moderate improvement 4.5 and 11 months later (middle panels, left side); SD-OCT demonstrated progressive thinning of the RNFL (middle panels, right side) and RGCL (bottom panels) in both eyes. Abbreviations: BCVA, best corrected visual acuity; L, left eye; R, right eye; RNFL, retinal nerve fiber layer; RGCL, retinal ganglion cell layer; mos, months.
Although there was a strong suspicion of CHM due to paternal family history and retinal appearance, the NGS gene panel testing did not reveal any point mutations in CHM. Because of this, copy-number variant analysis of CHM was further carried out. CHM carrier state was confirmed by a multiplex ligation-dependent probe amplification (MLPA) assay, which revealed a heterozygous deletion of all the exons (1–15) of the CHM.
Discussion
LHON is an inherited form of vision loss, which leads to the selective loss of retinal ganglion cells and axons, particularly of the papillomacular bundle. 11
Childhood-onset LHON is a rare condition that can be easily misdiagnosed as other types of optic neuropathies in its early stages. It is essential to discriminate between LHON and other optic neuropathies early on to avoid potentially long diagnostic delays, as different treatment modalities are available. 12
Despite all investigations for various causes of acute optic neuropathy being negative in our case, immunomodulatory therapies with high-dose systemic steroids (intravenous methylprednisolone) and IVIG were initiated. The lack of treatment response, which is typically expected in optic neuritis, along with VEP results that suggested neuropathy, raised the suspicion of LHON.
Radiographic features found in our patient, such as T2 hyperintensity in the posterior portion of both optic nerves and the optic chiasm, and enlargement of the chiasm, have been occasionally reported in LHON 13 but may be easily mistaken for demyelinating optic neuritis or chiasmal neuritis. Interestingly, the hyperintensity was more intense on the right side at the chiasm, despite the more severe pseudopapilledema of the left optic disc at presentation. This could be explained by the crossing fibers in the chiasm. Overall, the left eye had slightly better visual outcomes during the follow-up period.
Although the patient had RPE mottling and a family history of vision loss from a possible inherited retinal disease, the clinical presentation and test results did not suggest that retinopathy contributed to the vision loss. FAF irregularities in female carriers of CHM are specific and can help to diagnose the condition, even when the findings on funduscopy are subtle or missed. Given the patient's typical CHM carrier phenotype on FAF (Figure 4), an MLPA assay was performed, after the targeted NGS gene panel did not detect any pathogenic variants in the known causative genes associated with inherited retinal dystrophies. This confirmed the patient's carrier state.
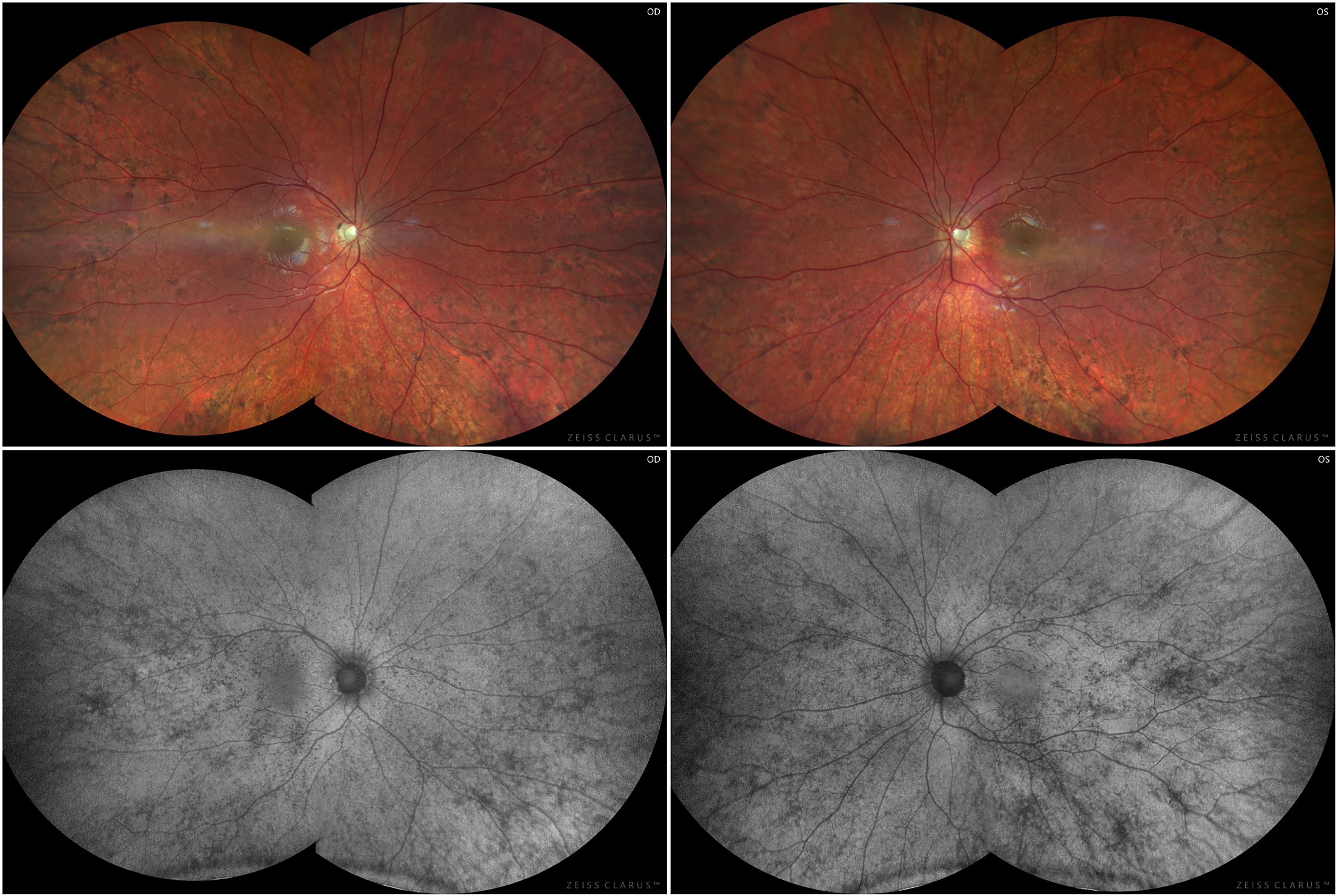
Ultrawide fundus photography 11 months after presentation demonstrated temporal optic nerve pallor in both eyes (top panels), with the corresponding ultrawide FAF images highlighting widespread hypo-autofluorescent fleck-like lesions (bottom panels).
Idebenone (Raxone, idebenone 150 mg tablets, Santhera Pharmaceuticals, Pratteln, Switzerland) is the first and currently the only approved treatment (European Medicines Agency (EMA) approval in the European Union and some other countries) for adults and adolescents with LHON since 2015. Even though there are controversies on the dosage of idebenone in patients under 12, growing evidence suggests its safety also in paediatric cases. 14 The treatment is known to be most effective when started early, as the visual loss in LHON is directly linked to the injury and death of retinal RGCL, which is particularly sensitive to intracellular metabolic defects because of its high energy demand and the long course of the cell axons. 15
A higher rate of recovery, both with idebenone treatment and spontaneously, has been reported in arLHON and in children with mtLHON compared to adult onset. Idebenone treatment was started 4.5 months after the first presentation in our patient. The treatment could have been initiated two months earlier if both nuclear DNA and mtDNA sequencing had been performed simultaneously. In our patient, significant spontaneous visual acuity improvement had occurred by the time of treatment initiation. During the 8-month treatment follow-up, the visual acuity further improved slightly.
In conclusion, this case report describes the clinical findings of a child of Eastern European ancestry with choroideremia carrier status who presented with vision loss due to arLHON. We emphasize that LHON should be considered as a possible diagnosis in children with sudden or gradual, simultaneous or sequential vision loss that does not respond to immunomodulatory treatment. Our case highlights the benefit of sequencing DNAJC30 in parallel with mitochondrial DNA for diagnosing LHON, especially in patients of Eastern European descent. Additionally, genomic rearrangement testing, which can detect larger deletions and duplications, should be considered in negative cases when a patient has a CHM carrier phenotype.
Footnotes
Acknowledgements
The authors thank Turman Eye Clinic for technical assistance and the patients’ family for their kind cooperation.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Statement on ethics
Written informed consent was obtained from the patient for the preparation of this work.