
Abstract
Introduction
Gyrate atrophy (GA) is a rare retinal dystrophy due to biallelic pathogenic variants in the ornithine aminotransferase (OAT) gene, causing a 10-fold increase in plasma ornithine levels. It is characterized by circular patches of chorioretinal atrophy. However, a GA-like retinal phenotype (GALRP) without elevated ornithine levels has also been reported. The aim of this study is to compare the clinical characteristics of GA and GALRP and to identify possible discriminators.
Methods
A multicenter, retrospective chart review was performed at three German referral centres on patient records between 01/01/2009 and 31/12/2021. Records were screened for patients affected by GA or GALRP. Only patients with examination results for plasma ornithine levels and / or genetic testing of the OAT gene were included. Further clinical data was gathered where available.
Results
Ten patients (5 female) were included in the analysis. Three suffered from GA, while seven had a GALRP. Mean age (± SD) at onset of symptoms was 12.3 (± 3.5) years for GA compared with 46.7 (± 14.0) years for GALRP patients (p = 0.002). Mean degree of myopia was higher in GA (−8.0 dpt. ± 3.6) compared to GALRP patients (−3.8 dpt. ± 4.8, p = 0.04). Interestingly, all GA patients showed macular oedema, while only one GALRP patient did. Only one patient with GALRP had a positive family history, while two were immunosuppressed.
Discussion
Age of onset, refraction and presence of macular cystoid cavities appear to be discriminators between GA and GALRP. GALRP may encompass both genetic and non-genetic subtypes.
Introduction
Gyrate atrophy (GA) is a rare, autosomal recessive disorder that is caused by biallelic pathogenic variants in the ornithine aminotransferase (OAT) gene, resulting in a deficiency of the enzyme ornithine ketoacid aminotransferase and a subsequent 10- to 20-fold increase in plasma ornithine levels.1–3 The estimated prevalence is approximately 1:1,500,000, 4 suggesting that approximately only about 500 patients in Europe and 56 in Germany may be affected by the disease. Although an accumulation of ornithine can be observed in many tissues, it primarily affects the eye, 5 in particular the retinal pigment epithelium (RPE). 6 The clinical hallmark feature of the disease is extensive atrophy of the neuroretina, RPE, and choriocapillaris (CC) in the midperiphery, which may also affect the centre of vision in later stages of the disease. 7 Patients usually also suffer from high myopia and early development of posterior subcapsular cataract. 3 A low-arginine diet has been shown to prevent retinal degeneration in a mouse model of the disease, 8 and is thus usually recommended in patients with GA. In addition, pyridoxine supplementation (variant-dependent) and/or lysine supplementation are discussed as viable options to lower plasma ornithine levels. 9 As a treatable entity, GA is thus an extremely rare, yet important differential diagnosis that should not be overlooked.
One of the most important differential diagnoses of GA is a gyrate-atrophy like retinal phenotype (GALRP) in patients with normal plasma ornithine levels. 10 Kellner and colleagues were among the first to report a father and two of his sons who had a phenotype resembling GA and normal plasma ornithine levels, suggesting a genetic background of this disease. Recently, a family affected by a missense variant in the C1QTNF5 gene, a gene previously described only for late-onset retinal degeneration (L-ORD), was reported to show a GALRP rather than a L-ORD phenotype. 11 Thus, there appear to be at least two distinct disease entities: treatable GA with high plasma ornithine levels and, in some cases monogenetic, GALRP with normal plasma ornithine levels that is as yet untreatable. The aim of this study was to examine patients with GA and / or GALRP presenting to three large eye centres and to describe similarities and differences that may help in the differential diagnosis to ensure that the diagnosis of GA is not overlooked.
Methods
Patients
This is a multicentre, retrospective, non-interventional study conducted at the Eye Centre of the University Hospital Freiburg, the Eye Centre of the St. Franziskus Hospital Münster and the Department of Ophthalmology of the University Hospital Cologne. Patients with a clinical diagnosis of GA or GALRP seen at the above mentioned centres between 01/01/2009 and 31/12/2021 were included in the study. Only patients on whom genetic testing was performed or for whom plasma ornithine levels were available were included in this study. Complete multimodal imaging was reviewed independently by two experienced retina specialists and phenotypes that were not consistent with GALRP were excluded. The remaining patients were included in the analysis. Ethics approval for a retrospective analysis of the data at the Freiburg site was obtained from the local Ethics committee (application number 22-1288-S1-retro) and written informed consent was obtained for patients from the Muenster and Cologne sites.
Imaging and data acquisition
Clinical data, including multimodal imaging and electrophysiology, were collected from all patients. This included a detailed history (e.g., age of onset of symptoms, age of presentation, family history suggestive of retinal dystrophies, systemic or ocular comorbidities) and clinical examination (including best-corrected visual acuity (BCVA), objective refraction, other ophthalmic pathology such as presence of macular oedema or pseudophakia). Patients usually underwent Goldmann visual field testing of isopters I.1, I.2, I.3, I.4 and III.4 or automated static perimetry (Humphrey 30-2). Imaging modalities included spectral domain optical coherence tomography (SD-OCT, Heidelberg Spectralis 2, Heidelberg, Germany), fundus autofluorescence (FAF, Heidelberg Spectralis 2, Heidelberg, Germany or Optos California, Optos, Marlborough, MA, USA), fundus photography (Carl Zeiss FF 450 Plus Fundus Camera, Carl Zeiss Meditec AG, Oberkochen, Germany), Optos images (Optos California, Optos, Marlborough, MA, USA) or Clarus images (Zeiss Clarus 500, Carl Zeiss Meditec AG, Oberkochen, Germany) where available. Electrophysiological testing (full-field electroretinogram (ffERG)) was performed according to ISCEV standards.
Plasma ornithine levels
Plasma ornithine levels were measured either at one of the eye centres, specialized metabolic clinics or by the patient's respective general practitioner.
Genetic testing
For patients 4, 5 and 7, molecular genetic screening of the OAT and the C1QTNF5 genes was performed by bidirectional Sanger sequencing of coding exons and exon/intron boundaries. Primers used for PCR amplification and Sanger sequencing are available upon request. Patients 3 and 10 had undergone next-generation sequencing (NGS) of a targeted sequencing panel for retinal disease as part of their standard of care at the respective institutions. Samples from all patients were recruited in accordance with the principles of the Declaration of Helsinki and were obtained with written informed consent accompanying the patients’ samples.
Confirmation of respective diagnosis
All patients included in this study had their plasma ornithine levels determined that were highly elevated in all GA patients and within normal limits for all GALRP patients. In 1 GA patient (patient 3) as well as 4 GALRP patients (patients 4, 5, 7 and 10), additional genetic testing performed. GA patients 1 and 2 were referred to specialist metabolic clinics, where the diagnosis was confirmed by multiple plasma ornithine measurements and enzymatic activity of ornithine ketoacid aminotransferase measured in fibroblast cell culture.
Statistical analysis
Decimal BCVA was converted to logarithm of the minimum angle of resolution (logMAR BCVA) to allow for statistical analysis using the formula logMAR BCVA = -log(decimal BCVA). 12 Spherical equivalents of the objective refraction values of each patient were calculated by adding the spherical power to half the cylindric power. 13 Statistical analysis was performed using R software. 14 One-sided student t tests for unpaired samples were performed on age of onset, age of presentation, logMAR BCVA and spherical equivalents for both groups. p values of 0.05 or less were considered statistically significant.
Results
Patients
Patients from the three eye centres with a diagnosis of GA or GALRP who had undergone either genetic testing and / or measurements of plasma ornithine levels to confirm the respective diagnosis were included in this study. A total of 10 patients were eligible. Three patients were diagnosed with GA based on repeated severely elevated plasma ornithine levels and / or biallelic pathogenic variants in the OAT gene, while seven presented with a GALRP. Of those seven, all showed normal plasma ornithine levels and four had undergone diagnostic genetic testing, which did not reveal any pathogenic mutations in either the OAT or the C1QTNF5 gene. The other three patients were either unable or unwilling to have genetic testing performed and the diagnosis was based on plasma ornithine levels within the normal range. A full summary of patient characteristics is presented in Table 1 (demographics and medical history) and 2 (ophthalmic examination). Five patients were male (2 GA and 3 GALRP), whereas five were female (1 GA and 4 GALRP). Mean age of onset of night blindness as a first symptom was significantly lower in GA patients compared with GALRP patients (12.3 (± 3.5) years vs. 46.7 (± 14.0) years, p = 0.002). The same applies to the mean age at first presentation at the respective institution, which was 24.3 (± 14.2) years for GA compared with 53.7 (± 11.2) years for GALRP (p = 0.004).
Demographic characteristics and patient history of participants.
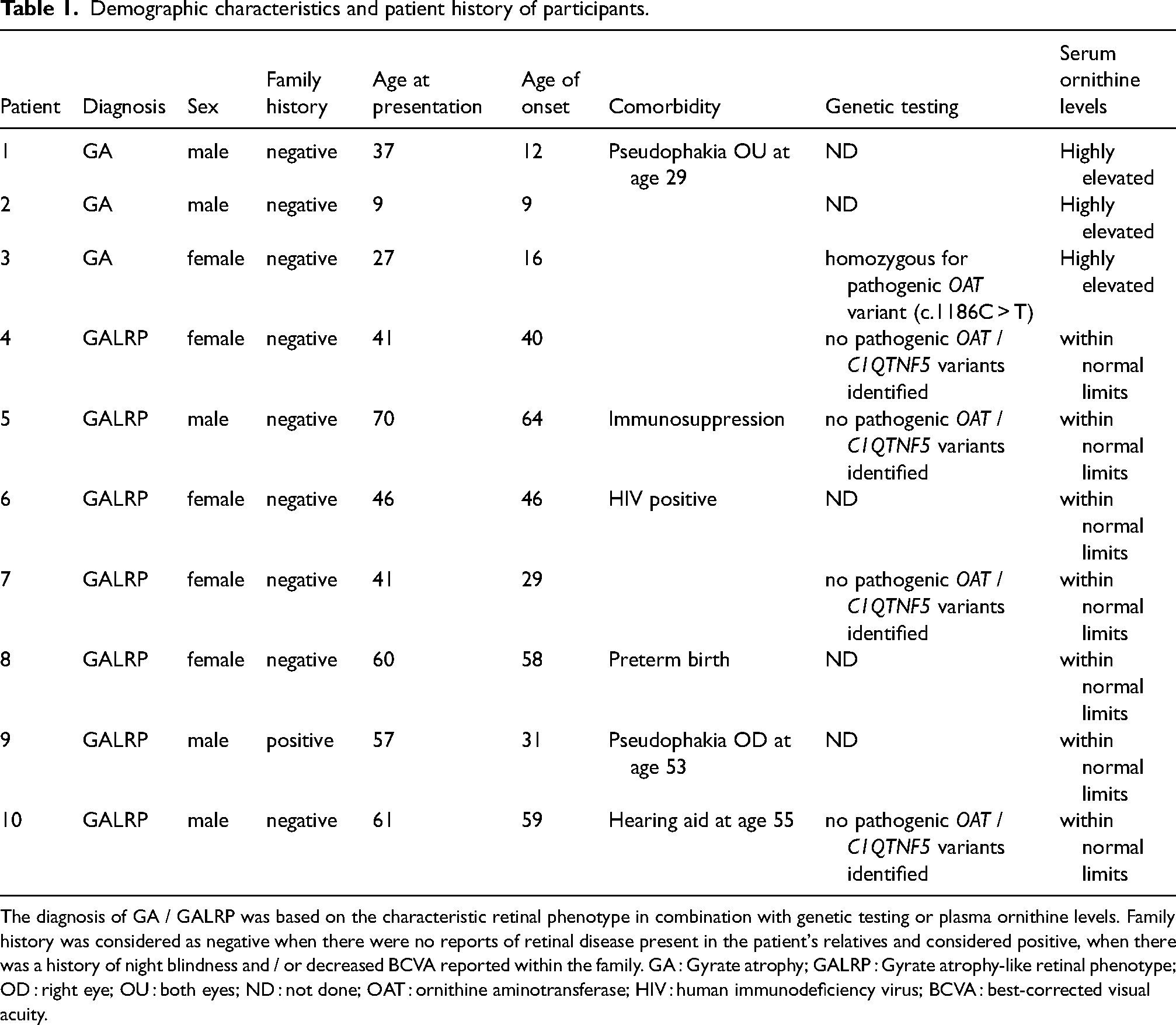
The diagnosis of GA / GALRP was based on the characteristic retinal phenotype in combination with genetic testing or plasma ornithine levels. Family history was considered as negative when there were no reports of retinal disease present in the patient's relatives and considered positive, when there was a history of night blindness and / or decreased BCVA reported within the family. GA : Gyrate atrophy; GALRP : Gyrate atrophy-like retinal phenotype; OD : right eye; OU : both eyes; ND : not done; OAT : ornithine aminotransferase; HIV : human immunodeficiency virus; BCVA : best-corrected visual acuity.
LogMAR BCVA was lower in GA patients (0.8 ± 0.6, ∼ decimal BCVA of 0.16) compared with GALRP patients (0.6 ± 0.5, ∼ decimal BCVA of 0.25), although this difference was not statistically significant (p = 0.07). Mean degree of myopia was statistically higher in the GA group (−8.0 dpt. ± 3.2) compared with GALRP patients (−3.8 dpt. ± 4.8, p = 0.04), although one patient in each group had already undergone cataract surgery. With regards to the degree of cataract, most patients showed an incipient cataract, with one patient in the GA and two patients in the GALRP group showing a clear lens (see Table 2). However, the age of initial presentation has to be considered when interpreting these results.
Functional characterization of patients.
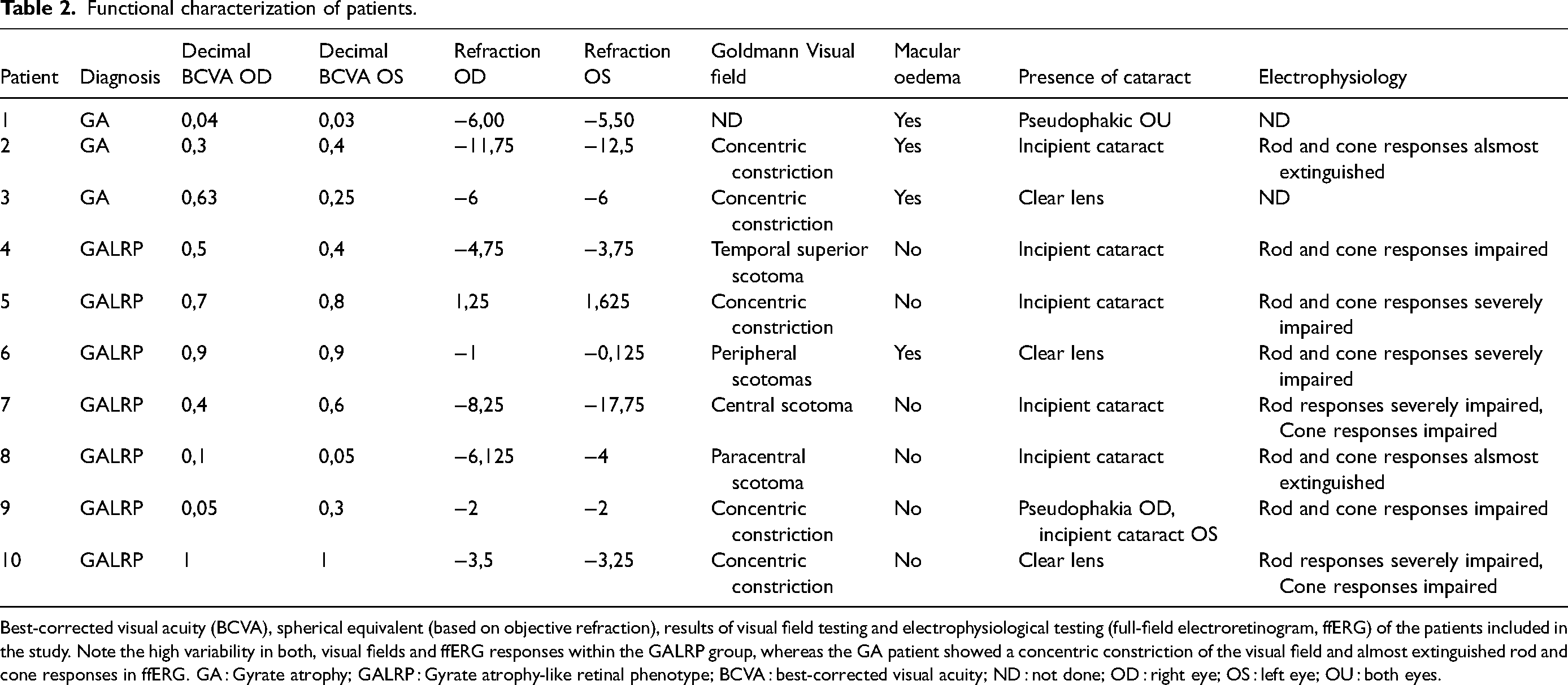
Best-corrected visual acuity (BCVA), spherical equivalent (based on objective refraction), results of visual field testing and electrophysiological testing (full-field electroretinogram, ffERG) of the patients included in the study. Note the high variability in both, visual fields and ffERG responses within the GALRP group, whereas the GA patient showed a concentric constriction of the visual field and almost extinguished rod and cone responses in ffERG. GA : Gyrate atrophy; GALRP : Gyrate atrophy-like retinal phenotype; BCVA : best-corrected visual acuity; ND : not done; OD : right eye; OS : left eye; OU : both eyes.
GALRP patients demonstrated considerably different clinical characteristics, including wide variability in the age at which night blindness occurred (29 to 64 years). Most patients reported no family history of retinal disease. One GALRP patient (Patient 9) reported that his sister also suffered from severe visual deterioration, but was never seen by an ophthalmologist and lived in Kazakhstan, so no clear link could be established.
Interestingly, two patients had some form of immunosuppression, one of them drug-induced due to a previous kidney transplant and one due to an HIV infection. One patient was a preterm infant; however, a history of retinopathy of prematurity was not reported.
Visual field testing
Visual field testing showed concentric constriction in GA patients, whereas this was observed in only three GALRP patients (Patient No 5, 9 and 10). Since there was less foveal sparing observed in GALRP, visual field defects more often affected the centre of vision. In all patients, both eyes showed a high degree of similarity.
Funduscopic appearance
Both diseases show a similar phenotype of extensive peripheral chorioretinal atrophy. Clinical phenotypes within the GALRP group appeared to be more heterogenous than within the GA group (Figure 1), with some patients showing central atrophy, whereas most GA patients showed foveal sparing. Both groups showed variability in the extent of atrophy and the degree of pigmentation. Interestingly, the optic disc was surrounded by chorioretinal atrophy in all three GA patients, whereas this was not the case in two GALRP patients (Pt. 5 and 6).
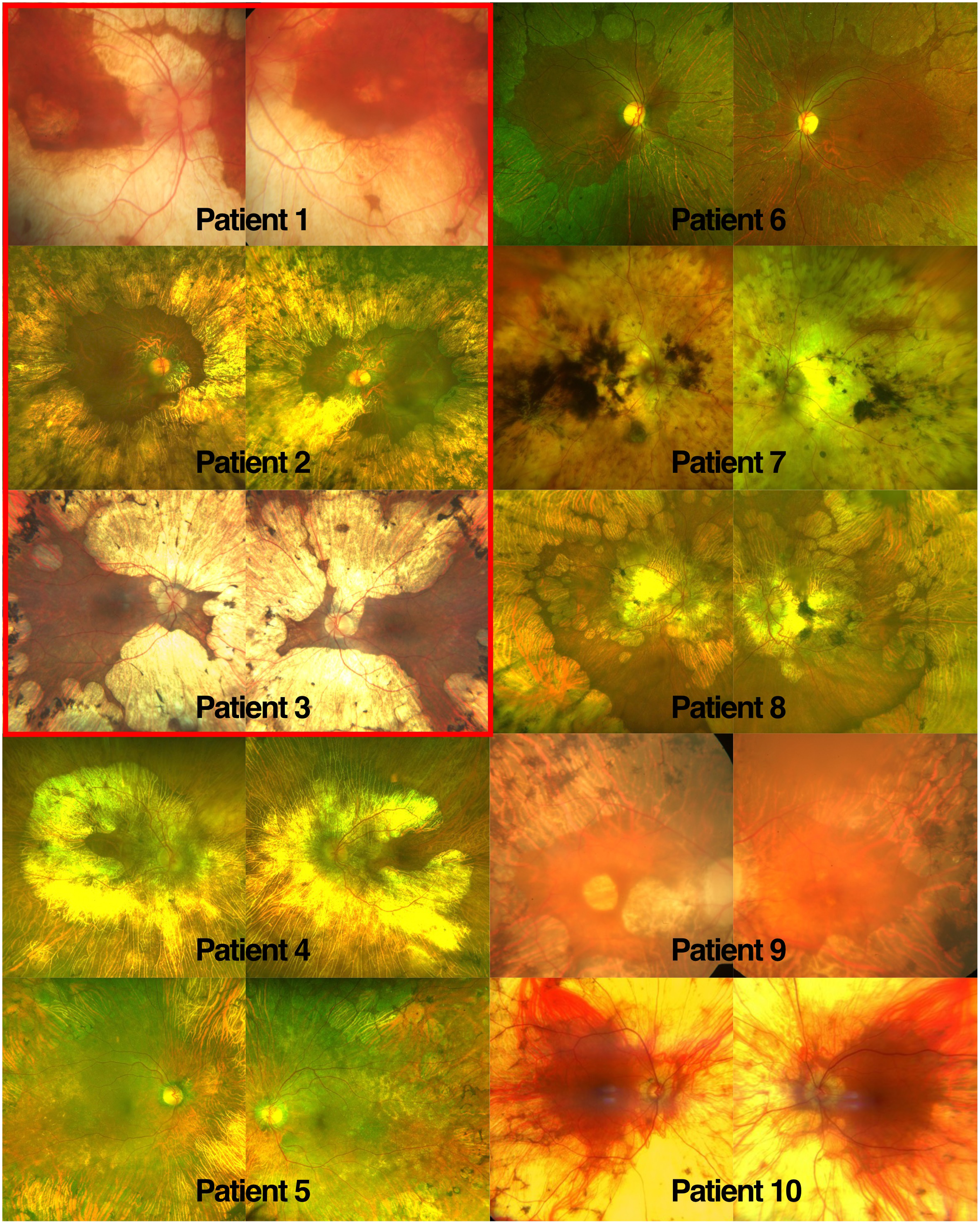
Retinal phenotype of GA and GALRP patients compared. Fundus images of the enrolled patients. Images from 1 to 3 belong to patients who suffer from Gyrate Atrophy, while images from 4 to 10 belong to patients who suffer from gyrate atrophy-like retinal phenotype. All patients showed extensive areas of chorioretinal atrophy with varying degrees of pigmentation within both the GA and the GALRP groups. Foveal sparing was present in two of the three GA patients (Pt. 2 and 3), whereas it was observed in four out of seven GALRP patients (Pt. 5,6, 8 and 10).
SD-OCT
SD-OCT revealed a macular oedema in all GA patients, whereas this was observed in only one patient with GALRP (Figure 2). The oedema of one GA patient (patient 2) was treated with topical dorzolamide 2% every eight hours and later with oral acetazolamide 250 mg every 12 h, but neither treatment was well tolerated and did not significantly reduce macular oedema. GA patients were also counselled on a low-arginine diet, which was followed by two of the three patients.
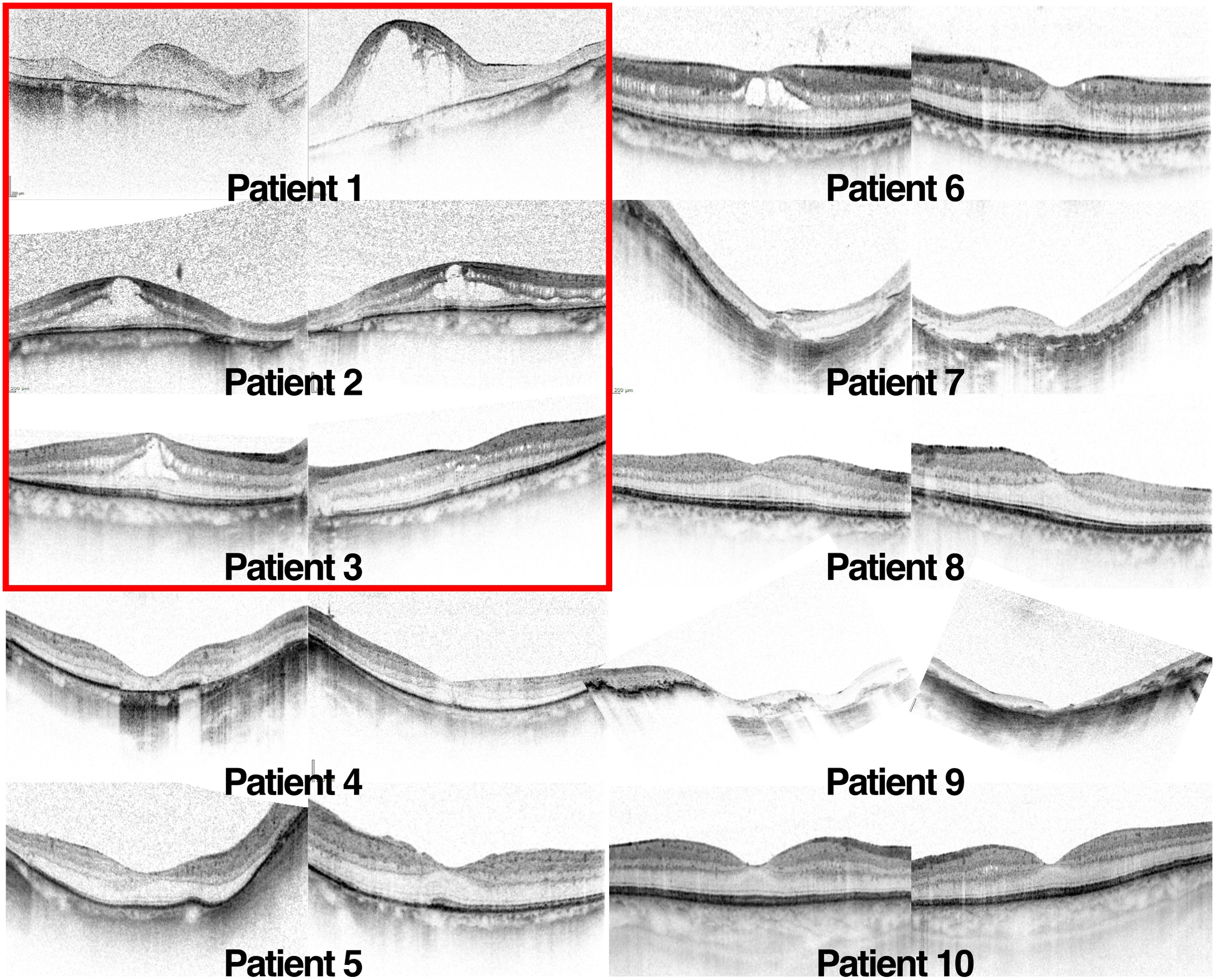
Optical coherence tomography of gyrate atrophy and gyrate atrophy-like retinal phenotype patients. All three GA patients (red box) showed a marked macular oedema with foveal involvement, whereas this was observed in only one GALRP patient (Pt. 6). However, macular atrophy was observed in three GALRP patients (4, 7 and 9), whereas it occured in only one GA patient (Pt. 1). Image quality and acquisition tended to be impaired due to the high degrees of myopia present in most of the patients included in this study.
FAF
FAF was severely reduced in the areas of chorioretinal atrophy in both GA and GALRP. In areas of coherent extensive atrophy, the outline of the underlying choroidal vessels was often visible (Pt. 2 - 4, 8 and 9). Patient 8 showed the highest FAF signal in the area of chorioretinal atrophy among all patients (Supplemental Figure 1), even though the funduscopic appearance did not differ significantly from the other patients.
Electrophysiology
FfERG was performed on one GA patient, who showed a severely impaired cone and rod function. The ffERG characteristics of GALRP patients varied from unremarkable to severely impaired cone and rod function (see Table 2). In general, rod responses appeared to be more severely affected than cone responses.
MfERG responses correlated well with the degree of central chorioretinal atrophy present and were generally well preserved when the central retina was spared.
Discussion
Our study highlights the phenotypic similarities between GA and GALRP (Figure 3) as well as the large heterogeneity especially within the GALRP group. As GA is a very rare but potentially treatable entity, a correct diagnosis for affected patients is of great importance. Therefore, the aim of this study was to identify key features that can aid the differential diagnosis of these two diseases.
Regarding the clinical presentation of the patients, we found that a higher mean age of onset of night blindness was observed in GALRP than in GA and that visual acuity was less severely affected. While the low age of onset of night blindness in GA is in line with previous reports, 3 some GALRP patients in our group had a higher age of onset compared with previous reports (Pt. 5, 8 and 10).10,11 Macular oedema was also less frequent in the GALRP group, similar to previous reports. 11 In contrast, all patients with GA had macular edema, which had also been observed in other GA patients. 15 Since only one case with GA revealed centre-involving photoreceptor atrophy on SD-OCT, the decrease in central vision in the other two GA patients was probably caused by a combination of the cystoid macular oedema, the high degree of myopia and / or the onset of cataract. Other macular changes previously reported to result in decreased central vision in GA, such as a macular hole, 16 a choroidal neovascularization, 17 or a significant epiretinal membrane, 18 were not observed in our cohort.
In addition to the retinal changes, high myopia and early development of cataract have also been described as key aspects of GA. 9 This was true for the degree of myopia in our cohort, which was higher in the GA group. However, GALRP patients also had a significant degree of myopia, although not as pronounced as in GA, which has not been reported to occur more frequently before. With regards to development of cataract, the results seem similar between both groups. It has to be taken into account, however, that the GALRP group was much older at the time of presentation, which could significantly impact these results. In the two cases where cataract surgery had occurred prior to initial presentation, it was performed at age 29 in the GA patient and at age 53 in the GALRP patient, also hinting at an earlier development of cataract in the GA group.
Regarding electrophysiological findings, there was considerable variation within the GALRP group, but patients were generally less severely affected than GA patients. This is in line with reports of diminished cone and rod ERG responses in GA, which may extend to near absent responses in later stages of the disease. 19 However, a case with an unremarkable ERG examination in GA has also been published. 20 In GALRP, one case report presented a patient with normal visual acuity and unremarkable ffERG results, 21 whereas a larger case series showed ERG responses ranging from mildly impaired to barely detectable. 10 A third report presented a 50 year-old with reduced cone response and no recordable rod responses. 22 ERG alone, therefore, may not be a good discriminator between these two entities.
With regards to family history, our cohort only included one GALRP patient reporting another potentially affected family member, who was, however, unable to participate in this study to confirm the phenotype. This contrasts with previous reports on GALRP, which often reported a positive family history. In one report, for example, 6 patients with a GALRP were analyzed. Three of the patients were related (a father and his two sons), suggesting an autosomal dominant inheritance pattern. The three other patients were unrelated. The two youngest patients had normal visual acuity, but already showed reduced ERG and EOG responses. A pathogenic mutation was, however, not identified. 10
Differentiating GA from GALRP is of particular importance due to the potential treatment of GA by a low arginine diet, which may slow progression.3,23 Patients are typically counselled to reduce intake of foods that are high in L-arginine such as red meat, chicken, turkey, fish (salmon, haddock), as well as nuts and seeds (almond, cashew, pumpkin seeds) and increase uptake of foods that have low arginine levels such as coffee, mollusk, clam, syrup, pudding, banana and dry mix. A long-term adherence to this diet can, however, be challenging. 24
Opinions on how to treat the macular oedema commonly found in GA differ widely. A resolution of macular oedema has been reported following a low arginine diet, 25 sub-tenon or intravitreal triamcinolone injection,26,27 or systemic carbonic anhydrase inhibitor and topical nonsteroidal anti-inflammatory drugs (NSAIDs). 28 Evaluating treatment response may be particularly difficult due to the heterogeneity of cystoid changes in GA. When no fluorescein angiography is performed (as was the case in our cohort), it can be difficult to differentiate macular oedema and other causes of cystoid retinal changes such as macular schisis. This could potentially explain the poor treatment response to topical or systemic carbonic anhydrase inhibitors seen in patient 2 of our study. Moreover, based on optical coherence tomography angiography studies, it has been suggested that an enlarged foveal avascular zone and RPE dysfunction may play pivotal roles in CME associated with GA unlike the CME seen in vascular retinal diseases,29,30 which could also impact treatment responses.
Recently, a novel mutation in the C1QTNF5 gene was identified that causes autosomal-dominant GALRP. 11 C1QTNF5 has previously been associated only with the phenotype of L-ORD characterized by subretinal pigment epithelial deposits, long anterior zonules and iris atrophy. 31 None of these signs were seen in the GALRP patients with the C1QTNF5 mutation, who instead showed the characteristic areas of extensive chorioretinal atrophy. 11 Based on data from the mouse model, it was hypothesized that a different location of the individual mutation may lead to this difference in retinal phenotype. 32 In patients with the novel C1QTNF5 mutation, visual problems appeared at about 40 years of age, and visual field defects were noted at about 50 years of age, which corresponds to the age of onset of some GALRP patients in this study. However, none of the GALRP patients that underwent genetic testing in our study harboured any sequence variants in the coding region of the C1QTNF5 gene or at the exon/intron boundaries.
We are aware that our study has some limitations. One is that GALRP is a disease entity defined only by the similarity in fundus appearance to GA. Thus, there is no definition of how closely the funduscopic phenotype must resemble GA for a patient to be diagnosed with GALRP. It is therefore possible that a GALRP with widespread atrophy may occur in several different disease entities with different aetiologies, which may explain the lack of a family history of retinal dystrophies in most of the patients included in this study. Moreover, this study was retrospective in design, and all patients were required to either undergo genetic testing or have their ornithine levels determined, which may have influenced patient selection. Since it was sufficient for the diagnosis of GA to be based on (multiple measurements of) plasma ornithine levels only, an erroneous lab test could have potentially led to a false group classification in those patients where no genetic testing was available. Lastly, the small sample size and the fact that only one GA patient underwent electrophysiology may impact the statistical significance of the comparisons and the conclusions that can be drawn from it.
In conclusion, our study suggests that GALRP represents either a heterogenous group of dystrophies caused by various as yet unknown genetic variants or that non-genetic factors are responsible for the phenotype, as suggested by the lack of a positive family history in many of our patients. Early disease onset, high myopia, early development of cataract and macular oedema are typical hallmarks of GA and rarely occur simultaneously in GALRP, which may aid in the differential diagnosis in patients with presumed GA. In cases of doubt, however, plasma ornithine levels and diagnostic genetic testing should be strongly recommended to ensure that the treatable entity of GA is not missed.
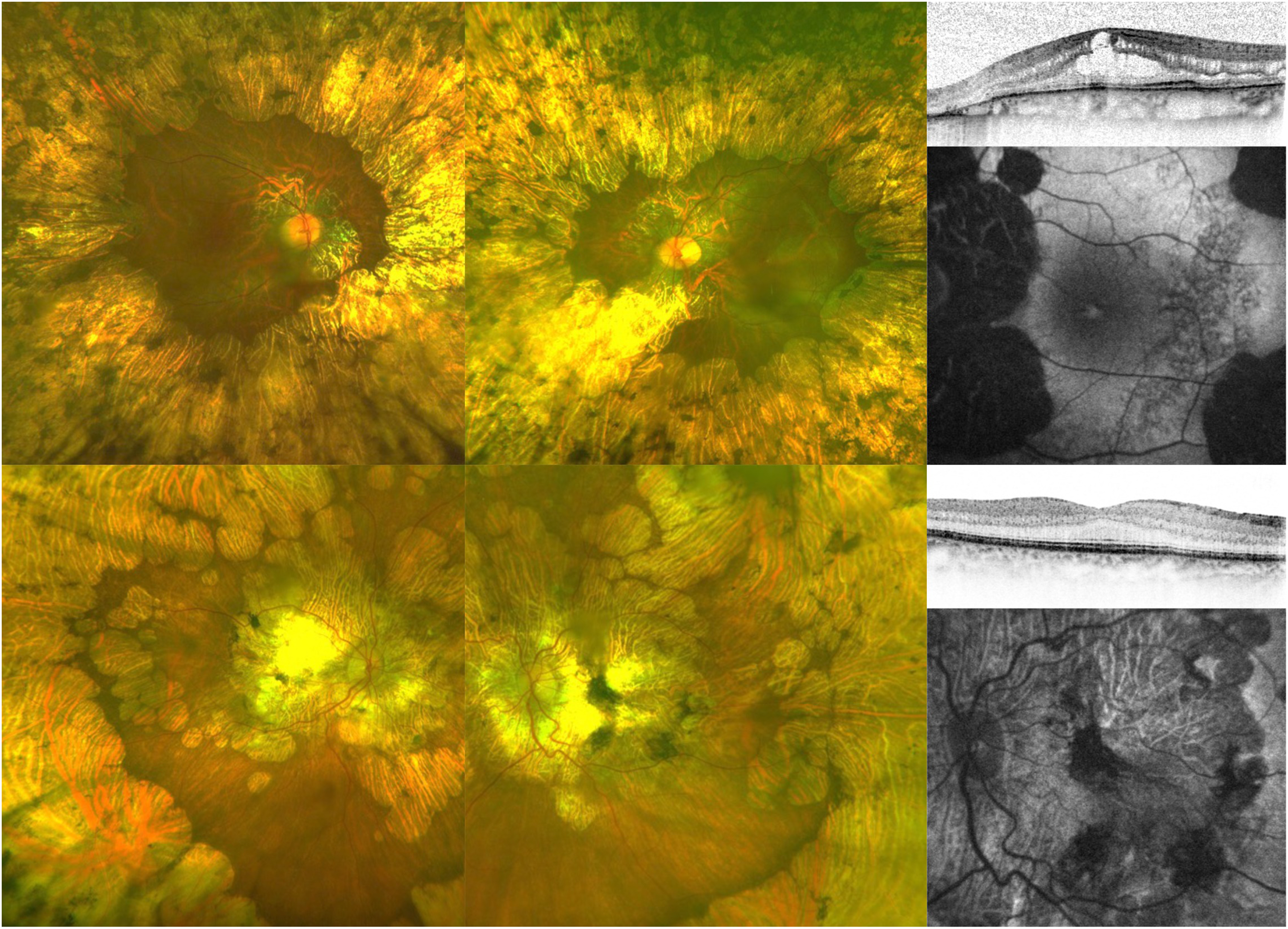
Comparison of typical wide-angle colour image, optical coherence tomography (OCT) and fundus autofluorescence images of gyrate atrophy (GA, top) and a gyrate atrophy-like retinal phenotype (GALRP, bottom). While the retinal phenotype can appear very similar, GA patients usually had a symmetric, concentric area of chorioretinal atrophy, whereas this was often more asymmetrical in GALRP patients. OCT revealed cystoid macular oedema in the GA patient, which is not present in the patient with GALRP.
Supplemental Material
sj-docx-1-ejo-10.1177_11206721231178147 - Supplemental material for Clinical characteristics of gyrate atrophy compared with a gyrate atrophy-like retinal phenotype
Supplemental material, sj-docx-1-ejo-10.1177_11206721231178147 for Clinical characteristics of gyrate atrophy compared with a gyrate atrophy-like retinal phenotype by L. Pauleikhoff, N. Weisschuh, A. Lentzsch, G. Spital, T. U. Krohne, H. Agostini and C.A.K. Lange in European Journal of Ophthalmology
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: L. J. B. Pauleikhoff was supported by the German Research foundation (grant number PA 4282/1-1). This funding organization provided unrestricted grants and had no role in the design or conduct of this research.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.