
Abstract
Background
Oculofaciocardiodental (OFCD) syndrome is a rare genetic disorder affecting ocular, facial, dental, and cardiac systems, being an X-linked condition caused by pathogenic variants in the BCL-6 corepressor gene (BCOR). We report a case series of three female patients with OFCD syndrome with severe glaucoma.
Results
Three female patients with OFCD syndrome with different variants involving BCOR gene, in heterozygosity: a seven-years-old girl with an insertion (c.2037_2038dupCT), a nine years-old girl with a microdeletion in the X (p21.2-p11.4)) spanning the BCOR gene; and a 25 years-old female with a deletion (c.3858_3859del). Systemic involvement is variable among patients ranging from one patient mainly with ocular and dental involvement to one with associated intra-auricular and intra-ventricular defects. All the patients presented with congenital cataracts diagnosed in the first days of life. Cataract surgery was performed without incidents between 6 and 16 weeks of age in all the patients. Postoperatively, the three patients developed ocular hypertension and glaucoma with the need for surgical interventions, including trabeculectomy, Ahmed valve implantation, and cyclophotocoagulation.
Conclusion
OFCD syndrome characterizes by a severe ocular involvement with glaucoma as a characteristic feature. Ocular hypertension after cataract surgery in these patients is challenging, almost always needing surgery during childhood. Therefore, we consider BCOR disruption may predispose to a higher incidence of glaucoma due to its aggressiveness and early onset on our case series. The awareness of these complications is crucial to an adequate follow-up of the patients.
Introduction
Oculofaciocardiodental (OFCD) syndrome is a rare genetic disorder described for the first time in 1980 by Hayward , 1 constituting an X-linked dominant syndrome caused by null variants in the BCOR gene. 2
BCOR gene, located in the X chromosome (Mendelian Inheritance in Men (MIM): 300485; cytogenetic location: Xp11.4), 3 encodes a protein that functions as a corepressor for BCL6, which itself is a transcription repressor. 2 BCL6 plays an essential role in embryogenesis and cell differentiation, 3 having a crucial role in the formation of the eye. 4
Besides OFCD syndrome, BCOR variants are also associated with severe X-linked microphthalmia syndrome, also known as Lenz syndrome. 2 Although the molecular mechanisms are not completely understood, OFCD syndrome is typically caused by BCOR null variants , 2 while Lenz syndrome is typically characterized by missense variants.2,3 OFCD syndrome is inherited in an X-linked dominant way, affecting exclusively females and presumed to be lethal in males. 2 On the other hand, Lenz syndrome is an X-linked recessive microphthalmia that affects males, suggesting that the severity of null variants in males is not compatible with life.2,3
OFCD is characterized by ocular, facial, dental, and cardiac anomalies, in a variable spectrum of severity. Considering ophthalmologic features, congenital cataracts are a consistent finding in all the patients described.3,5 Microphthalmia, iris synechiae, colobomas, ptosis, secondary glaucoma, and retinal detachment have also been reported. 3 However, these are believed to be non-consistent findings. Nevertheless, the rarity of this syndrome precludes a complete characterization of these patients.
We report a case series of three patients with OFCD syndrome with different BCOR pathogenic variants and severe glaucoma, suggesting BCOR disruption may have a role in the development of glaucoma. This is a new concept regarding the manifestations and pathophysiology of the disease. The awareness of these complications is crucial to an adequate follow-up of the patients.
Cases description
The pre-birth history of the girls is characterized by normal supervised pregnancies, followed by eutocic or cesarian deliveries, without complications.
OFCD syndrome was suspected in all the girls in the first months of life due to the presence of congenital cataracts associated with cardiac, dental or facial anomalies. Genetic analysis revealed three different pathogenic variants involving BCOR gene confirming the diagnosis of OFCD syndrome. Variable systemic manifestations occurred in the three girls during their growth. Genotype and phenotype descriptions of the girls are summarized in Table 1.
Summary characterization of the patients.
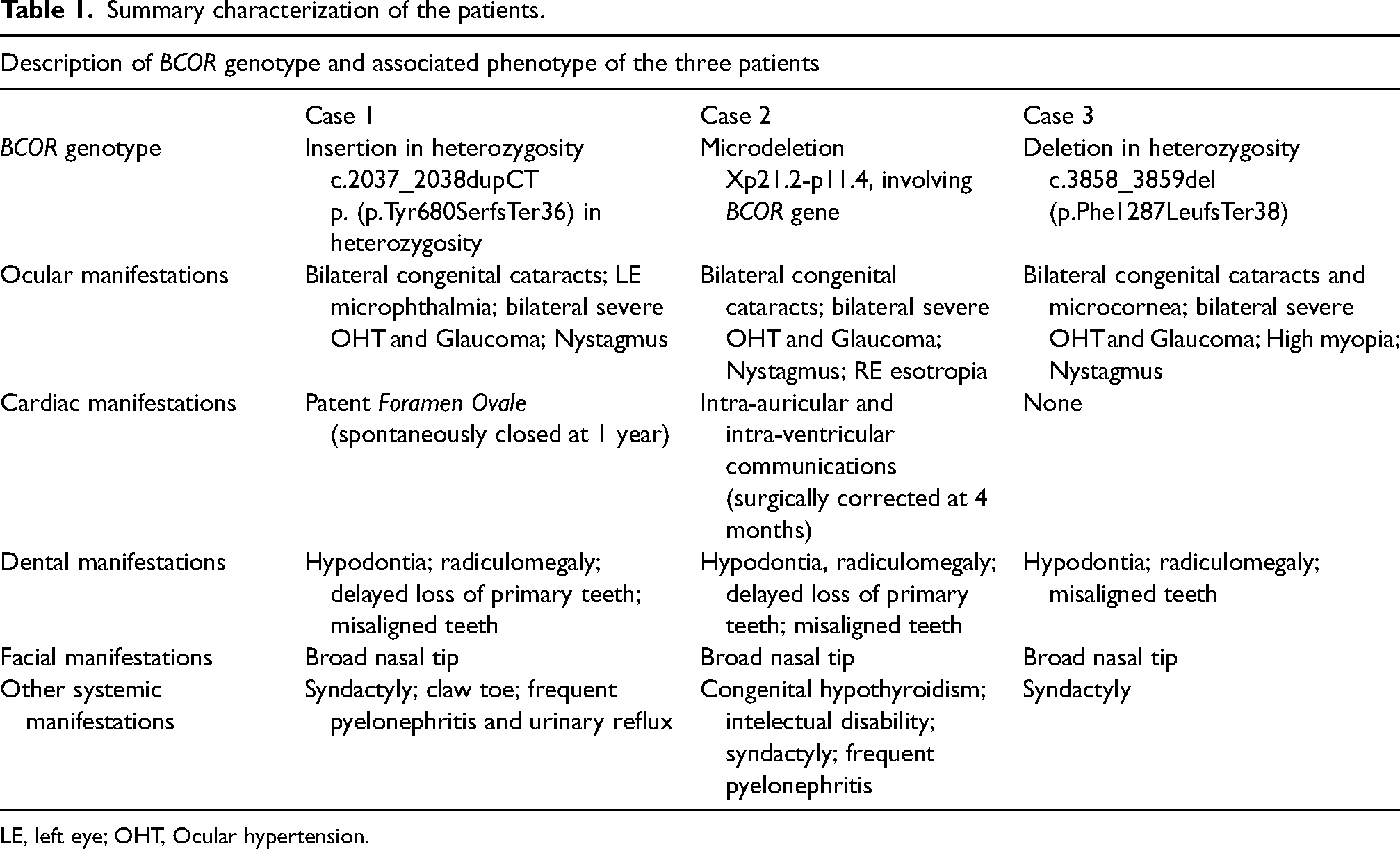
LE, left eye; OHT, Ocular hypertension.
Case 1
The first case refers to a seven-year-old girl with bilateral congenital cataracts and left eye (LE) microphthalmia present at birth.
Bilateral lensectomy with anterior vitrectomy, without intraocular lens (IOL) implantation, was performed at 7 weeks of life, without complications. However, during the first year of life, the girl experienced uncontrolled bilateral intra-ocular hypertension (OHT) with the consequent development of corneal edema and Haab striae. Medical therapy revealed ineffective, leading to bilateral trabeculectomy performed at 3 months. Right eye (RE) needling was needed one month later; tough ineffective. Cyclophotocoagulation at 5 months followed by FP8 Ahmed valve implantation with tube valve placement on the anterior chamber at 7 months were also performed in the RE.
After all the surgical approaches, clinical stability was ensured with bilateral intra-ocular pressure (IOP) below 25 mmHg under topical therapy (timolol 0,5% and dorzolamide 2%) until 7 years, when de novo uncontrolled bilateral hypertension relapsed. A second FP7 Ahmed valve in the RE, with drainage from the anterior chamber, and a first FP7 Ahmed valve in LE, with tube placed in the ciliary sulcus were performed. Both surgeries were done in combination with pars plana vitrectomy for tube clogging prevention.
After the surgeries, IOP oscillates between 15 and 25 mmHg on both eyes, under the association of timolol 0,5% with dorzolamide 2%. No complications were noted after surgeries.
Best-corrected visual acuity (BCVA) is 20/200 on the RE and 20/100 on the LE. Biomicroscopy reveals well-positioned valves and tubes, with no conjunctiva erosion, transparent corneas and aphakia (Figure 1). Microphthalmia without other remarkable findings is also present in the LE. Reduction of the neuroretinal rim area with an increased cup-disc ratio is evident at fundoscopy (Figure 1).

a. External appearance of case number 1. Evidence of dental misalignment and hypodontia. b. Anterior segment photographs. RE at left and LE at right. Evidence of tube drainage in the nasal side of the anterior chamber on the RE. RE mydriasis as consequence of multiple surgeries. c. Retinography of the RE at the last follow-up visit, showing enlarged cup-disk ratio. Deficient fixation, miosis and age preclude LE retinographies RE, Right eye; LE, left eye.
Case 2
A ten-year-old girl was submitted to bilateral lensectomy with anterior vitrectomy without IOL implantation at 6 weeks. The postoperative period elapsed without complications.
Bilateral IOP started to raise during the second year of life. Topical medication was insufficient, leading to bilateral FP8 Ahmed valve implantation, with tube placement in the ciliary sulcus, assisted by vitrectomy for tube clog prevention. No complications were noted after surgeries. IOP fluctuates between 20 and 30 mmHg on the RE and between 12 and 16 mmHg on the LE, both under topical medication (Timolol 0,5%, Dorzolamide 2% and Latanoprost 0,05 mg/mL on RE; Timolol 0,5% and Dorzolamide 2% on LE).
At the last follow-up visit, BCVA was 20/200 on the RE and 20/160 on the LE. RE esotropia and nystagmus due to deficient binocular development are evident. The slit-lamp evaluation shows correct position of valve and tube, with no conjunctival exposition or cornea damage and a good filtration bubble. Both eyes maintain aphakia. Nystagmus and low collaboration due to intellectual disability prevent retinographies acquisition; however, an enlarged cup disc is evident in fundoscopy.
For the reasons stated above, no high-quality images with minimal publication criteria are available for this case.
Case 3
The third case refers to a 25-year-old female. Congenital cataracts and microcornea were noted in the first weeks of life. Bilateral lensectomy without IOL implantation was performed at 4 months. LE re-intervention was needed at 2 years for excision of an inflammatory pupillary membrane. Due to very low vision and deficient binocular vision development, nystagmus and strabismus resulted. Furthermore, eye elongation started early resulting in bilateral high myopia.
According to the previous cases, OHT started at an early age. Although initially controlled on medical therapy, FP7 Ahmed valve implantation with tube placement on the anterior chamber was needed on the RE at 8 years and on the LE at 20 years. The postoperative period of the RE was complicated by a choroidal detachment followed by recurrent retinal detachments. A total of four vitrectomies were performed, the last one with silicone tamponade. No complications were noted after LE valve implantation.
Currently, BCVA is light perception on RE and counting fingers on LE. Band keratopathy resulted in RE, despite good valve placement without conjunctival exposition. In the LE, a good filtration bubble is evident without valve or tube exposition and correct tube placement in the anterior chamber (Figure 2).

a. Face photography of patient number 3 at the last follow-up visit. Evidence of a long narrow face, low fixation of RE, with slight right exotropia and broad nasal tip with separated nasal cartilages characteristic of OFCD syndrome. To note gingival hyperplasia in the superior arcade. Dental implants are present, supplanting the previous dental anomalies. b. Biomicroscopic aspect of the RE showing band keratopathy in the inferior third of the cornea and complete anterior segment disorganization. c. Biomicroscopic aspect of the LE evidencing significant miosis, superior iridectomy and aphakia. d. Detail of the anterior segment of the LE showing valve tube under conjunctiva without exposition or other complications. e, f and g. Retinographies at 15 (E, RE) and 21 yo (F, RE; G, LE) evidencing pallid and tilted discs with peripapillary atrophy and significative myopic chorioretinal atrophy. Deficient fixation and media preclude better and more recent retinographies. RE, right eye; LE, left eye; yo, years old.
LE IOP maintains below 25 mmHg under timolol 0,5%, dorzolamide 2% and brimonidine 2 mg/mL. Poor optical media and low fixation difficult fundoscopy, however, progressive cupping of the disc with peripapillary atrophy and diffuse myopic chorioretinal atrophy were noted during the follow-up (Figure 2).
Discussion and conclusions
OFCD constitutes a rare syndrome characterized by a highly variable phenotype.3,5 Regarding ophthalmologic findings, congenital cataracts are considered a ubiquitous finding. 2 Microphthalmia, iris synechiae, colobomas, ptosis, secondary glaucoma, and retinal detachment are other reported findings.1,2
We present three patients with OFCD syndrome with severe ophthalmologic commitment. All three girls present congenital cataracts, in accordance with the literature. Furthermore, besides microphthalmia in one girl and high myopia and microcornea in another one, all the patients present severe OHT requiring early surgical interventions for pressure control. The early and severe OHT in this case series may suggest that BCOR pathogenic variants may also have a role in the development of glaucoma in these patients.
The development of glaucoma in post-congenital cataract surgery is a well-known complication.6–8 The reported incidence varies among the studies, reflecting differences in study populations, glaucoma definitions, cataract surgery techniques, and follow-up period. The Infant Aphakia Treatment Study revealed an incidence of glaucoma of 9% at 1-year, 17% at 5 years and 22% at 10 years. 6 A recent revision of 1268 eyes found that the 5-year cumulative incidence of glaucoma or glaucoma suspect after congenital cataract surgery was 46% (95% CI, 28%-59%) in participants with bilateral aphakia and lower in patients with pseudophakia. However, the percentage of glaucoma surgeries needed was much lower. 7
Although the pathophysiology is not completely understood, several mechanisms for the development of glaucoma after congenital cataract surgery have been hypothesized. Younger age at surgery, persistent fetal vasculature, fetal nuclear cataracts, microphthalmia or microcornea, retained lens material, chronic inflammation, and reoperations have been associated with increased risk.6–8 The effect of primary IOL implantation is controversial, with published studies supporting and failing to demonstrate a protective effect. 9
In our case series, cataract surgery was performed between 6 and 16 weeks without IOL implantation and all the eyes of the patients developed glaucoma early in life. IOL implantation was not performed due to the low age of the patients and concomitant ocular comorbidities associated. Aphakia, microphthalmia in one girl, microcornea and reoperation post cataract surgery in another may be considered reasons for higher glaucoma in our case series. However, severe HTO developed bilaterally regardless of surgeries or anatomic anomalies. Furthermore, a recent meta-analysis does not show evidence of a protective effect of IOL implantation in glaucoma development. 9 Moreover, OHT was noted very early and severely in the follow-up of the girls, differing from the previous literature.
Therefore, we hypothesize that BCOR disruption may potentially have a role in glaucoma development. BCL-6, the protein encoded by BCOR, is responsible for embryogenesis, having a role in eye development. 4 We believe BCOR pathogenic variants may have the potential to cause minor eye dysgenesis besides congenital cataracts, and possibly, predisposing to the development of OHT and the subsequent optic neuropathy. Notwithstanding, further studies need to confirm this association.
Zhu and colleagues described a boy with multiple birth anomalies including cardiac septal defects and congenital glaucoma associated with a de novo missense variant in BCOR gene. 10 Another report also describes the presence of posterior embryotoxon in several patients with BCOR pathogenic variants implying it is a possible manifestation of part of the spectrum of eye anomalies associated with this gene variants. 2 Although these reports are more closely related to the spectrum of Lenz syndrome, we believe BCOR pathogenic variants in OFCD syndrome also predispose to some common phenotypic manifestations. Furthermore, some authors describe an overlap between both phenotypes.3,10
Therefore, although not clinically evident in our cases, molecular ocular dysgenesis may be present and contribute to glaucoma development. This potential causal relation was not previously described and may potentially contribute to a better understanding of the pathophysiology of this syndrome. The awareness of this potential relationship is crucial for a more stringent follow-up of the patients and a more guided treatment. This is particularly important since glaucoma management in children and specifically in these patients may be challenging. Very scarce data exist regarding the best surgical approach for glaucoma after congenital cataract surgery. Surgeries described in these patients include trabeculotomy, trabeculectomy, glaucoma drainage devices, and endoscopic diode cyclophotocoagulation, reflecting the absence of a uniform initial approach due to the lack of evidence. Furthermore, surgical procedures are more demanding in children, who frequently need vitrectomy support to lower the complication rate. A prompt diagnosis relied on high suspicion and allied to regular follow-up is essential for the timely and wise management of these patients. Although this syndrome reveals very rare, the new genetic sequentiation panels have found more frequently BCOR variants. 5 Therefore, the incidence of these variants may be underestimated and be more frequently found in the future, bringing awareness of the potential manifestations of a need.
Notwithstanding, several limitations of this work need to be stated. First, only three patients are presented limiting the power of the data. The rarity of this syndrome precludes bigger cohorts. Furthermore, the different approaches in the three girls hamper comparisons. Moreover, the absence of gonioscopy precludes glaucoma classification and leads to possible missing information regarding angle anatomy and potential anomalies. The execution of gonioscopy in very young patients with poor visual capacities and significant anomalies in the anterior segment was not possible. Therefore, we must keep in mind that this is a limitation of the work that prevents us from obtaining potentially significant findings. Consequently, more research in this area and bigger multicentric case reports are needed for stronger conclusions.
Despite limitations, and in conclusion, we hypothesize BCOR pathogenic variants may predispose to a higher incidence of glaucoma due to its aggressiveness and early onset of the disease on our case series. The knowledge of this complication as a potential finding of this syndrome is crucial to an adequate follow-up and management of the patients.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
All the reported data required for this publication were collected as part of routine diagnosis and did not require specific ethical committee approval.
Informed consent for use of data and personal images was obtained from the patients and parents of the patients.