
Abstract
Objectives
The aim of this study was to isolate feline dental pulp stem cells (fDPSCs) and characterize their clonogenic and proliferative abilities, as well as their multipotency, immunophenotype and cytogenetic stability.
Methods
Dental pulp was isolated by explant culture from two cats <1 year old at post mortem. Their clonogenicity was characterized using a colony-forming unit fibroblast assay, and their proliferative ability was quantified with a doubling time assay in passages 2, 4 and 6 (P2, P4 and P6, respectively). Multipotency was characterized with an in vitro trilineage differentiation assay in P2, and cells were immunophenotyped in P4 by flow cytometry. Chromosomic stability was evaluated by cytogenetic analysis in P2, P4 and P6.
Results
The fDPSCs displayed spindle and epithelial-like morphologies. Isolated cells showed a marked clonogenic capacity and doubling time was maintained from P2 to P6. Trilineage differentiation was obtained in one sample, while the other showed osteogenic and chondrogenic differentiation. Immunophenotypic analysis showed fDPSCs were CD45−, CD90+ and CD44+. Structural and numerical cytogenetic aberrations were observed in P2–P4.
Conclusions and relevance
In this study, fDPSCs from two cats were isolated by explant culture and immunophenotyped. Cells displayed clonogenic and proliferative ability, and multipotency in vitro, and signs of chromosomic instability were observed. Although a larger study is needed to confirm these results, this is the first report of fDPSC isolation and in vitro characterization.
Introduction
Adipose tissue (AT)-derived mesenchymal stromal/stem cells (AT-MSCs) are commonly chosen for feline stem cell therapy owing to their proliferative ability and ease of obtainment. 1 The published sources for feline MSCs are bone marrow, adipose tissue, amniotic membrane, umbilical cord, spermatogonia, peripheral blood and Wharton’s jelly.2–8 Despite displaying characteristics in common, cells derived from different tissues present different phenotypes that can reflect variations in their functional properties. 9 The ectomesenchymal origin of dental pulp stem cells (DPSCs) could imply different characteristics. These cells have been studied for the treatment of oral, neurologic, ocular, muscular and systemic diseases in humans and experimental animal models. 10 Obtaining sufficient numbers of MSCs for therapy involves in vitro culture, and expansion of cells takes prolonged exposure to proliferation stress, which can produce chromosomal aberrations throughout successive passages.11–13 The objective of this study was to isolate and characterize feline DPSCs (fDPSCs).
Materials and methods
With informed consent, maxillary permanent canine teeth were extracted post mortem from two female mixed breed cats (8 and 10 months of age, respectively) that died as a result of polytrauma after presenting at seperate veterinary emergency practices in a critical condition. The cadavers were refrigerated at 4°C and teeth were extracted between 6–12 h after death, disinfected with iodopovidone and alcohol (1:1), rinsed with phosphate buffered saline (PBS) and the dental pulp (DP) was extracted using an endodontic barbed broach (Maillefer [ethical committee approval: CEUA-FVET-N°1377]). The isolation was carried out by explant method (ExM) 14 and the fDPSCs were cultured as previously reported. 15 Briefly, the DP was cut into 1 mm pieces and placed in six-well plates. The culture medium was Alpha MEM (Gibco) supplemented with 20% heat-inactivated fetal bovine serum (FBS; Capricorn), 100 U/ml penicillin G and 100 U/ml streptomycin (Capricorn). Plates were incubated at 37°C in a humidified atmosphere of 5% CO2. Once the first adherent cells were observed, the FBS concentration was reduced to 10%. Media were replaced every 3–4 days, and cells were harvested using 0.25% trypsin-EDTA (Sigma) when they reached 70–80% confluence. Every assay was performed on fresh cells except for immunostaining. Cells used for immunostaining were cryopreserved in 95% FBS and 5% dimethyl sulfoxide and frozen at −80°C until use.
Morphology, clonogenic capacity, doubling time and cytogenetic stability were characterized in passages 2, 4 and 6 (P2, P4 and P6, respectively). Morphology was evaluated by optical microscopy. A colony-forming unit (CFU) fibroblast assay (CFU-F) was performed to assess clonogenic capacity by seeding 1 × 103 cells/well, culturing for 14 days, fixing the cells with cold methanol and staining them with Giemsa. 16 Colony-forming efficiency was calculated using the following formula: [number of colonies/number of seeded cells] × 100. 17 Proliferative capacity was assessed using a doubling time (DT) assay by seeding 1 × 104 cells/well, harvesting at 80% confluence and counting the cells in the Neubauer chamber. DT was calculated using the following formula: DT = duration × log(2)/[log(concentration final)−log(concentration initial)]. 18 Means and SDs were calculated using GraphPad Prism 9.
Trilineage differentiation capacity was evaluated in P2. Differentiation culture media was adapted from previously described protocols. 16 Three different media were added to the culture medium without antibiotics: (1) 60 μM dexamethasone (Sigma), 10 mM beta (β)-glycerophosphate (Sigma) and 1.7 mM ascorbic acid (Sigma) for osteogenic differentiation; (2) 500 μM isobutylmethylxanthine (Sigma), 60 μM indomethacin (Sigma), 1 μM dexamethasone (Sigma) and 50 μg/ml insulin–transferrin–selenium A (Gibco) for adipogenic differentiation; and (3) 1.7 mM ascorbic acid (Sigma), 10 ng/ml transforming growth factor-β (Sigma) and 6.25 µg/ml insulin–transferrin–selenium A (Gibco) for chondrogenic differentiation. Cells were cultured for 21 days, and the medium was changed every 3–4 days and fixed with 4% paraformaldehyde. The presence of mineralized matrix was confirmed by Alizarin Red S staining (Biomedicals), the presence of lipid droplets was confirmed by Oil Red O staining (Sigma) and the presence of cartilaginous matrix was confirmed by Alcian Blue staining (Biomedicals). Control cultures were maintained under the same conditions. Means and SDs were calculated.
Immunophenotype was evaluated at P4 by flow cytometry, as previously described. 17 Cryopreserved cells were thawed, and rapidly washed with PBS and resuspended in PBS (Mg++ and Ca++ free) containing 5 mM EDTA and 1% FBS (FACS EDTA). Next, 1 × 106 cells/tube were immunostained by incubating them at room temperature for 15 mins with allophycocyanin-conjugated anti-CD44 (clone IM7; Leinco Technologies), fluorescein isothiocyanate-conjugated anti-CD45/LCA (CD45RO, clone UCHL1; Acris Antibodies) and phycoerythrin-conjugated anti-CD90 (THY1, clone 5E10; antibodies-online) antibodies. Then, cells were washed and resuspended in the minimum volume of FACS EDTA. At least 20,000 events per sample were acquired on a FACS Canto II system (BD Biosciences) using FACS DIVA software for acquisition. Cells were gated based on forward/side scatter patterns. Offline analysis was performed using FlowJo software (Tree Star).
Cytogenetic stability was studied during P2–P6. Briefly, cells were cultured to 50–60% confluence, where 0.1 µg/ml colcemid (PAA) was added to the culture and incubated for 2 h under standard culture conditions. Cells were harvested, centrifuged and treated with hypotonic KCl solution (0.075 M) for 15 mins and fixed with methanol-acetic acid (3:1). Chromosome smears were prepared on slides with cold methanol, then heat-fixed, stained with 3% Giemsa and observed under an optical microscope (Olympus BX 60). Analysis, interpretation and karyotype were based on the international feline chromosome nomenclature standard, and structural alterations were classified as previously described.18,19
Results
Isolation and morphology
Isolation of fDPSCs was successful using the ExM. The first plastic adherent cells for samples A and B were observed after 9 and 15 days of culture, respectively (Figure 1a). Cells generally showed a spindle-shaped morphology, although cells with a large, rounded cytoplasm with epithelial-like shape were also observed in both samples (Figure 1).

Microscopic image of feline dental pulp stem cells (fDPSCs) (× 4 objective). (a) fDPSC explant with cells adhered to the plastic. The dental pulp explant is indicated with a single asterisk and the lines marked on the bottom of the well to facilitate adherence of the explant are indicated with a double asterisk, cells with an epithelioid appearance are indicated with a white arrow and cells with a spindle-like appearance with a black arrow. (b) fDPSCs in passage 2 (× 4 objective), (c,e) fDPSC of sample A in passage 2 (× 20 and × 40 objective) and (d,f) fDPSC of sample B in passage 2 (× 20 and × 40 objective)
CFU-F
The fDPSCs were able to form colonies (Figure 2). Colony formation efficiency during P2, P4 and P6 for sample A was 8.5%, 1.4% and 8.8%, respectively, and was 10.6%, 9.3% and 7.6% during P2, P4 and P6 for sample B, respectively.
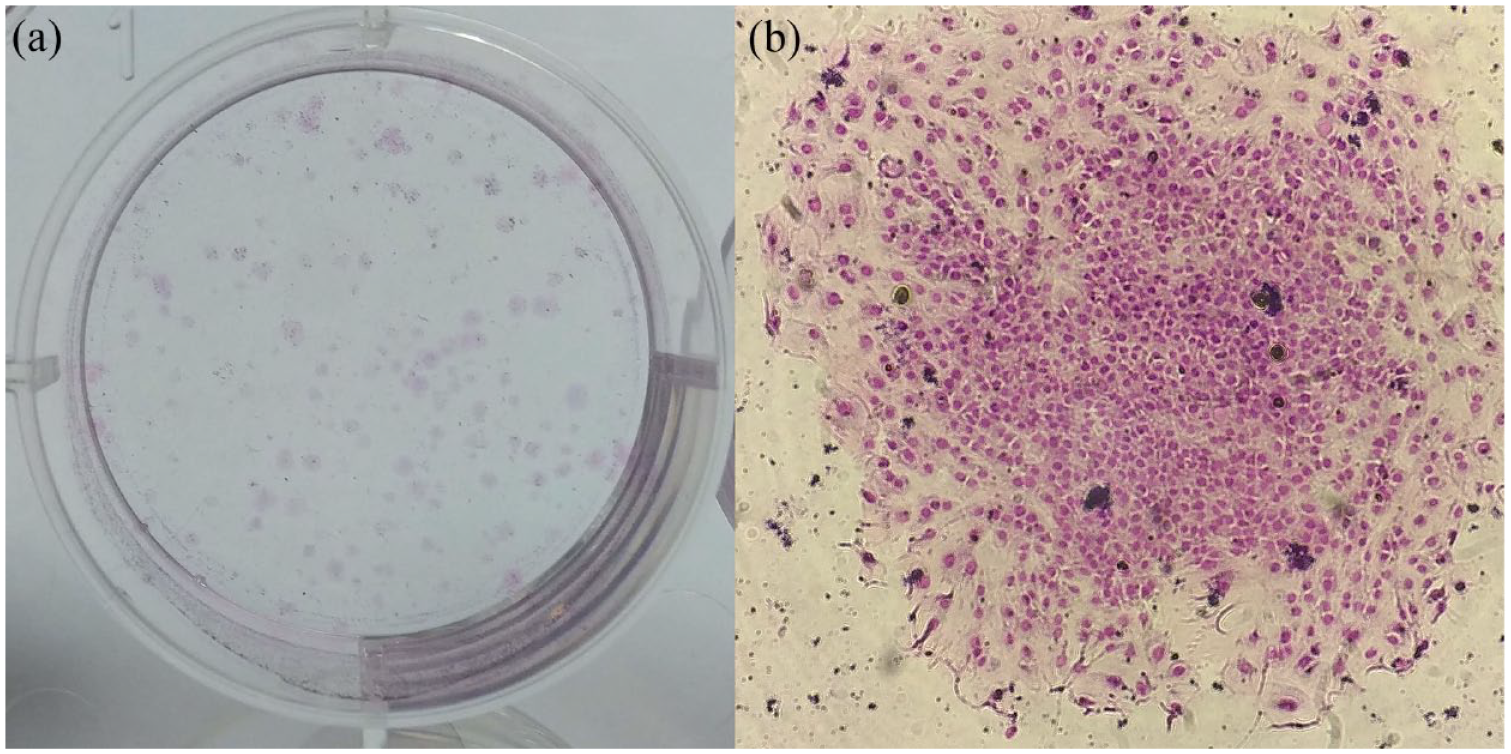
Feline dental pulp stem cell (fDPSCs) colony-forming unit fibroblast assay. (a) Macroscopic image of colonies. (b) Microscopic image (× 20 objective) of an fDPSC colony
Doubling time
The DT in P2, P4 and P6 for sample A was 2.5, 6.7 and 9.4 days, respectively, and for sample B was 5.1, 12.3 and 10.2 days, respectively. Additionally, sample A was analyzed in P8, obtaining a DT of 9.9 days. The mean number of days to reach 80% confluence in each passage was 21, 43.5 and 25, respectively.
Trilineage differentiation assay
Complete differentiation was obtained in P2 for sample B (Figure 3). Sample A showed differentiation to osteogenic and chondrogenic lineages. Osteogenic differentiation was observed after 17.6 ± 4.5 days and chondrogenic differentiation after 16.3 ± 4.6 days, while adipose differentiation was obtained after 10 days.

Trilineage differentiation of feline dental pulp stem cells in passage 2. Microscopic images of (a) osteogenic differentiation stained with Alizarin red showing calcium deposits (× 10 objective), (c) chondrogenic differentiation stained with Alcian blue showing matrix rich in glycosaminoglycans (× 10 objective) and (e) adipogenic differentiation stained with Oil Red O showing lipid vacuoles (× 40 objective). (b,d,f) Undifferentiated controls at (b,d) × 10 objective and (f) at × 40 objective
Immunophenotyping
One replicate of each sample at P4 was immunophenotyped. Both samples were CD45 negative, CD90 and CD44 positive, and showed a few cells with higher fluorescence intensity for CD44 (<1%; Figure 4).

Immunophenotypic analysis of feline dental pulp stem cells (fDPSCs). (a) Representative histograms of CD90, CD44 and CD45 expression of fDPSCs (dashed line on each histogram represents non-stained cells). (b) Dot plots representing the gating strategy employed: cells were gated based on forward scatter (FSC) and side scatter (SSC) properties, single cells were then selected and CD90, CD44 and CD45 expression analyzed. Representative dot plot of CD44 and CD90 expression of fDPSC is shown. FITC = fluorescein isothiocyanate
Cytogenetic analysis
Fourteen metaphases were analyzed in P2, showing a mitotic index of 0.14%, and 47 metaphases were observed in P4 with a mitotic index of 0.65%. Structural and numerical aberrations by passage are shown in Table 1 and a normal tentative karyotype of fDPSCs in Figure 5. One metaphase showed a trisomy (2n = 39, XX) presumably of chromosome C1 in P2, and one metaphase showed diplochromosomes in P4. No metaphases were observed in P6.
Total number of altered metaphases per passage of both samples of feline dental pulp stem cells (analyzed metaphases: P2, n = 14; P4, n = 47)


Cytogenetic study of feline dental pulp stem cells (fDPSCs). (a) Normal 38, XX metaphase of fDPSCs. (b) Proposed normal karyotype (2n = 38, XX) stained with Giemsa of fDPSCs from a female cat
Discussion
In this study, fDPSCs from two cats were isolated for the first time and their clonogenic, proliferative, multipotency characteristics and cytogenetic stability characterized. We observed spindle-shaped cells adhering to plastic and with clonogenic capacity in vitro. There are no previous reports of fDPSCs available for comparison, but our results were similar to descriptions on the morphology and clonogenic capacity of DPSCs from other species such as humans, rats, dogs and ferrets.20–23
Most studies of human DPSCs utilize enzymatic isolation,20,24 whereas this study used ExM. The primary benefit of ExM is the ability to achieve an effective isolation with low-volume DP samples, resulting in a low-cost method for fDPSCs isolation. Previous reports have described no differences in multipotency and proliferation of DPSCs isolation between the enzymatic method and ExM. 14 It has been shown that the enzymatic method gives rise to more heterogeneous morphology than ExM, probably because endothelial cells are released, while the explant only allows the adherence of cells capable of migrating.14,20 We observed a variety of morphologies in the isolated fDPSCs, consistent with previous observations of fibroblastic-like and epithelial-like cells in human DPSC cultures. 14 The heterogeneity of human-derived DPSCs has been previously described; 25 however, further characterization of the heterogeneity of fDPSC is required. The DT obtained was high, showing a slow growth rate compared with feline MSC from other sources.4,26,27 The mean efficiency of fDPSC colony formation was 9.6% in P2 and 8.2% in P6, which is similar to human DPSCs in early passages, where 2–7.7% efficiency has been reported. 20
Trilineage differentiation was obtained in one sample, while the other showed osteogenic and chondrogenic differentiation. Previous studies showed difficulties in adipogenic differentiation of DPSCs. 20 The concentration of ascorbic acid typically used in chondrogenic and osteogenic in vitro differentiation protocols is between 50 and 250 µM.4,28,29 However, we used a concentration 10 times higher in this study, based on previous experiments in our laboratory that showed faster results in osteogenic and chondrogenic differentiation staining with this protocol (data not shown). Differentiation of fDPSCs took 2 weeks, which is considerably shorter than the 3–6 weeks reported for human DPSCs.20,24 The shorter time frame for our protocol provides considerable time savings.
The immunophenotypic analysis of the fDPSCs is consistent with immunophenotyping of other sources of feline stromal/stem cells.1,3,8 A small population of cells (<1%) was observed to have a higher fluorescence intensity for CD44 that may be related to odonto/osteoblastic lineage population. This marker is strongly expressed in cells undergoing mineralization such as ameloblasts, odontoblasts and osteoblasts, and it has been shown that odontoblastic differentiation of DPSCs is mediated by CD44. 30 Further analysis of this population is required to better understand this effect.
In this study, the cytogenetic stability of fDPSCs was assessed, and structural alterations were observed in 71% of analyzed metaphases in P2 and in 25% of analyzed metaphases in P4. No metaphases were observed in P6, despite the proliferation tests showing that clonogenic capacity was maintained. This difference could be the result of DT increase, which implies a reduction in the mitotic index and, therefore, in the number of metaphases. Although the number of metaphases analyzed is not enough for conclusive analysis, it has been reported that the method of cell harvesting, as well as high percentages of oxygen or glucose during culture, can influence the number of aberrations found.31–33 This could be relevant in DPSCs since oxygen tension in DP tissue is lower than in culture conditions. 34 The decrease in aberrations between P2 and P4 could be explained by the fact that MSCs tend to lose abnormal karyotypes as the number of passages increases. 35 The literature on the cytogenetic stability of MSCs is still controversial: some studies have detected cytogenetic instability, but others have shown stable karyotypes during MSCs culture.35–38 Cell alterations and cell senescence affect the therapeutic potential of MSCs, highlighting the importance of clearly understanding the in vitro behavior of these cells. 39 Further studies conducted on a larger number of samples are necessary to analyze the significance of the presence of cytogenetic alterations during culture of fDPSC and their implications for MSC therapy.
Similar to recent studies on feline Wharton jelly MSCs, 8 this study evaluated a small number of samples, obtaining original results. Further studies on a larger, more diverse number of samples is needed to confirm the findings and further characterize fDPSCs. Of note, the samples used in this study were obtained post mortem from two young female cats. The young donor age may have affected the proliferative capacity noted in this study, as it has been reported for feline AT-MSCs that younger donors have faster proliferation than older animals. 40 Similar to the present study, post-mortem isolation of chimpanzee DPSCs has been reported, and cell multipotency and clonogenic and proliferative capacity have also been confirmed. 41 The impact of donor characteristics and post-mortem collection of DPSCs was not evaluated in this study, but would be important information for the application of fDPSCs as feline dental procedures that involve tooth extractions or vital pulpotomy, which could be potential sources of dental pulp obtained from routinely discarded material.
Conclusions
This is the first report of fDPSC isolation and their in vitro characterization. In this study, the ExM was successfully used for the isolation of fDPSCs. These cells showed clonogenic ability, and DT was maintained in P2, P4 and P6. One of the samples achieved trilineage differentiation, while the other achieved osteogenic and chondrogenic differentiation. Immunophenotyping showed that the cells were CD45 negative, and CD44 and CD90 positive. Some signs of chromosomic instability were observed. Further research on a larger number of samples is needed to fully characterize this population of cells.
Footnotes
Acknowledgements
The authors thank Guillermo Grazzioli DDS, and Alejandro Francia DDS (Dentistry Faculty, UdelaR) for their contribution to obtaining the dental pulp stem cells and isolation protocol.
Author note
The results of this study were presented as an e-poster in the XI Brazilian Association of Cell and Gene Therapy Meeting – ABTCel-Gen, ISCT South & Central America Regional Forum, III Stem Cell and Cell Therapy Meeting in Veterinary Medicine, I Symposium on Stem Cells and Biomaterials in Dentistry online, 28 April to 1 May 2021.
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
The work described in this manuscript involved the use of non-experimental (owned or unowned) animals. Established internationally recognized high standards (‘best practice’) of veterinary clinical care for the individual patient were always followed and/or this work involved the use of cadavers. Ethical approval from a committee was therefore not specifically required for publication in JFMS. Although not required, where ethical approval was still obtained, it is stated in the manuscript.
Informed consent
Informed consent (verbal or written) was obtained from the owner or legal custodian of all animal(s) described in this work (experimental or non-experimental animals, including cadavers) for all procedure(s) undertaken (prospective or retrospective studies). No animals or people are identifiable within this publication, and therefore additional informed consent for publication was not required.