
Abstract
Objectives
This study was designed to confirm the efficacy and tolerability of a daily dose of 7.0 mg/kg (3.2 mg/lb) ciclosporin (CsA) in the treatment of feline hypersensitivity dermatitis (HD), as this includes some of the most frequently suspected skin diseases in cats and recent publications have reported the successful use of CsA in the treatment of feline HD.
Methods
In total, 217 cats with feline HD were treated daily for 42 days with a target dose of 7 mg/kg CsA (n = 144) or a placebo control (n = 73) administered either in the food or directly in the mouth following feeding. Clinical and dermatological evaluations were conducted on days 0, 21 and 42, or study exit. Safety was evaluated through physical examinations, clinical pathology and the monitoring of adverse events (AEs).
Results
Administration of CsA at 7.0 mg/kg produced a significant improvement in the total lesion score (P <0.0001). The average reduction from visit 1 to visit 3 was 65.1% in the CsA group (9.2% for the placebo). In addition, owners assessed 78.3% of the cases in the CsA group as a success. Statistically significant recoveries were also seen in extent of lesions, investigator assessment of overall improvement, and mean improvement in both the investigators’ and owners’ assessment of pruritus. Mild gastrointestinal disorders were the most common AEs but did not require cessation of therapy.
Conclusions and relevance
Results confirm that 7.0 mg/kg CsA dosed daily in food or orally for up to 6 weeks is effective and well tolerated by cats with feline HD.
Introduction
Hypersensitivity dermatitides is a term used to describe a wide variety of allergic diseases such as flea bite hypersensitivity dermatitis, cutaneous adverse food reactions, urticaria, angioedema and atopic dermatitis. 1 The International Committee on Allergic Diseases of Animals recently adopted the term ‘feline atopic syndrome’, which specifically excludes parasitic causes but includes environmental allergen causes, and at least some manifestations of asthma, and food reactions, as such manifestations may occur concurrently in a single cat. 2 Non-flea, non-food hypersensitivity dermatitis (HD) has been traditionally called ‘atopic dermatitis’, but this terminology is no longer recommended, as cats with non-flea, non-food HD often do not share the features that characterize human and canine atopic dermatitis. 3 Cats presenting with HD have pruritus associated with one or more of four typical lesion patterns: head and/or neck excoriations, symmetrical self-induced alopecia (SA), eosinophilic disease (eosinophilic plaques [EPs] or granulomas, indolent ulcers) or miliary dermatitis (MD). The diagnosis of feline HD is difficult and based on the exclusion of all other potential causes of pruritus.4,5 Because of the complexity of the diagnosis, few studies have evaluated the frequency of feline skin allergic disorders. However, a retrospective study evaluating a large population of cats examined in the dermatology service of an animal hospital over a 15 year period, found that allergies accounted for 32.7% of all feline dermatoses and that 10.3% of the cats were specifically diagnosed with atopic dermatitis as formerly defined. 3
There are several treatment options for non-flea, non-food HD in cats. 4 Glucocorticoids are frequently used, but may not be well tolerated for long-term administration owing to the frequency of adverse effects. Antihistamines, allergen-specific immunotherapy and/or essential fatty acids may also be recommended, but their efficacy has been poorly documented in well-controlled clinical studies. Several publications have, however, reported the successful use of CsA to treat cats with non-flea non-food HD.6–8 A randomized, double-blinded, placebo-controlled study assessing CsA given at two different doses in 100 cats demonstrated the efficacy and tolerability of CsA given at 7.0 mg/kg (3.2 mg/lb) once a day for up to 6 weeks in the treatment of feline HD. 9 The objective of the present study was to confirm, in a larger cat population, the clinical response and tolerability of CsA administered orally for up to 6 weeks at the dose of 7.0 mg/kg (3.2 mg/lb) in the control of feline non-flea, non-food HD.
Materials and methods
Study design
This study was conducted in two phases. Phase 1, reported here, was designed to confirm the field efficacy of CsA for the control of HD and administered at a target dose of 7 mg/kg (3.2 mg/lb) body weight. The second phase (Phase 2), a non-randomized, open-label study, which immediately followed this Phase 1 study, explored the effect of a dose-tapering regimen on the clinical response to CsA.
Phase 1 was a randomized, double-blinded, placebo-controlled, multi-center trial conducted in the USA (23 sites) and Canada (one site), according to the procedures and principles of good clinical practices using board-certified dermatologists. All procedures were reviewed and in compliance with Novartis Animal Health animal welfare guidelines and the US Department of Agriculture’s Animal Welfare Act (2009). In addition, cat owners signed informed consent for their cat to be included in the study and were free to withdraw their animals at any point. Cats withdrawn because of a lack of efficacy following a minimum of 10 days of treatment, cats completing the trial without experiencing a serious adverse event and cats that experienced good owner treatment compliance were eligible to participate in Phase 2. 10
Randomization and blinding
Owners and investigators were blinded to treatment. The test articles (CsA and placebo) had exactly the same appearance and packaging along with identical administration procedures. An individual with no responsibility in efficacy or safety evaluations acted as a dispenser to manage all test material-related activities. Cats included in the study were randomly assigned to treatment on day 0 in a 2:1 ratio (two cats treated with CsA for every one treated with the placebo). Individual randomization lists were prepared for each investigational site. No separate randomization schedule was prepared for sex, age or body weight.
Animal selection
Recruited cats were client owned, at least 6 months of age and weighed a minimum of 1.4 kg. They must have had a history of year-round pruritus and at least one of the following lesions: MD; facial or neck excoriation; SA; and/or eosinophilic plaque.
Cats received a thorough physical examination, including clinical pathology. Flea-bite hypersensitivity was ruled out as cats were required to have had treatment with a flea adulticide for at least 1 month prior to inclusion (a flea adulticide was not administered in two sites, one in Colorado and one in Nevada, as they were not considered to be flea endemic; the cases at these two sites represented 12% of the cases evaluated for efficacy). Primary bacterial and fungal infections were eliminated via appropriate cytology, culture and treatment prior to enrollment. Six week food trials with novel ingredients were used to eliminate food allergies. In addition, cats had to be feline leukemia virus (FeLV) and feline immunodeficiency virus (FIV) negative and have their Toxoplasma serology status assessed prior to inclusion. In cats that had not previously been biopsied, skin biopsies were collected at the discretion of the investigator as an aid to the diagnosis of possible neoplastic and immune-mediated conditions. Other exclusion criteria included pregnancy, lactation or animals intended for breeding; requirement for vaccinations during the study; active systemic infection, evidence or history of any type of malignancy; uncooked home-prepared diet (owing to variability in constituents and the potential for transmission of Toxoplasma species); and lesions localized only to the mouth, oral cavity and upper lip. Owners were also instructed not to change the home management or diet of their cat during the study unless requested by the investigator for reasons unrelated to the allergic condition.
A sufficient washout period was required for the following therapies: systemic (long-acting) glucocorticosteroids – 56 days; essential fatty acids – 56 days (or the same regimen had to be maintained throughout the study); systemic (short-acting) glucocorticosteroids and megestrol acetate – 28 days; topical and ophthalmic glucocorticosteroids, antihistamines, serotonin reuptake inhibitors and topical calcineurin inhibitors – 14 days; shampoos – 14 days (or the same regimen had to be maintained throughout the study only for shampoos not containing antifungals, antibiotics or corticosteroids); ketoconazole, itraconazole, erythromycin and phenobarbital – 7 days. If the response to allergen-specific immunotherapy was considered unsuccessful by the investigator, and the cat qualified for inclusion, a 14 day withdrawal period was required following the last injection.
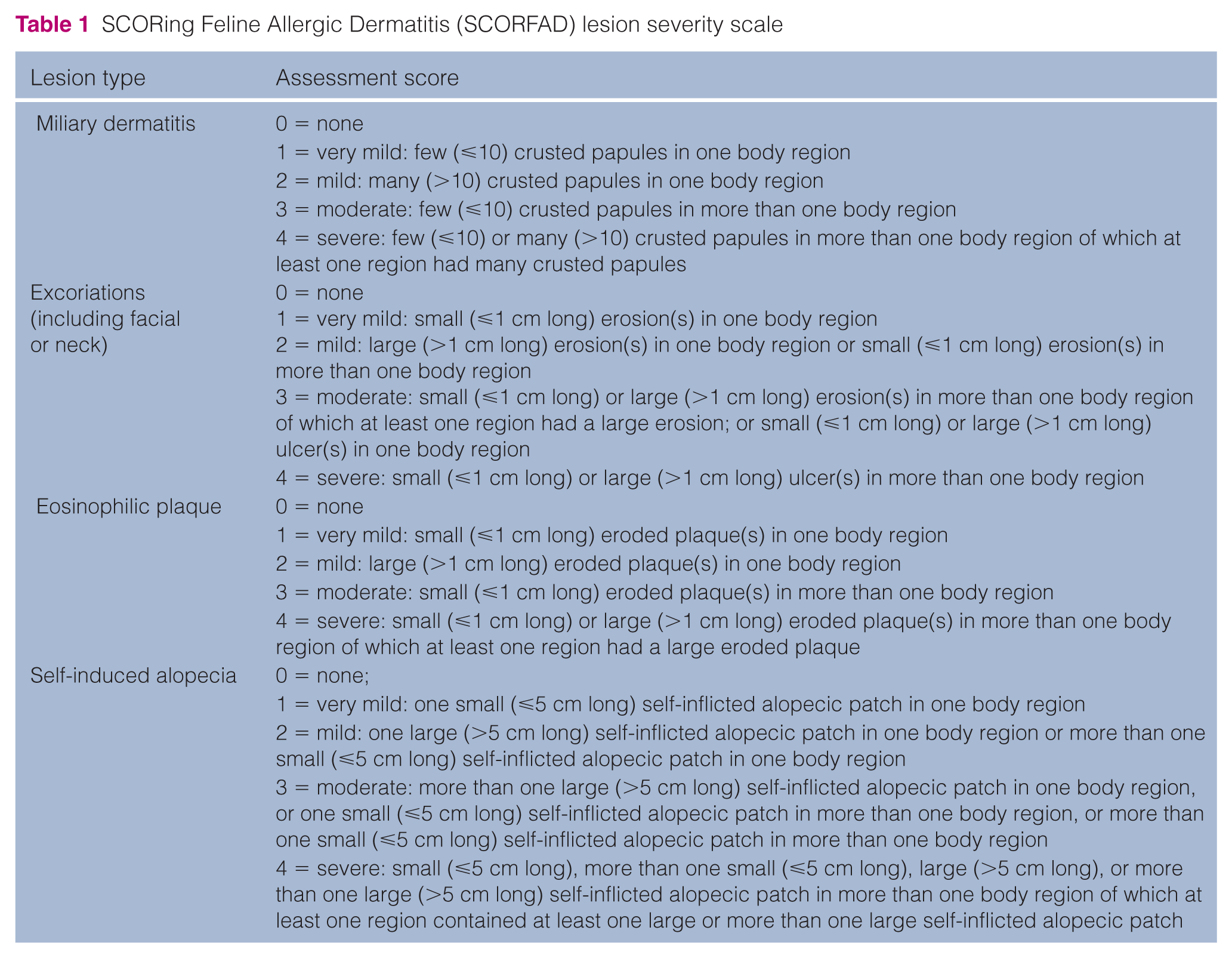
To be included, a minimum total lesion score of 2 using SCORFAD (SCORing Feline Allergic Dermatitis; as described below)9,11 was required with the total lesion score being the sum of each individual lesion score (for MD, facial or neck excoriation, SA, EP), each ranging from 0–4 (Table 1).
SCORing Feline Allergic Dermatitis (SCORFAD) lesion severity scale
Dosage and administration
Cats were orally treated either with CsA or placebo. The CsA was provided by Novartis Animal Health as a 100 mg/ml microemulsion liquid formulation contained in a 50 ml glass bottle (Atopica for Cats; ciclosporin oral solution, USP). The target dosage of CsA was 7.0 mg/kg (3.2 mg/lb) body weight administered up to 42 days (dose range of 6.2–8.0 mg/kg) according to the body weight obtained on day 0. The corresponding volume of CsA or placebo was delivered by using a 1.0 ml syringe graduated in 1 lb increments and calibrated to deliver the required volume of treatment according to the cat’s body weight. The placebo formulation was the treatment medication without the active ingredient, was similar in appearance to the active formulation and was presented in identical bottles with similar labeling, to preserve blinding. Initial treatment was administered via mixing with a small quantity of the cat’s normal daily food ration; owners were instructed to withdraw the cat’s food for a sufficient period prior to dosing to encourage the cat to eat the medicated feed. If the cat completely or partially refused the medicated food for two consecutive days, the owner was instructed to administer the treatment directly into the cat’s mouth using the syringe immediately after feeding.
For ethical reasons, cats considered as not responding to treatment and which had received treatment for at least 10 days could be withdrawn.
Concomitant treatments
All concomitant treatments administered during the trial were recorded. Prohibited treatments included topical and systemic glucocorticosteroids, megestrol acetate, antihistamines, essential fatty acids (when not used for at least 57 days prior to day 0 and maintained with the same treatment regimen throughout the study), topical calcineurin inhibitors, clomipramine, amitriptyline and fluoxetine, drugs interfering with CsA such as ketoconazole, itraconazole, erythromycin and phenobarbital, if given for more than 7 days during the study, allergen-specific immunotherapy and shampoos, except where the same regimen was maintained throughout the study. Shampoos that contained antifungals, antibiotics or corticosteroids were not allowed. With the exception of those interfering with CsA metabolism, such as erythromycin (macrolides), the use of topical or systemic antibiotics (eg, beta-lactams, cephalosporins and fluoroquinolones) were permitted. In cases of severe facial/neck pruritus, at the discretion of the investigator, an Elizabethan collar could be used at study initiation and up to a maximum of 10 days thereafter. Obligatory treatment included a flea adulticide for the duration of the study except in regions not considered flea endemic areas.
Efficacy evaluations
The study consisted of four visits in which the prestudy visit determined inclusion suitability. Clinical and dermatological evaluations were conducted on the day of inclusion (day 0), day 21 (± 3 days) and day 42 (± 3 days) or final visit (FV). Treatment was initiated on day 1. Each case was to be evaluated by the same individual (investigator and owner) throughout the study.
For each lesion (MD, facial or neck excoriation, SA, EP), the lesion extent and severity was measured using the SCORFAD, a scoring system previously described and partially validated.9,11 The SCORFAD is a numerical rating scoring system to assess the severity of excoriations, SA, MD and EPs in each of 10 body areas, resulting in a total lesion score (TLS) of 0–16 per area. Each lesion type was scored independently with a numerical rating scale with five degrees of severity (0 = none; 1 = very mild; 2 = mild; 3 = moderate; 4 = severe). The 10 body regions evaluated were the head, neck, dorsal and lateral thorax, rump and tail, flanks, sternum and axilla, abdomen, perineum, front limbs and paws, hindlimbs and paws. The TLS was calculated as Excoriation + MD + EP + SA. The change in TLS = (TLS on day 0) – (TLS at FV [day 42 or FV]).
The owner evaluated the overall clinical improvement independent of the investigator on day 21 and day 42 or FV, using a numerical rating scale (NRS) from 0 to 4 (Table 2), compared with baseline (day 0). The variable was then evaluated as a binomial success/failure where a score of 0 or 1 (excellent or good, respectively) constituted a treatment success and a score of 2, 3 or 4 (fair, poor or worse, respectively) a treatment failure.
Assessment of overall clinical improvement used by owners and investigators

The two co-primary endpoints were defined as the numerical change in TLS from day 0 to day 42 or FV and the binomial success/failure of the owner’s assessment of overall clinical improvement. The overall criteria for product effectiveness were based on the superiority of both co-primary endpoints in favor of the CsA group, as well as a 50% or greater reduction of the CsA-treated group TLS population mean at baseline compared with the population mean of the treated group TLS at the final assessment (day 42 or FV).
The following variables were secondary endpoints: investigator assessment of overall clinical improvement assessed at day 21 and day 42 (or FV) using the same NRS as the owner’s (Table 2); and pruritus assessed independently both by the investigator (investigator assessment of pruritus) and the owner (owner assessment of pruritus) on day 0, day 21 and day 42 (or FV). The owner assessment of pruritus evaluated pruritus using a visual analog scale based on the owner’s observations of their cat’s behavior during the 3 days before the visit. A line was marked on a linear scale from ‘0’ to indicate that the cat was comfortable, with normal grooming behavior, to ‘100’ to indicate that the cat was uncomfortable and was grooming all the time. The investigator assessment of pruritus used a NRS from 0 to 4 (Table 3). In addition, treatment acceptance was assessed daily by the owner.
Investigator assessment of pruritus

Safety evaluations
Before including a case in the study, the investigators evaluated clinical pathology (hematology and clinical chemistry) completed between day −14 and day −2, and the results of the pre-screen for FeLV, FIV and toxoplasmosis (IgG and IgM titers). Clinical pathology samples were analyzed by a central laboratory (Antech Diagnostics). In addition, a thorough clinical examination with body weight was performed on day 0 and repeated at each subsequent visit. At study exit or in cases of premature withdrawal, a follow-up physical examination, clinical pathology evaluation and toxoplasmosis screen was performed. Adverse events (AEs) were monitored by the owner and the investigator throughout the study. An AE was considered to be any observation that was unfavorable or unintended and occurred after the administration of either CsA or placebo. All suspected AEs were documented whether or not considered to be treatment related. A causality assessment for each AE was performed prior to unblinding.
Statistics
Sample size calculations, based on results from a previously published study, assuming that the mean baseline scores would decrease as seen in this study on day 43, indicated that at least 21 control and 42 treated animals were needed to obtain a significant difference at P <0.05 with 80% power. The minimum sample size included 100 CsA- and 50 control-treated cats to evaluate sufficiently the clinical safety of CsA treatment.
The experimental unit was the individual cat. Groups were compared using two-sided tests with a 0.05 level of significance. All cases receiving treatment were included in the safety evaluation. At least two evaluable cases per site were required for site inclusion in the efficacy evaluations.
For the TLS, according to predefined rules, missing values at study exit were imputed using the last observation carried forward method, the baseline observation carried forward method or complete exclusion from the effectiveness database.
For the owner assessment of overall clinical improvement, in case of missing values, predefined rules allowed the case to be considered as a failure or to be excluded from the effectiveness dataset.
Change in the TLS was analyzed using ANOVA, including treatment, site and the interaction treatment by site effects. The results of the owner assessment of overall clinical assessment were individually dichotomized as success or failure, and analyzed (SAS, Version 9.1.3 PROC GLIMMIX; SAS Institute) with the same effects as above.
Quantitative secondary variables (owner assessment of pruritus, number of regions with lesions) were evaluated using a repeated-measures analysis of variance (RMANOVA; SAS, Version 9.1.3 PROC MIXED, SAS Institute).
Qualitative secondary variables (investigator assessment of pruritus, investigator assessment of clinical improvement) were also analyzed (SAS, Version 9.1.3 PROC GLIMMIX; SAS Institute).
Hematology and clinical chemistry variables were statistically evaluated using ANCOVA with the pretreatment value used as a covariate. Values were also classified as high, normal or low based on laboratory-provided normal ranges. A Cochran–Mantel–Haenszel test was performed on the tendency of animals to move towards the category high or leave this category.
Body weight was analyzed using a RMANOVA model, including treatment, time, site, treatment*time, treatment*site and treatment*time*site. Change in body weight between study exit and baseline was analyzed using an ANOVA.
Results
Study population and demographics
Among the 217 cats enrolled in the study (144 in the CsA group and 73 in the placebo group), 164 completed the study normally and 53 had a premature withdrawal. The reasons for withdrawal included safety for three cats (two in the CsA group and one in the placebo group; each cat exhibited gastrointestinal signs that resolved following cessation of treatment), lack of efficacy for 41 cats (12 in the CsA group and 29 in the placebo group), and miscellaneous other reasons for nine cats – four cats that the owners were unable to medicate (three in the CsA group and one in the placebo group) and five cats where there was a lack of owner compliance/loss to follow-up (two cats in the CsA group and three treated with the placebo). Cases with protocol deviations were excluded from the effectiveness dataset such that effectiveness was evaluated in 181 cats (120 cats treated with CsA and 61 that received the placebo). All cases were included in the safety analysis.
The population consisted of male (42%) and female (58%) cats that ranged in age from 11 months to 16 years, with a mean ± SD age of 6.1 ± 3.6 years in the CsA group and 6.7 ± 3.3 years in the placebo group. Study animals weighed between 1.95 and 9.91 kg (4.3–21.8 lb). Fifteen different breeds were represented in the study, with 71% being domestic shorthairs. Cats were fed a commercial diet, with only two cats receiving a homemade diet. The majority of cats (88%) were described as living indoors only, with the remaining 12% living both indoors and outdoors. Ninety-four percent of the cats were located in suburban areas.
Efficacy results
Primary efficacy variables
Mean TLS values are presented in Table 4. At inclusion, the majority of the cats had a TLS ⩾6 (64.1 % of cats in the CsA group and 67.2 % of cats in the placebo group). The mean ± SD TLS decreased from baseline to visit 3 by 4.73 ± 3.54 in the CsA group and 0.69 ± 2.83 in the placebo group. The difference between groups was statistically significant (P <0.0001) in favor of the CsA-treated group. The corresponding average reduction in the TLS was 65.1% in the CsA-treated group compared with 9.2% in the placebo-treated group. At visit 3, a TLS ⩾6 was still observed in 13.3 % of cats in the CsA group and 55.7% of the cats in the placebo group. Conversely, 46.7% of the cats in the CsA group had a TLS ⩽1 vs 3.3% only in the placebo group at visit 3.
Summary statistics for the total lesion score (TLS), overall assessments, owner assessment of pruritus, and number of regions with lesions

Statistically significant at P ⩽0.01
The model did not converge
Visit 1 = day 0; visit 2 = day 21; visit 3 = day 42 or final visit if exited early; CsA = ciclosporin
At study exit, the owners considered the clinical response successful in 78.6% of the cases in the CsA-treated group vs 26.2% of the cases treated with the placebo. Table 4 shows that the difference was statistically significant (P <0.0001) for the owner assessment of overall clinical improvement.
Secondary efficacy variables
At study exit, the investigator overall assessment of overall clinical improvement was significantly better (P <0.0001) in the CsA group, with mean ± SD values of 1.03 ± 1.12 compared with 2.68 ±1.20 in the placebo group (Table 4). Although not statistically analyzed, numerical values at visit 2 were improving in the CsA group. A statistically significant difference was also found between treatment groups in the owner assessment of pruritus at visit 3 (P <0.0001). Although not statistically significant, mean values were better in the CsA group (0.93 ± 1.13) compared with the placebo group (2.58 ± 1.15) at visit 3 for the investigator assessment of pruritus.
As shown in Table 5, at inclusion, the majority of cats were presented with fair-to-severe pruritus (84% were assessed as >50% by the owner and 80% were scored a 3 or a 4 by the investigators). However, at visit 3, in the CsA-treated group, 78% of the cats were considered comfortable by the owner (score ⩽50%) when only 25% of the cats in the placebo group had a comparable score. These results were confirmed by the investigators who evaluated 72% of the cats as a 0 or a 1 (comfortable or tolerable and calm) in the CsA group and only 15% in the placebo group.
Owner and investigator assessments of pruritus score by treatment and visit

CsA = ciclosporin
While the number of regions with lesions was not statistically different between treatment groups at day 0, a statistically significant difference was seen at visit 2 and at visit 3 (P = 0.0020 and P <0.0001, respectively). At inclusion, the majority of cases had lesions distributed over several areas of the body. A total of 37% of the cats had lesions in 2–3 body regions, 43% had lesions distributed over 4–6 regions and 15% had lesions present in ⩾7 body regions (Table 6). At study exit, 61% of the CsA-treated animals had lesions in one or no body regions with only 12% having lesions in four or more body regions. In comparison, 8% of the placebo control animals had lesions in one or no body regions, while 52% had lesions in four or more body regions.
Frequency of number of body regions with lesions by treatment and visit

CsA = ciclosporin
Concomitant treatments
CsA was used in conjunction with various medications such as parasiticides, antimicrobials, nutritional supplements, topical skin cleansers and drying agents for allergic skin disease. Of the 144 cats treated with CsA, 26 (18%) were on concurrent systemic antimicrobials and six (4.2%) were on concurrent topical otic products (ear cleaners, antimicrobials). Of the 73 cats in the placebo group, 12 (16.4%) were on concurrent systemic antimicrobials and four (5.5%) were on concurrent topical otic products. A total of 67 cats treated with CsA were also treated with a macrocyclic lactone (primarily selamectin) as a flea adulticide.
Acceptance
Out of the 69 cats in the CsA-treated group and that completed the study normally, 35% accepted dosing in the food at day 42 (vs 42% in the placebo group), while the remaining cats were treated by mouth. Owners were able to medicate 97.9% of the CsA-treated cats.
Safety results
No distinct breed, age, dose or sex predilections for AE reporting was noted. A total of 179 adverse drug reports were described in 115 cats (84 animals in the CsA-treated group and 31 in the placebo group). Digestive tract disorders were the most frequently reported AEs (40% of the cats in the CsA-treated group and 39% in the placebo group) and included, primarily, vomiting, diarrhea and hypersalivation. Five of the 179 reports were considered clinically serious (four CsA-treated animals and one placebo control cat). Those clinically serious reports in cats with a possible relationship to treatment with CsA included such clinical signs as weight loss, lethargy and lymphopenia. Three of the cats reported to have clinically significant AEs completely recovered for a satisfactory outcome (two CsA and the one placebo treated cat); the remaining two cats’ conditions (with reported weight loss and lethargy) were considered static at study exit. No cats died and none of the cats were reported to have any neurologic adverse effects. Fourteen cats (10 in the CsA group and four in the placebo group) were reported to have positive Toxoplasma titers (either IgM or IgG or both) during the study.
Follow-up titers showed that some animals returned to a negative status, while some titers persisted. All positive titers occurred in the absence of clinical signs of disease.
Cats treated with CsA tended to lose weight with a weight change of −0.12 ± 0.27 kg (–0.26 ± 0.60 lb) in the CsA-treated group between inclusion and study exit vs 0 ± 0.21 kg (–0.01 ± 0.47 lb) in the placebo group. Evaluation of clinical pathology (hematology and clinical chemistry) indices showed no overall clinically relevant abnormalities associated with CsA treatment. However, there were statistically significant increases in several clinical chemistry parameters, including total bilirubin, glucose, urea nitrogen and creatinine in CsA-treated cats compared with placebo control animals; all means remained within the normal ranges for cats so that any biological relevance is uncertain.
Discussion
Recently, several studies have reported the use of CsA in cats with feline HD as a promising alternative therapy to existing drugs used to control this disease.6–8 However, published literature reports the use of differing doses of CsA, making the interpretation of efficacy and safety results difficult. In addition, well-controlled clinical trials with a large number of cats with HD were needed to confirm the efficacy and safety of such a treatment. A placebo-controlled, double-blinded, dose-determination clinical trial recently published established that 7.0 mg/kg (3.2 mg/lb) CsA dosed daily was efficacious and well tolerated in the treatment of feline HD. 9 The study was conducted in 88 cats, 33 of which received CsA at a dose of 7.0 m/kg. The results of this current prospective, randomized, double-blinded, placebo-controlled clinical trial, with 217 cats (including 145 cats treated with 7.0 mg⁄ kg [3.2 mg/lb] CsA daily) diagnosed with non-flea, non-food HD confirmed these previous findings.
The study population of this trial represented a wide range of demographic and geographic conditions for cats with feline HD. Evaluation of treatment efficacy included both investigator and owner assessments. Feline HD, like atopic dermatitis in dogs, 12 is a disease with a significant impact on the quality of life of both the animal and owner. Therefore, efficacy was based both on clinical dermatological observations and daily observations as assessed by the pruritus score provided by the owner and the owner assessment of the overall clinical improvement.
To demonstrate success, three criteria were predefined: a statistically significant superiority for the two co-primary endpoints, the TLS and the owner assessment of the overall clinical improvement, together with a change between study exit and inclusion in the TLS of at least 50%. A 50% reduction in the canine atopic dermatitis extent and severity index appeared to be a relevant marker of treatment success in dogs with atopic dermatitis. 13 This 50% reduction was also shown to be a marker of treatment success in cats (SCORFAD). 11 All three of these predefined endpoints were achieved in this trial. Results showed that there was a 65.1% reduction in the TLS in the CsA-treated group, representing a significant improvement (P <0.0001) from visit 1 to visit 3. In addition, owners assessed 78.6% of the cases in the CsA group as a success at the end of the treatment period (P <0.0001 difference between treatment groups). This was also comparable with the investigator overall assessment, which noted that 72% of the cats treated with CsA appeared to be comfortable. Efficacy in this trial was also confirmed through the reduction in pruritus, as determined by both the owner and by the investigator. These results are comparable with those obtained in the trial conducted by King et al, 9 who also demonstrated a significant improvement in TLS and pruritus, which correlated with clinical improvement as assessed by both the owner and investigator. After a maximum duration of treatment of 6 weeks, approximately half of the cats treated with CsA (46.7%) had a TLS ⩽1 meaning that at least 3/4 lesions evaluated in this trial (MD, excoriations including facial or neck, SA and EPs) were absent, with a one lesion maximum assessed as very mild. In addition, the improvement in the extent of the signs was significant. Indeed, at inclusion, about half of the cats had extensive lesions in 4–6 body regions, with only 5.5% of the cats with lesions in one body area, while at study exit 61% of the CsA-treated cats had no lesions or lesions present in only one body region. Therefore, treatment with CsA was shown to decrease the severity, extent and discomfort of feline HD.
Feline hypersensitivity dermatitis is a lifelong disease and therefore compliance can be a challenge to maintain adequate disease management. The method and ease of administration and the resulting efficacy of the treatment are key elements of treatment compliance. After 6 weeks of treatment with CsA, 35% of the cats were still receiving their treatment mixed with food. At study exit, the majority of cats were dosed by mouth using a syringe; only 2.1% of the cats treated with CsA were unable to be medicated. These results are also comparable with those of King et al, 9 who reported that the majority of cats were either dosed by mouth or received the medication mixed with food, with only one cat withdrawn because it was not possible to administer medication.
Safety was evaluated through physical examinations, clinical pathology (including a screen for FeLV, FIV and Toxoplasma gondii) and the monitoring of AEs by owners and investigators. As in dogs, the most frequent AEs associated with CsA treatment in cats were gastrointestinal, mostly vomiting, diarrhea and hypersalivation. There were approximately equal numbers of gastrointestinal events in the CsA and placebo groups. A true placebo (formulation excipients without the active ingredient CsA) was used in this trial, suggesting that the gastrointestinal signs reported may be, in part, due to the formulation rather than to the CsA itself. There were four serious reports in four animals treated with CsA. One cat was reported to have lymphopenia (1073 changed from study entrance value of 1886; normal range 1200–8000/µl). Two cases experienced body weight loss (23.6% loss and 14.3% loss, respectively); however, in both cases the weight loss was confounded by a prescribed weight loss diet and hyperthyroidism, respectively. Another case had reported weight loss of 11.4% and lethargy of unknown cause. The majority of clinically significant AEs completely recovered. Overall, cats treated with CsA tended to lose weight during this study. This is comparable with the results reported by King et al; 9 however, in that study the reported weight loss occurred with initial treatment and reversed over the final 3 weeks of the 6 week trial. (Phase 2 results, where animals enrolled into Phase 1 and continued on tapered CsA therapy for an additional 12 weeks, also showed that there was no statistically significant change in body weight when measured from visit 1 [in Phase 1] to the FV of Phase 2, although two animals were reported with persistent progressive weight loss [weight loss >20% of initial body weight] that resulted in hepatic lipidosis.) Evaluation of clinical pathology indices showed no overall clinically relevant abnormalities associated with CsA treatment.
From these data, CsA treatment appears to be well tolerated in cats when used to control feline HD. These results are consistent with findings from Roberts et al, 14 which showed that CsA was safe and well tolerated by cats, even at multiples of the recommended dose of 7 mg/kg, given daily over a 6 month period. However, as CsA is known to be immunosuppressive, animals with significant systemic disease, including neoplasia or serious infectious disease, should not receive CsA for the control of this skin disease.
Conclusions
This study confirms that CsA given at a dose of 7.0 mg/kg (3.2 mg/lb) daily is efficacious and well tolerated in the treatment of feline non-flea, non-food HD.
Footnotes
Acknowledgements
We thank all the study investigators who contributed cases: Drs Beale, Bloom, Burkett, Byrne, Cannon, Daigle, DeManuelle, Friberg, Grieshaber, Griffin, McKeever, Messinger, Rosales, Schick, Spiegel, Stokking, Tapp, Tater, Thomas, Trimmer, Vitale, Waisglass, White and Williamson. We also thank the cats and cat owners; and the team at Novartis Animal Health.
Conflict of interest
ESR, CS, JS, LR and SK were full-time employees of Novartis Animal Health. CF and CG have received related and unrelated consultancy fees, lecture fees or investigator fees from Novartis Animal Health.
Funding
This study was funded by Novartis Animal Health US.