
Abstract
The pharmacokinetics of terbinafine was studied in six healthy fasted cats following a single intravenous and oral administration at a dose of 10 mg/kg and 30 mg/kg, respectively, according to a two-period crossover design. Plasma terbinafine concentrations were determined using a reverse phase liquid chromatographic method. The pharmacokinetic parameters were calculated by non-compartmental analysis with WinNonlin 5.2.1 software. After intravenous administration, the terminal half-life and area under the curve from time 0 to infinity were 10.40 ± 4.56 h, 15.20 ± 3.61 h·µg/ml, respectively. After oral dosing, the mean maximum concentration was 3.22 ± 0.60 µg/ml, reached at 1.33 ± 0.41 h. The terminal half-life, area under the curve from time 0 to infinity and apparent volume of distribution were 8.01 ± 3.46 h, 13.77 ± 4.99 h·µg/ml, 25.63 ± 6.29 l/kg, respectively. The absolute bioavailability of terbinafine hydrochloride tablets after oral administration was 31.00 ± 10.85%. Although bioavailability was low, excellent penetration at the site of infection and low minimum inhibitory concentrations values provided terbinafine with good efficacy against dermatophyte infections.
Introduction
Dermatophytosis is a common and infectious disease in cats. The fungi that cause these infections are known as dermatophytes. Cats are vulnerable to dermatophytes, which can also be easily transmitted to humans, threatening human health. 1
Terbinafine is a new allylamine antifungal agent with a unique mechanism of fungistatic action. It can selectively inhibit squalene epoxidase, thereby resulting in the reduction of ergosterol, which interferes with membrane function and cell growth.2–4
Over 20 different species of dermatophytes can cause clinical disease in cats, but Microsporum canis, Microsporum gypseum and Trichophyton mentagrophytes are the most commonly isolated pathogens.5,6 Compared with other agents, such as natifine, itraconazole and fluconazole, terbinafine usually shows excellent antifungal activity against these three pathogens.6–12 Although terbinafine is not licensed for use in veterinary medicine, it is often used by veterinary practitioners to treat fungal infections of dogs and cats.
The pharmacokinetics of terbinafine has been evaluated in humans, horses, dogs, penguins and hawks.13–17 Although some studies on the determination of terbinafine in cat plasma and hair have been described,18–20 little is known about the pharmacokinetics of terbinafine in cats. Given the use of terbinafine in cats, the purposes of this study were to assess the pharmacokinetics of terbinafine in cats after intravenous (IV) and oral administration, and to determine the oral bioavailability of terbinafine hydrochloride tablets.
Materials and methods
Animals
Six adult domestic mixed breed cats (two females and four males) weighing 3.51 ± 0.75 kg (mean ± SD) were used in the study. They were kept in individual cages in a room where temperature and humidity could be controlled. Two weeks prior to the study, cats were confirmed to be healthy by physical examination and were dewormed with fenbendazole 50 mg/kg (Jindun Animal Pharmaceutical) for three consecutive days. Food was withheld for 12 h before the experiment and until 4 h after drug administration. At other times, they were fed a standard dry cat food and water was provided ad libitum. The study was approved by the Institutional Animal Care and Use Committee of South China Agricultural University.
Drugs
Terbinafine hydrochloride tablets (Lamisil Tablets, 250 mg; Novartis) were used.
Powdered terbinafine hydrochloride (>99% purity; East-Asia Pharmaceutical) was dissolved in a sterile ethanol solution (50%, v/v) to a concentration of 50 mg/ml. The solution was filtered through a 0.22 µm membrane. Owing to the poor solubility of terbinafine hydrochloride in water, 50 mg/ml was the maximum concentration that remained in solution; higher doses would result in precipitation. The solution was made 24 h before the study and stored at 4°C until use.
Drug administration
A two-period crossover design with a washout of 2 weeks was used in this study. Six cats were randomly divided into two groups: an IV group and an oral group. For the oral group, terbinafine hydrochloride tablets were divided according to the individual dosage, placed into gelatin capsules and given at a dose of 30 mg/kg. Following each dose, 2 ml of tap water was administered orally to ensure that the capsules passed to the stomach. An IV dose of 10 mg/kg was administrated through the cephalic vein over 1 min. After 2 weeks washout period, the experiment was repeated by change of treatment. Each cat was observed daily for abnormal clinical signs.
Blood sampling
For sample collection, a 20 gauge, 20 cm catheter (Target Medical Technologies) was placed in a jugular vein 2 days before the study according to a technique previously described. 21 The cats were sedated with ketamine (5 mg/kg, IM) and zolazepam (5 mg/kg, IM) for jugular catheter placement. After IV and oral administration of terbinafine, blood samples (1.5 ml) were collected at 0, 0.083, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12 and 24 h, and were placed immediately into heparinised blood collection tubes. At each collection the catheter was flushed by injection of 1.5 ml 0.9% saline and then 200 µl of heparin solution (10 IU/ml in 0.9% saline). The samples were centrifuged at 3000 × g for 10 mins and stored at −20°C until analysis.
Terbinafine determination
The analytical method was derived from a fully validated method described previously. 22 Briefly, terbinafine was extracted from plasma (0.5 ml) by n-hexane and back extracted into a mixture of 0.5 M sulphuric acid 2-propanol (85:15 v/v) prior to injection onto the high performance liquid chromatography (HPLC) system. Chromatographic separation of terbinafine was carried out on an Acquity UPLC (Waters Corporation). The column was a reversed-phase Hypersill BDS C18 (5 µm, 250 mm × 4.6 mm). The mobile phase was an aqueous solution of 0.012 M triethylamine/0.020 M orthophosphoric acid with acetonitrile (55:45 v/v). Detection was performed by using ultraviolet detector at 224 nm with a flow rate of 1 ml/min, the injection volume was 20 µl and the column temperature was maintained at 24°C.
Acetonitrile from Fisher Scientific was HPLC grade, and HPLC-grade water was obtained by using a Milli-Q water purification system (Millipore). Other reagents used in this study were of analytic grade purchased in China.
The concentration of terbinafine in each sample (eg, calibrators, quality control and unknowns) was determined by an external standard method. The limit of quantification for terbinafine in feline plasma was 0.05 µg/ml, which was the lowest concentration that could be accurately back-calculated on the linear calibration curves.
Pharmacokinetic analysis
Non-compartmental pharmacokinetic analysis was performed with the aid of WinNonlin 5.2.1 (Pharsight Corporation). The main pharmacokinetic parameters included area under the curve (AUC), terminal half-life (t1/2), total body clearance for IV administration (ClB) and apparent volume of distribution per fraction of the dose absorbed (Vd/F). The area under the curve (AUC0–∞) was calculated by the trapezoidal rule with extrapolation to infinity. The absolute oral bioavailability (F) of terbinafine hydrochloride tablets was calculated using the following equation:

All pharmacokinetic parameters were expressed as an arithmetic mean ± SD.
Results
Terbinafine assay
Terbinafine is stable and potent in stored solutions at 4°C and feline plasma at -20°C.
Because the concentrations were expected to be different between IV and oral administration, two standard curves were made. One was confirmed for each assay (r2 ≥ 0.9994)within the concentration range of 0.05–5 µg/ml, the other was confirmed for each assay (r 2 ≥0.9973) within the concentration range of 1–20 µg/ml. Precision analysis showed that the intra-assay coefficient of variation ranged from 2.14% to 3.77% and the inter-assay coefficient of variation was 2.52% to 4.04%. The recoveries ranged from 76.4% to 83.5%.
Clinical effects
After IV injection, five of the cats entered a period of lassitude, but they recovered quickly within 15 mins. Haemolysis of the plasma was noted during the first 3 h, but after 3 h the samples appeared clear. No adverse effects were observed after oral administration.
Pharmacokinetic analysis
The mean plasma concentrations of terbinafine versus time after IV and oral administration are presented in Figure 1 and Figure 2, respectively. The pharmacokinetic parameters are summarised in Table 1. The terminal half-life of terbinafine after oral administration was 8.01 ± 3.46 h, which was shorter than IV dosing (10.40 ± 4.56 h). The Vd/F (corrected by oral bioavailability) was 25.63 ± 6.29 l/kg. The absolute bioavailability (F) of terbinafine hydrochloride tablets was 31.00 ± 10.85%.

Mean plasma concentrations of terbinafine after IV (10 mg/kg) administration in cats (n = 6)
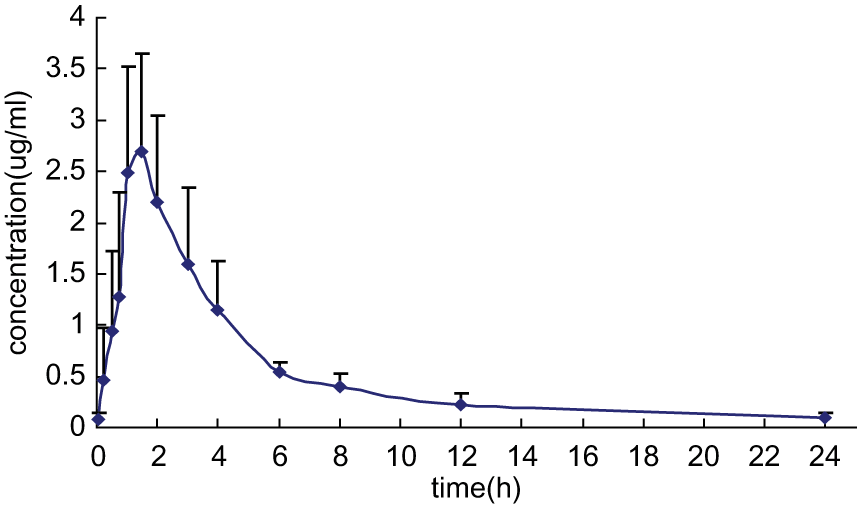
Mean plasma concentrations of terbinafine after oral (30 mg/kg) administration in cats (n = 6)
Pharmacokinetic parameters determined following a single IV (10 mg/kg) and oral (30 mg/kg) administration of terbinafine in cats (n = 6, mean ± SD)

tmax = time to maximum plasma concentration, Cmax = maximum plasma concentration, AUC0-24 = area under the curve from time 0 to 24 h, AUC0-∞ = area under the curve from time 0 to infinity, ClB = total body clearance, Vd/F = apparent volume of distribution, area method per fraction of the absorbed dose, t1/2 = terminal half-life, F = the absolute oral bioavailability
Discussion
Many studies have assessed the pharmacokinetics of terbinafine following oral administration in humans.15,16,23–25 Related studies have also been carried out in animals, including African penguins and red-tailed hawks,13,14 horses and Greyhounds. 17 However, to date, no information about the pharmacokinetics of terbinafine after IV or oral administration in cats has been reported.
In humans, after a single 250-mg oral dose, terbinafine is well absorbed (>70%). A peak plasma concentration of 0.8–1.5 µg/ml is reached within 2 h. It is extensively metabolised by the liver and approximately 70% of the drug is eliminated in the urine. 26
The main pharmacokinetic parameters of terbinafine after oral administration in cats compared with other animal species are presented in Table 2. Absorption of terbinafine from the gastrointestinal tract was more rapid in cats (1.33 h), Greyhounds (2.0 h) and horses (1.7 h) than in red-tailed hawks (3.4 h) and African penguins (7.0 h). Following an oral dose of 30 mg/kg, maximum plasma levels were 3.22, 4.01 and 1.2 µg/ml in cats, Greyhounds and red-tailed hawks, respectively. The half-life of terbinafine was not significantly different among cats (8.01 h), Greyhounds (8.6 h) and horses (8.1 h), while it is significantly longer in red-tailed hawks (17.5 h) and African penguins (17.0 h). The apparent volume of distribution across animals tested was larger than total body water, which is comparable to what has been reported in humans. 26 The large volume of distribution of terbinafine is a result of its extensive distribution to adipose tissue, dermis, epidermis and nail, 27 and the slow redistribution of the drug from peripheral tissues into the blood because of high tissue binding.
Non-compartmental pharmacokinetic parameters (mean) of terbinafine given orally to different animal species(arithmetic mean)

tmax = time to maximum plasma concentration, Cmax = maximum plasma concentration, t1/2 = terminal half-life, Vd/F = apparent volume of distribution, area method per fraction of the absorbed dose
The half-life of terbinafine after oral administration (8.01 ± 3.46 h) in cats was shorter than for IV administration (10.40 ± 4.56 h) and the oral bioavailability was low (31.00 ± 10.85%). This may be attributed primarily to first-pass metabolism of terbinafine, which has been described in humans,15,17 Greyhounds and horses. 17 Food can improve the extent of absorption of terbinafine and increase the Cmax in humans. 25 In our study, terbinafine was administered without a meal. Comparison of oral bioavailability of terbinafine in cats with that of other animals is difficult as no data have been published following IV administration of terbinafine in animals or humans.
Although the bioavailability of terbinafine is low, it is important to recognise that the concentrations of antifungal drugs, such as terbinafine, at the target site of infection are much more useful than plasma concentrations. Terbinafine can extensively distribute to the site of infection and remain active to achieve a clinical cure. Terbinafine can persist in the hair of cats at concentrations above the minimum inhibitory concentration (MIC) for several weeks after a short-term therapy (14 days) at a daily dose of 34–45.7 mg/kg. 18 The mean MIC values of terbinafine against the most common pathogens M canis, M gypseum and T mentagrophytes are 0.01 µg/ml, 0.012 µg/ml and 0.007 µg/ml, respectively. 28 The low MIC values provided terbinafine with good efficacy against fungal diseases despite its low bioavailability.
To the our knowledge, the pharmacodynamic parameter (T >MIC, AUC/MIC or Cmax/MIC) most predictive of antibiotics efficacy has not been established in humans and animals, and this strategy is rarely applied to antifungal agents. Therefore, we are unable to know whether terbinafine acted by a concentration-dependent killing mechanism or a time-dependent killing mechanism. As strong plasma protein binding (>99%) is observed in human, dog and rabbit plasma, 26 tissue or cellular concentrations would be the most appropriate for determining the required kinetic parameters for drugs which are highly protein bound with a free fraction of <20% in plasma. 29 Dosage recommendations for terbinafine by the pharmacodynamic parameter in plasma may be not suitable.
Terbinafine hydrochloride is freely soluble in methanol and methylene chloride, soluble in ethanol and slightly soluble in water. Methanol and methylene chloride are not suitable for IV administration because of their great toxicity. Ethanol was the final choice, but it was not a good delivery system. A similar situation appeared in the pharmacokinetics of genistein in cats: the author concluded that haemolysis might lead to a slight underestimation of the Vdss, but the effect was thought to be mild. 30
To date, three formulations of terbinafine have been developed in humans: orally-administered tablets and topically-administered cream and spray. Because of the low bioavailability of these formulations, new pharmaceutical formulations, such as IV formulation and oral suspension, need to be explored to improve the solubility of terbinafine and enhance absorption and bioavailability.
In conclusion, in this study, we first described the pharmacokinetic properties of terbinafine after single IV and oral administation, and determined the absolute bioavailability of terbinafine hydrochloride tablets in cats. The results contribute further data to the kinetic disposition of terbinafine in cats.
Footnotes
Acknowledgements
We thank the staff at the Animal Hospital of South China Agricultural University, including Qiping Zhong and Bonian Zhai for providing technical services in collecting blood samples.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
The authors do not have any potential conflicts of interest to declare.
Accepted: 22 February 2012