
Abstract
Fluid dynamics is a ubiquitous problem that arises in different branches of science and industry. It is usually tackled by numerically solving continuum Navier-Stokes type equations. Molecular dynamics has been not a feasible tool to approach fluid dynamics until very recently due to its disproportional computational complexity for relevant system sizes. In this paper, we propose a new type of boundary conditions for molecular dynamics simulations of stationary fluid flows and present its possible GPU-based implementations in OpenMM and LAMMPS. We examine the performance and scalability of the proposed implementations. The benchmarking results show promising performance that makes it possible to reach turbulence in atomistic models of stationary fluid flows using modern supercomputers.
1. Introduction
For the numerical study of laminar and turbulent fluid flows, the generally accepted method of their simulation is numerical solution of the Navier-Stokes equations. Attempts to numerically solve the Navier-Stokes equations date back to the very dawn of the computer era (Kawaguti (1953); de G. Allen and Southwell (1955); Simuni (1964); Chorin (1968)). Since then this approach and its different variations have remained dominant.
Unfortunately, Navier-Stokes equations have fundamental limitations, since the premise that the system can be represented as a continuous field does not hold at the atomistic scale where many important physical processes take place, see e.g. Hadjiconstantinou (2006); Grinberg et al. (2011); Sergeenko et al. (2022). The onset of turbulence is always analysed as a multiscale phenomenon (e.g. see Chashechkin and Mitkin (2001)) and its fundamental understanding in fluid flows should include the atomistic level description at the Kolmogorov’s length scale (Kolmogorov (1941)). The development of high performance computing systems brings the promise that such an atomistic-level description of turbulent fluid flows will come true, and in this paper, we describe our step to this goal.
Classical molecular dynamics (MD) simulation method is a key research tool in many areas of science and engineering. Nowadays MD is one of the major consumers of supercomputer resources worldwide. MD tools that enable ultra-long MD trajectories (up to tens of milliseconds) and extreme MD system sizes (up to trillions of atoms) are important avenues of development for high performance computing methods (Begau and Sutmann (2015); Tchipev et al. (2019); Shaw et al. (2021)). Shortly after the Nvidia CUDA technology had been introduced in 2007, hybrid MD algorithms that use GPU accelerators appeared and showed their promising performance. Currently, GPU-accelerated hardware provides the most efficient and affordable way of doing MD studies as shown in Kutzner et al. (2015, 2019); Stegailov et al. (2019); Kondratyuk et al. (2021) and makes various applied MD studies feasible, e.g. see Nikolskiy and Stegailov (2020); Nguyen-Cong et al. (2021); Antropov and Stegailov (2023); Fominykh et al. (2023); Kondratyuk et al. (2023).
The emergence of parallel distributed memory supercomputing systems stimulated the development of parallel algorithms for MD calculations. Among others, LAMMPS (Thompson et al. (2022)) is an MD package that has developed into a versatile simulation MD toolkit and is widely used nowadays on high performance computing systems. Since the emergence of general-purpose computing on graphics processing units (GPGPU), LAMMPS has been supplemented with GPU offloading capabilities as described in Brown et al. (2011, 2012); Brown and Yamada (2013). Newer MD libraries like HOOMD (Anderson et al. (2008); Glaser et al. (2015)) and OpenMM (Eastman et al. (2013, 2017)) use GPU-oriented MD algorithms that are designed in a way that keeps the amount of CPU-GPU communication to an absolute minimum. This is also a feature of the Kokkos-based variant of LAMMPS (Thompson et al. (2022)) aimed at performance portability, GPU acceleration including.
In this work, we extend our preliminary results (Pavlov et al. (2023)) on the novel kind of boundary conditions for MD of fluid flows, demonstrate a working OpenMM and LAMMPS/Kokkos implementations of such boundary conditions and compare the performance of OpenMM and LAMMPS/Kokkos for very large system sizes.
2. Related work
The first attempts to apply MD for studying eddy formation in fluid flows were carried out by Rapaport and Clementi (1986) in a two-dimensional system and for a small (according to modern standards) number of particles (N ≈ 104). With the rise of supercomputing capabilities in 2000s, MD calculations at extreme length scales were considered in Kadau et al. (2010) as a tool that is able to complement Navier-Stokes-based continuum fluid-simulation methods. MD models revealed the influence of the wall-fluid interactions on slip flow of simple fluids Priezjev (2007) and the structure of oscillatory Couette flows Priezjev (2013). A special MD package ls1 mardyn focused on extreme length scales is under development (Tchipev et al. (2019)) and is being used for the corresponding multiscale simulations (Hitz et al. (2020, 2021)).
Among the most remarkable results published recently we would mention the work of Komatsu et al. (2014) and the work of Smith (2015) that compare MD and Navier-Stokes descriptions of special turbulent flow patterns. Smith (2015) provides the detailed description of MD modelling using the SGI Altix ICE 8200 EX supercomputer with manycore CPUs and Infiniband interconnect. Below, we compare the data of Smith (2015) with our MD calculations for a similar fluid using modern multi-GPU systems.
The coupling between CFD and MD algorithms is under active development: for example, Grinberg et al. (2011) discuss the coupling of Navier-Stokes equations with particle-based models. Smith et al. (2020) described such a coupling for OpenFOAM and LAMMPS. A multiscale 3D model based on the dissipative particle dynamics with special inflow/outflow boundary conditions was implemented in LAMMPS for blood flow simulations by Lykov et al. (2015).
The success of using Kokkos library for performance portable code development aimed at molecular dynamics is exemplified by LAMMPS and other projects, e.g. the recent work on Multi-Particle Collision Dynamics of Halver et al. (2023).
3. Software
As a part of this work, we implemented a new kind of boundary conditions using the OpenMM and LAMMPS packages. Below, we describe both packages briefly.
3.1. OpenMM
OpenMM (Eastman et al. (2017)) is an open source toolkit for molecular dynamics. With its main focus being computational biology, it still performs remarkably well on other systems. This is achieved because unlike the high-level OpenMM Application Layer Python API OpenMM team, 2023a, which is built around domain-specific constructs such as force fields, residue chains and topologies, OpenMM Library Level C++/Python API OpenMM team, 2023b is completely divorced from such constructs and provides a lower-level access to the underlying structures that can be used in a broader range of applications.
OpenMM supports four platforms: Reference, CPU, CUDA, OpenCL. There also exists a separately-developed plugin StreamHPC (2023) that adds HIP support to the list. CUDA, HIP and OpenCL are GPU-oriented platforms, and they provide the best performance when compared to the CPU and Reference platforms which don’t use GPU acceleration. The underlying algorithms that are used in these platforms are almost identical, with most of the code having been merged into a meta-platform called Common Compute. CUDA, OpenCL and HIP platforms operate in mostly the same way.
When parallelizing over a big amount of processing units, there are three wide classes of approaches as described by Plimpton (1995): atom-decomposition, force-decomposition and domain-decomposition. For distributing workload inside a GPU, OpenMM uses a force-decomposition-like algorithm as described in Eastman and Pande (2009): for N atoms, the NxN force matrix is divided into ‘tiles’ of size 32x32. Then, only the tiles that might contain non-zero forces are marked as ‘interacting’ based on comparing the tiles’ coordinate bounds. Then, the list of interacting tiles is traversed to calculate forces and energy. Once the tiles’ bounds change to a point that new interactions might appear, the list of interacting tiles is rebuilt.
One serious downside of this algorithm is its complexity: traversing an NxN force matrix has the complexity of
3.2. LAMMPS
LAMMPS is an open-source package for classical molecular dynamics computations written in C++. It is designed to be compiled and run on local computers as well as on supercomputers, and allows the simulation of many millions of particles. It implements a large number of models of interaction potentials, ranging from the most popular to very rare ones.
The source code of LAMMPS is very extensible, allowing both very easy addition of new interaction potentials and fine-tuning of the numerical integration process by making changes to individual steps. The latter is achieved with the help of a so-called “fix” mechanism (Thompson et al. (2022)).
To optimize the computation of the short-range potential, LAMMPS implements domain decomposition: it splits the simulation box into sub-domains (one per MPI rank) and distributes the computation for atoms within all of them. Moreover, LAMMPS creates Verlet neighbor list (Eijkhout et al., 2016, ch. 7.1.2) for each particle inside the sub-domains which stores atoms near that particle within radius r
c
. For even more optimization, LAMMPS uses the same list at multiple time steps, and the clipping radius is increased to r
v
= r
c
+ 2nd, where d is the estimated maximum distance a particle can travel in a time step, and n is the number of steps at which the neighbor lists are updated. To perform neighbour list construction in linear time, LAMMPS bins each sub-domain’s particles into a uniform cell grid (this is known as the linked cell method). Applying both of these optimizations when calculating the short-range potential yields the asymptotic complexity of
LAMMPS is also capable of optimizing the computation of long-range potentials, either using FFT or the multipole method, but we have not used the long-range potential in our simulations.
The general support of heterogeneous computing in LAMMPS is based on the Kokkos library, which allows writing performance-portable code and is designed for almost all existing supercomputer systems (Edwards et al. (2014)). To use it, you can write the code once, and then you just have to specify the required target platforms in compilation flags. The Kokkos library is integrated into the LAMMPS package as the Kokkos module. In addition to this module in LAMMPS, there is another older module called GPU that uses an offload strategy of GPU acceleration. One can anticipate that LAMMPS/Kokkos will perform better on modern DGX-like GPU servers due to less amount of data transfers between CPUs and GPUs.
4. Flow Boundary Conditions
When running MD simulations, it is not feasible and currently not possible to simulate a macroscopic system due to its enormous amount of particles. So it is common practice to simulate a small system as a part of the whole by putting it in periodic boundary conditions (PBC). However, if one was to simulate a fluid flow using just the PBC, the collective kinetic energy of the flow would dissipate over time, eventually bringing the flow to a halt. In real-world scenarios, whenever there is a stationary flow of some kind, there is also an energy source that keeps pumping kinetic energy into the system to counteract this energy dissipation. This means that a simple NVE simulation with PBC is unfit for these kinds of models.
This problem can be addressed in a number of ways: for example, by introducing moving walls to create a Couette flow (like in Smith (2015)), or by reintroducing particles that left the cell with a reset velocity (like in Rapaport and Clementi (1986)). In this paper, we expand upon the latter method. The Flow Boundary Conditions (FBC) that we introduce are a special kind of boundary conditions that resembles PBC, but resets particle velocities whenever they cross one of the periodic boundaries.
4.1. Derivation of Flow Boundary Conditions for an ideal gas
For simplicity, we assume that all particles in the system are identical, and that the system is an ideal gas. Let us consider the Boltzmann equation for a single-particle probability density function f (
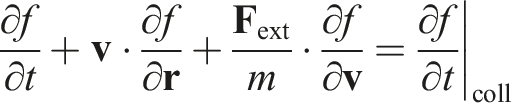
We wish to create a stationary flow by using some sort of boundary conditions. Since the flow is stationary, the desired probability density function fobc is also stationary, so
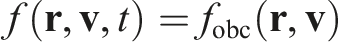
Also, there are no external forces, therefore
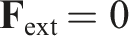
The right-hand side of the equation represents the collision term. It would normally represent changes in the probability density function due to particle collisions. However, since we assume that the gas is ideal, the collision term represents only the particle interactions with FBC. The analytical form of this term shall dictate the exact behavior of FBC. Let us denote it as s (
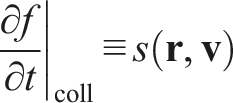
Then, by substituting all of the above into the initial Boltzmann equation, we get
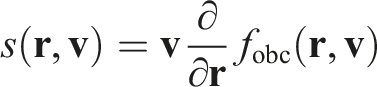
Let us say we want to simulate a part of an infinite stationary flow that has the probability density function f(
Let us assume the left-side boundary is situated at the plane z = z1. Particles to the left of the boundary are not simulated directly, but their impact must be somehow approximated. We can outline two kinds of impact that these particles make: first, they interact with particles on the right side, and second, the left and the right side exchange particles. The former impact is sufficiently approximated by PBC. The latter one, however, is not: in the case with stationary fluid flow, it is impossible to maintain the pressure gradient with just PBC. The solution we propose is to make FBC emit particles into the right side of the left-side boundary, instead of them naturally coming from the left side (and vice versa for the right side). To do this on the left side, we want to remove all particles with z < z1 and v
z
> 0 from the initial probability density function f(
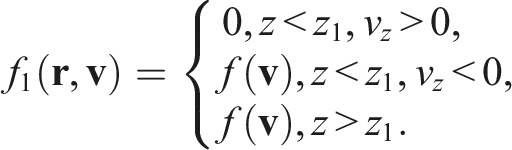
It could be achieved by multiplying the initial distribution by some a1 (z, v
z
):
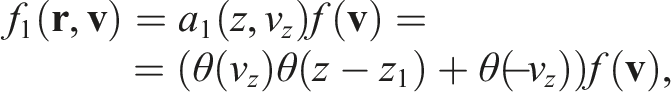
By analogy, for the right-side boundary we can define a2 (z, v
z
):
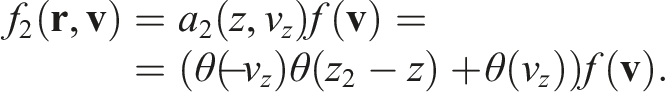
By multiplying the initial distribution f(
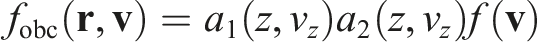
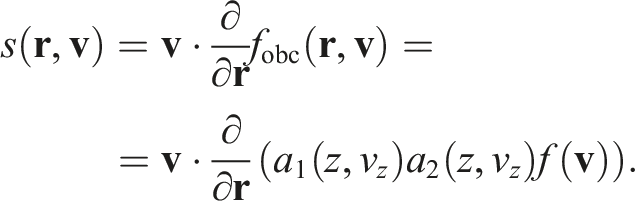
It is worth noting that
Then, finally,
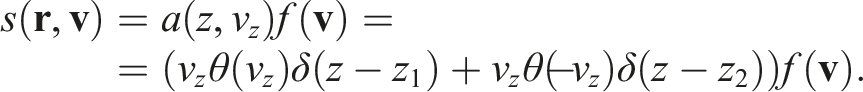
The resulting distribution fobc (
4.2. Flow Boundary Conditions algorithm
The nature of the collision term
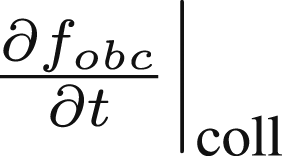 dictates how often, on which side and with what velocity particles are emitted. Our implementation does not change the amount of particles in the system, it only resets their velocities when they hit the boundary, effectively re-emitting them from an appropriate side with an appropriate velocity, according to the distribution. There is an emergent property that since the flow is stationary, the amount of particles that leave the sector z1 < z < z2 equals the amount that is emitted. It eliminates the need to change the amount of particles over the course of the simulation.
dictates how often, on which side and with what velocity particles are emitted. Our implementation does not change the amount of particles in the system, it only resets their velocities when they hit the boundary, effectively re-emitting them from an appropriate side with an appropriate velocity, according to the distribution. There is an emergent property that since the flow is stationary, the amount of particles that leave the sector z1 < z < z2 equals the amount that is emitted. It eliminates the need to change the amount of particles over the course of the simulation.The first step of the algorithm is detecting which particles crossed the border. The simplest way to do it is by iterating over every particle and checking whether or not this event occurred. Once a particle crosses the border, the first thing that needs to be computed is which side it should be re-emitted on (the possibility of the particle being re-emitted is one of the distinguishing traits of our approach; it was not accounted for by Rapaport and Clementi (1986)). The probability of a particle being re-emitted from a certain side can be derived as follows:
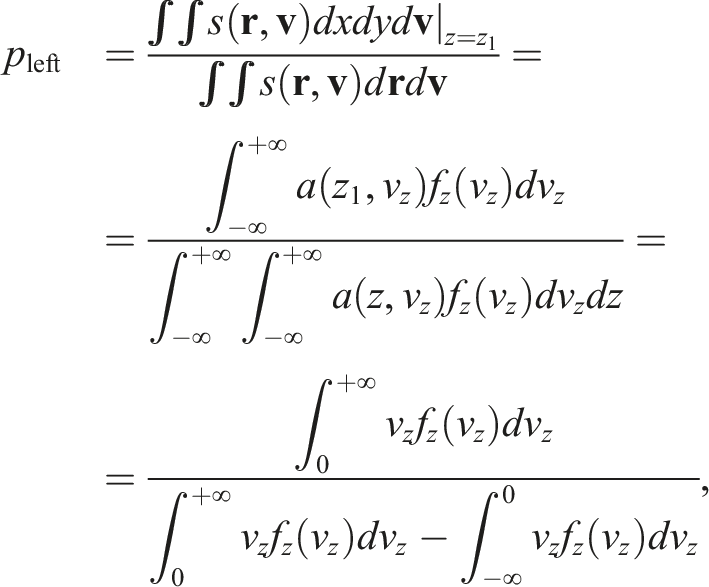
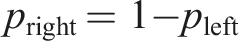
It is worth noticing that the above formula is agnostic to whether the particle left the cell upstream or downstream. In practice, due to the periodic conditions, the re-emission of a particle does not actually require its position to be changed. The re-emission is effectively just a re-assignment of velocity without any necessary modification to the particle position. Since such a mechanism can be represented as a plane that changes the velocity of any particle that passes through, there is no need to even introduce any extra ghost atoms.
From now on, let us assume that the particle is emitted from the left side. The new velocities on x and y need only be picked from f
x
(x) and f
y
(y) distributions (which are just Maxwell distributions in our case). The v
z
can be inferred from the following equation, where ξ0 ∈ (0, 1) is a uniformly-distributed random number:
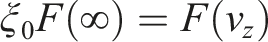
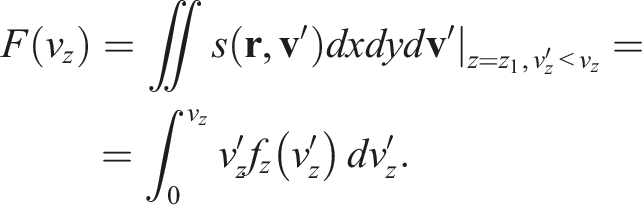
Now, we want the underlying distribution f(
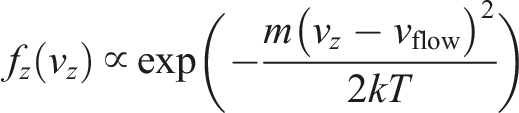
Then F (v
z
) can be computed in terms of error functions, allowing us to solve the equation numerically. We use the Newton’s method to find the root of the equation. From now on, v
i
is the ith approximation on the velocity v
z
:
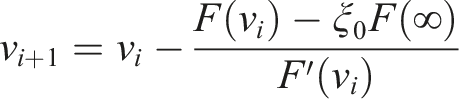
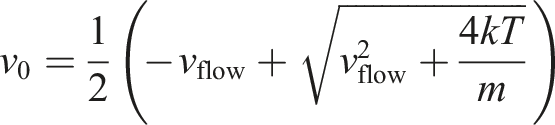
4.3. OpenMM implementation
The OpenMM implementation of the FBC (Pavlov, 2022a; Pavlov et al., 2023) was our earlier attempt in implementing the aforementioned algorithm. It was implemented as the
The OpenMM implementation also supports having multiple FBC planes inside the simulation box (see the discussion below). Additionally, it supports having no additional internal planes with only the box boundary acting as an FBC plane.
Currently, the master branch of OpenMM uses 32-bit counters for storing tile indices. This makes it impossible to correctly run systems bigger than
4.4. LAMMPS implementation
We have created a new fix called
The walls divide the simulation box along some axis into segments. In order to keep track of the events of an atom crossing a wall, our algorithm checks the segment index from the previous step and compares it to the current segment index. To keep track of the segment index, we have added a per-atom index array in this fix. This array is also involved in exchanging atom data between MPI processes, and for now there seems to be no apparent way to avoid this extra work.
The syntax of our fix is:
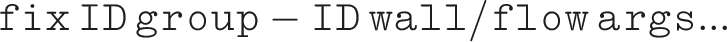
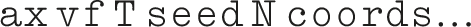
The fix gets the flow direction from the sign of
4.5.1. Kokkos-accelerated version
We also have implemented a Kokkos version of this fix in LAMMPS. In general, its idea and structure is exactly the same as the regular version, which is what Kokkos is designed to do.
However, there was a pitfall worth mentioning. Before the 22 August 2023 release, LAMMPS was not able to use GPU-aware MPI capabilities to exchange data when there is at least one Kokkos-accelerated fix that needs to communicate data. In that case, LAMMPS disabled GPU-aware MPI and exchanged all data (including per-atom arrays that are not part of the fix) through the host. As we said above, our fix contains an additional per-atom array, which participates in the exchange between processes. Fortunately, there is a Pull Request (Taniguchi (2019)) that has been merged recently, which allowed us to optimize away the need to copy data to host and back when a direct communication between GPUs is possible. The LAMMPS/Kokkos benchmark results presented in this paper have been obtained with this new capability.
5. Grid aggregation
When processing trajectory data from simulations, it is not always feasible to process it on per-particle basis, and some kind aggregation is necessary. Here, we calculate average velocities on a grid of Ncells = N x × N y × N z cells.
One of the principal metrics in analyzing fluid flows is vorticity:
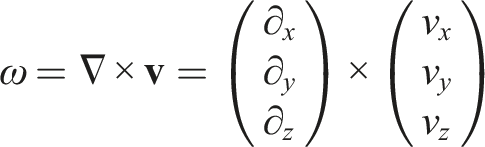
Then, by abuse of notation, we can derive a numerical approximation of vorticity for our grid:
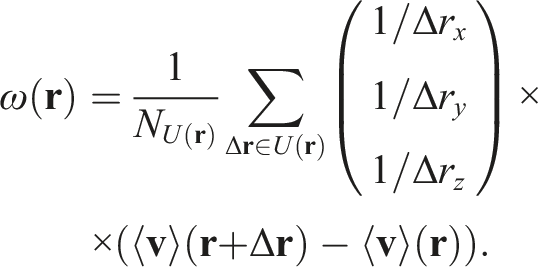
However, the images obtained using this approximation can be noisy. Indeed, it is not always correct to assume that it is possible to measure a continuous field of values on an atomic level. The next step is quantifying errors of values obtained from averaging on grid cells. We use σ to denote the absolute error of a value and ϵ to denote its relative error.
The single-axis Maxwell velocity distribution is:
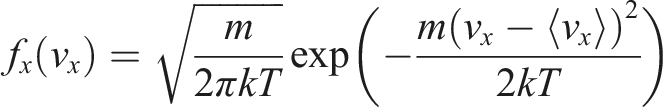
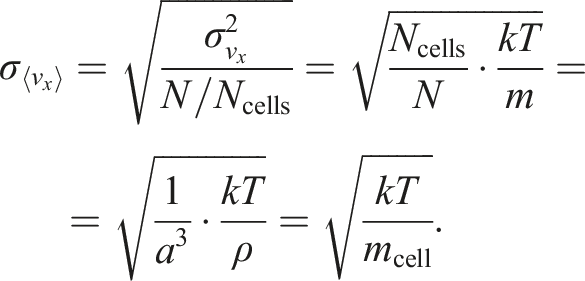
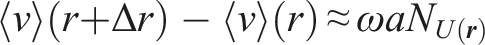
Now, the error of vorticity is:
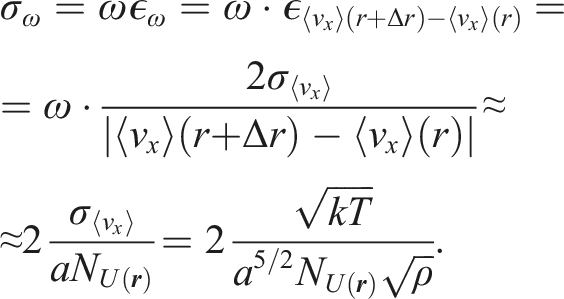
5.1. Distributed grids aggregation in LAMMPS
Since the 22 December 2022 release LAMMPS has a special mechanism called distributed grids. They consist of uniformly distributed cells covering the simulation box. There are also commands that utilize these grids to perform different operations.
To perform grid aggregation we have used such a distributed grids mechanism with different cell resolutions and the
6. Simulation results
6.1. Performance comparison
Specifications of the multi-GPU systems considered in this work.
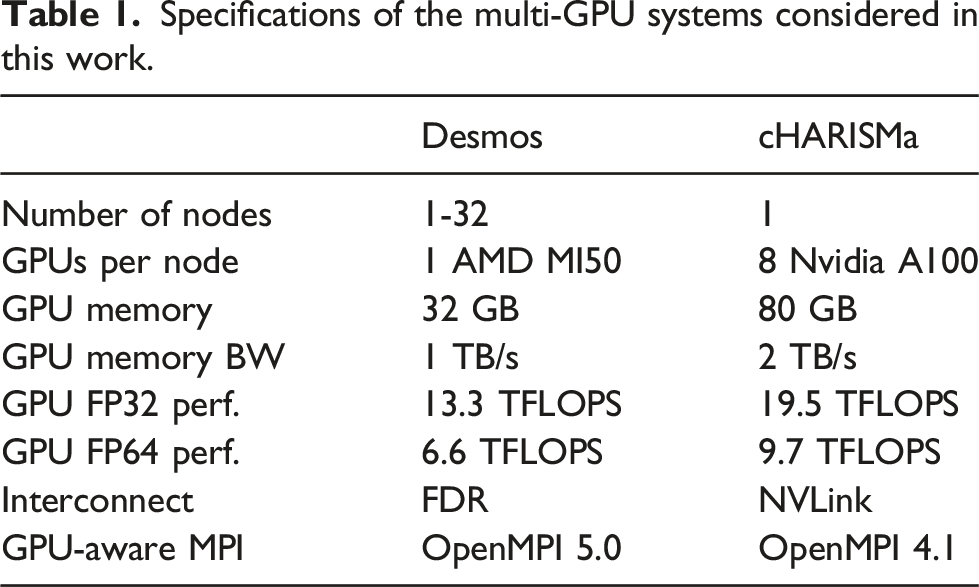
OpenMM and LAMMPS are built in the “Release mode”. Precision is set to mixed in OpenMM, and to double in LAMMPS/Kokkos (due to the fact that the mixed precision is not supported by LAMMPS/Kokkos).
The systems that we are running benchmarks on are LJ systems with the main parameters borrowed from Smith (2015) unless specified otherwise. Those systems have the relative dimensions of approximately 10:2:1 with a cylindrical obstacle in the upstream part.
We have measured the performance of both our FBC implementation in OpenMM and our Kokkos accelerated LAMMPS implementation of Benchmark results for the Lennard-Jones fluid. The performance of MD calculations is shown as Natoms · Nsteps per unit of wall-clock time (higher is better). Results for A100 and MI50 GPUs are presented. The rhombs show the data for OpenMM, the dots show the data for Kokkos backend of LAMMPS on MI50 and A100 GPUs. The performance of 256 Intel Xeon Nehalem cores used in Smith (2015) for a 300 million atom simulation is shown as a star.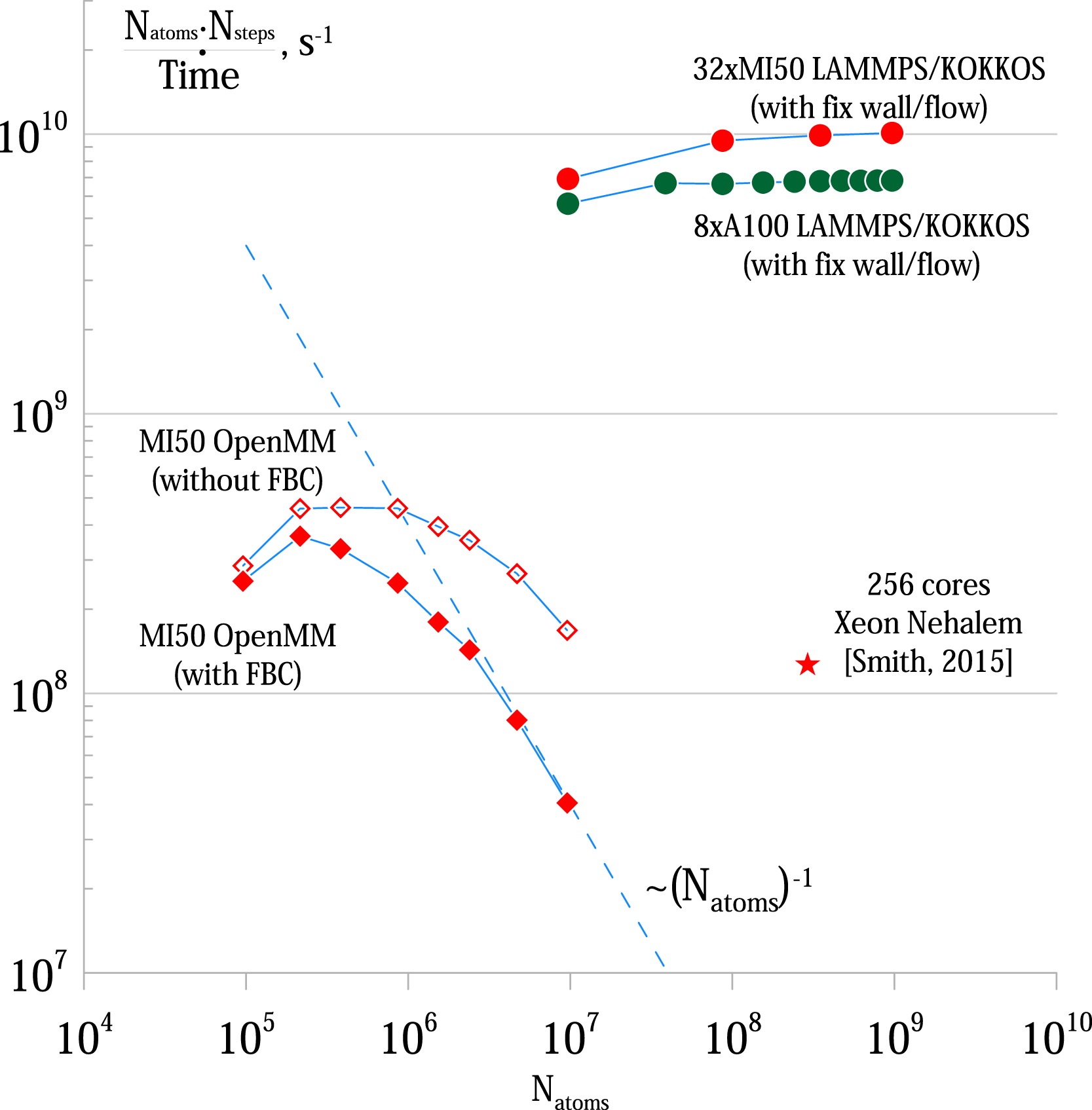
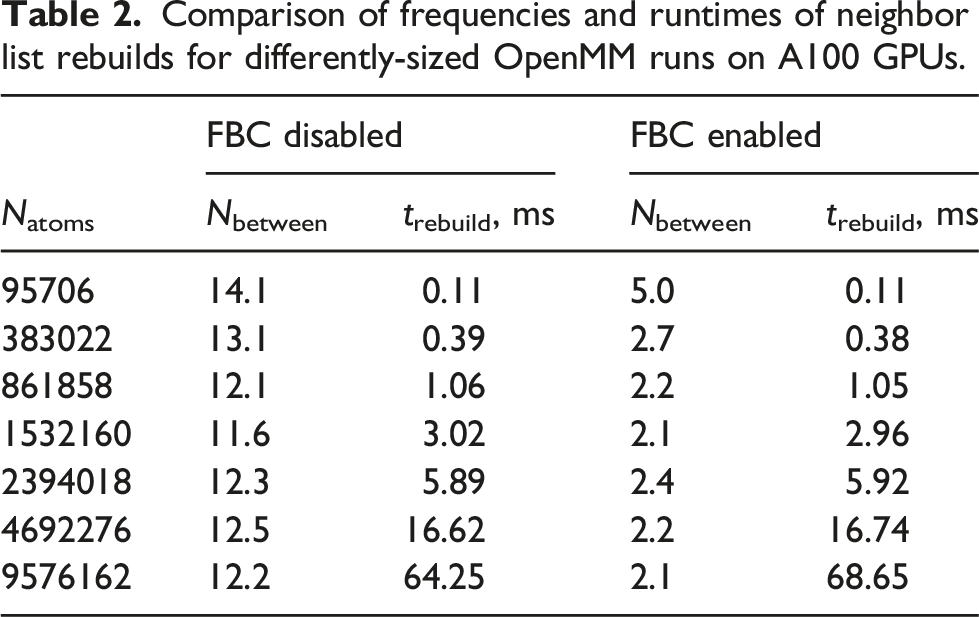
Comparison of frequencies and runtimes of neighbor list rebuilds for differently-sized OpenMM runs on A100 GPUs.
We also measured the strong scaling for a typical LAMMPS/Kokkos simulation with Strong scaling of wall/flow simulations using the LAMMPS/Kokkos package. Performance for 10000 steps with Natoms = 60480000 (higher is better).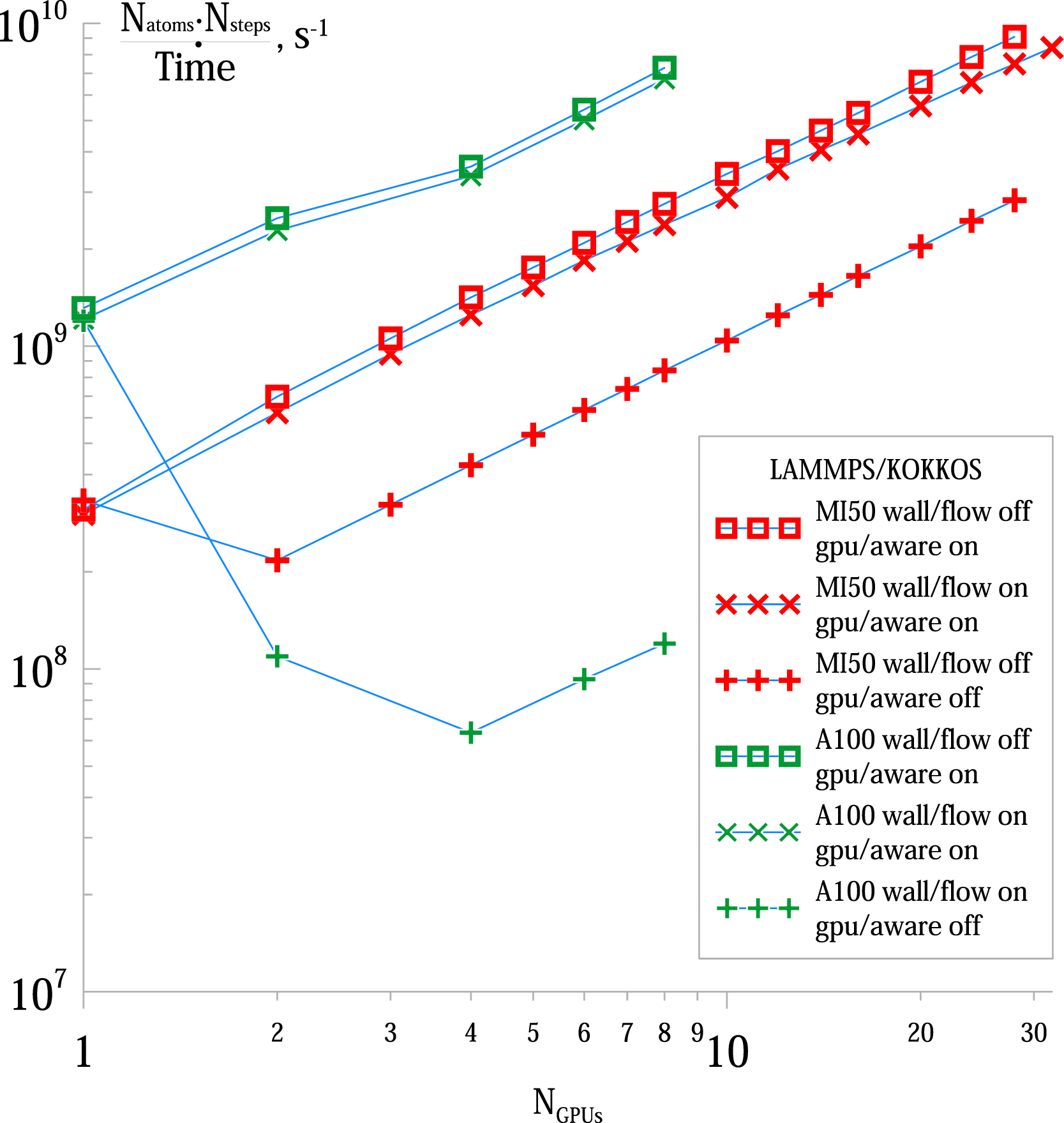
These benchmarks demonstrate that on bigger systems LAMMPS/Kokkos allows for excellent scaling for the entire range of our sample. We did not test the scaling of OpenMM because it does not implement domain decomposition and the only multi-GPU parallelization it can offer is offloading parts of the array of interacting tiles to other GPUs when computing forces. However, the most expensive part at these system sizes — the force matrix traversal — still happens on one GPU and is not parallelized.
We have performed a tracing analysis of LAMMPS/Kokkos on the Desmos supercomputer with Score-P ver. 8.1 (Knüpfer et al. (2012)) compiled with Kokkos and HIP profiling support. Tracing has been done with Illustration of the 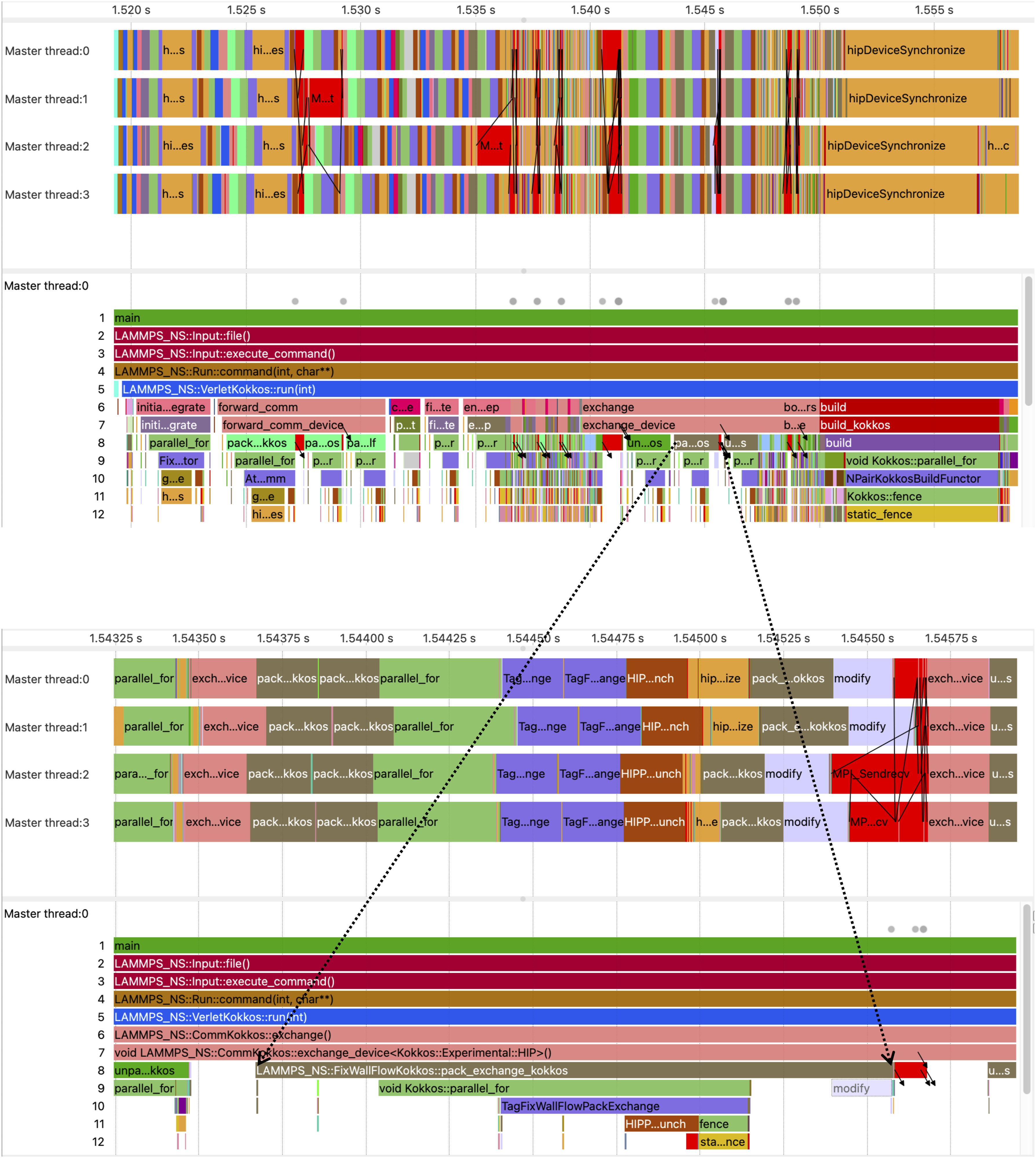
Our trace analysis corroborates that the relative performance plunge with two or more GPUs compared to a single GPU happens due to the communication overhead, as can be seen on Figure 4. An acute difference can be seen in the amount of time spent inside the Juxtaposition of two traces: at the top is the trace of LAMMPS/Kokkos running with GPU-aware communication, at the bottom is the similar trace obtained without GPU-aware communication. The process timelines are shown on the left. Both runs were carried out on four Desmos supercomputer nodes with one MI50 GPU per node. For the sake of comparison of the traces, their alignment begins with the beginning of the 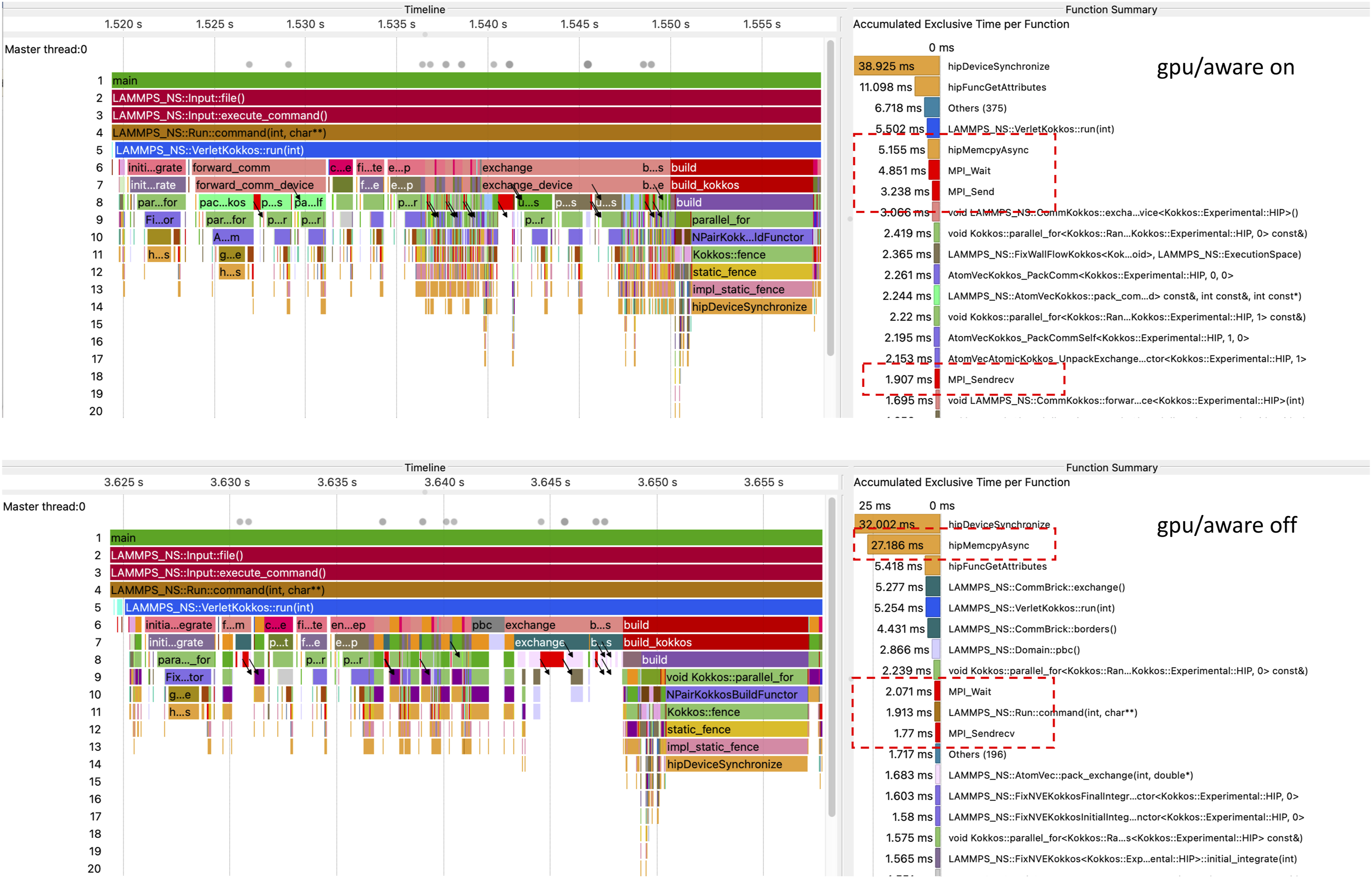
6.2. Additional Flow Boundary Conditions planes
In our initial implementation, the only plane that was reassigning the particle velocities was the periodic boundary plane at the edges of the simulation box. This, however, proved insufficient due to the non-ideal nature of the fluid.
One undesirable effect cased by there being insufficient planes that can be observed is a “vorticity leakage”. An example of it can be seen on Figure 5. Adding extra FBC planes helps mitigating this issue and makes the flow more homogeneous. Side-by-side comparison of the vorticity plots of two identical systems with side ratio of 25:10:1 after some time has passed and stationary flow established. Here, Natoms ≈ 3.25 × 106 and vflow = 3 with a cylindrical obstacle with r = 35. The left one has no extra FBC planes (one in total), the right one has five FBC planes. A much stronger vorticity leakage can be observed in the left system.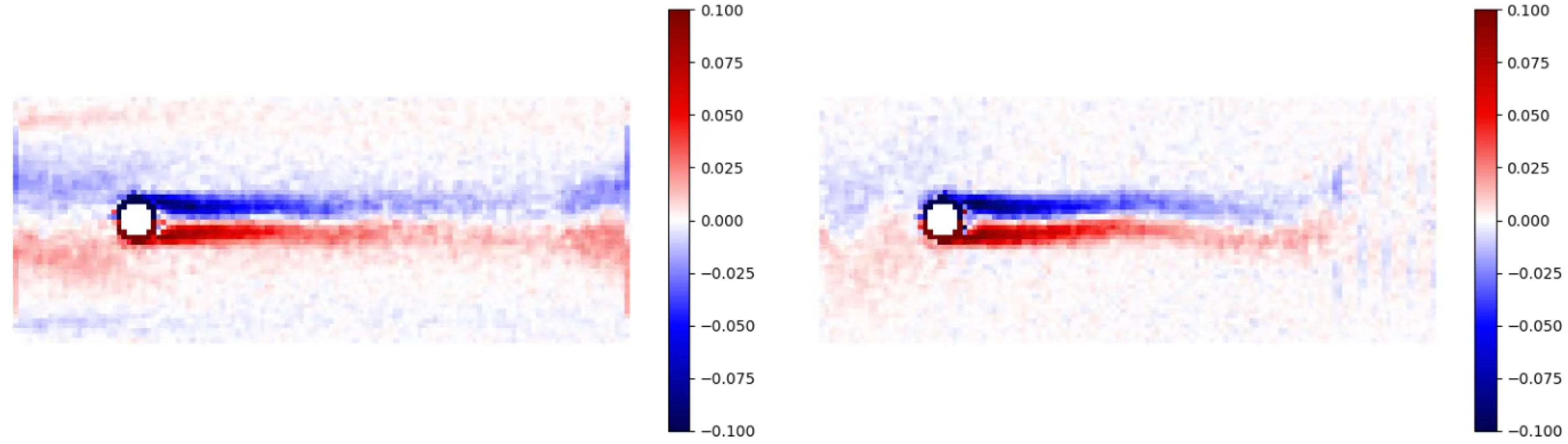
Another issue that can arise is that at higher flow velocities and higher system sizes particles can start accumulating near a downstream boundary. This particular effect is demonstrated on Figure 6. A system with Natoms = 41 × 106 and vflow = 0.9. There are 4 additional FBC planes at the right side (five in total).
6.3. Calibration of flow velocity and tempterature
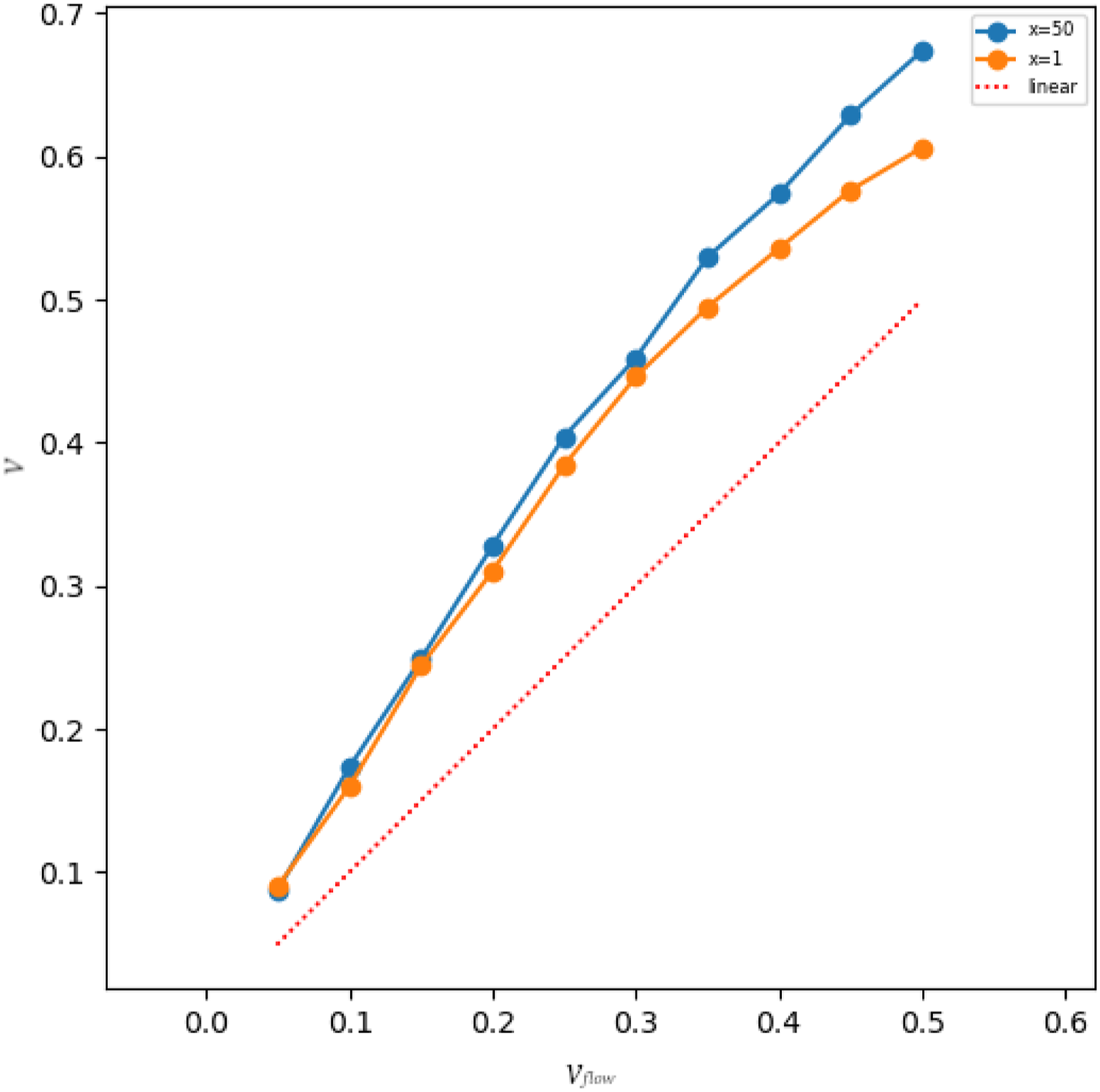
Since the premise upon which the distributions were derived was that the system consisted of ideal gas only, in more realistic scenarios this premise is certainly violated. This means that after putting a system into proposed FBC and letting the flow establishing after some time, the temperature and the actual flow velocity may not match the target ones. Figures 7 and 8 demonstrate how the achieved actual temperature and flow velocity differ from the target depending on the target flow velocity. Temperature over time for a system with Natoms = 2.1 × 106 for different target vflow values. Target temperature is T0 = 0.4 in all cases. Established velocity in the relative x = 1 and x = 50 positions (the left part and the middle part of the simulation box) depending on the target flow velocity in the system with Natoms = 2.1 × 106 for different target vflow values. Target temperature is T0 = 0.4 in all cases.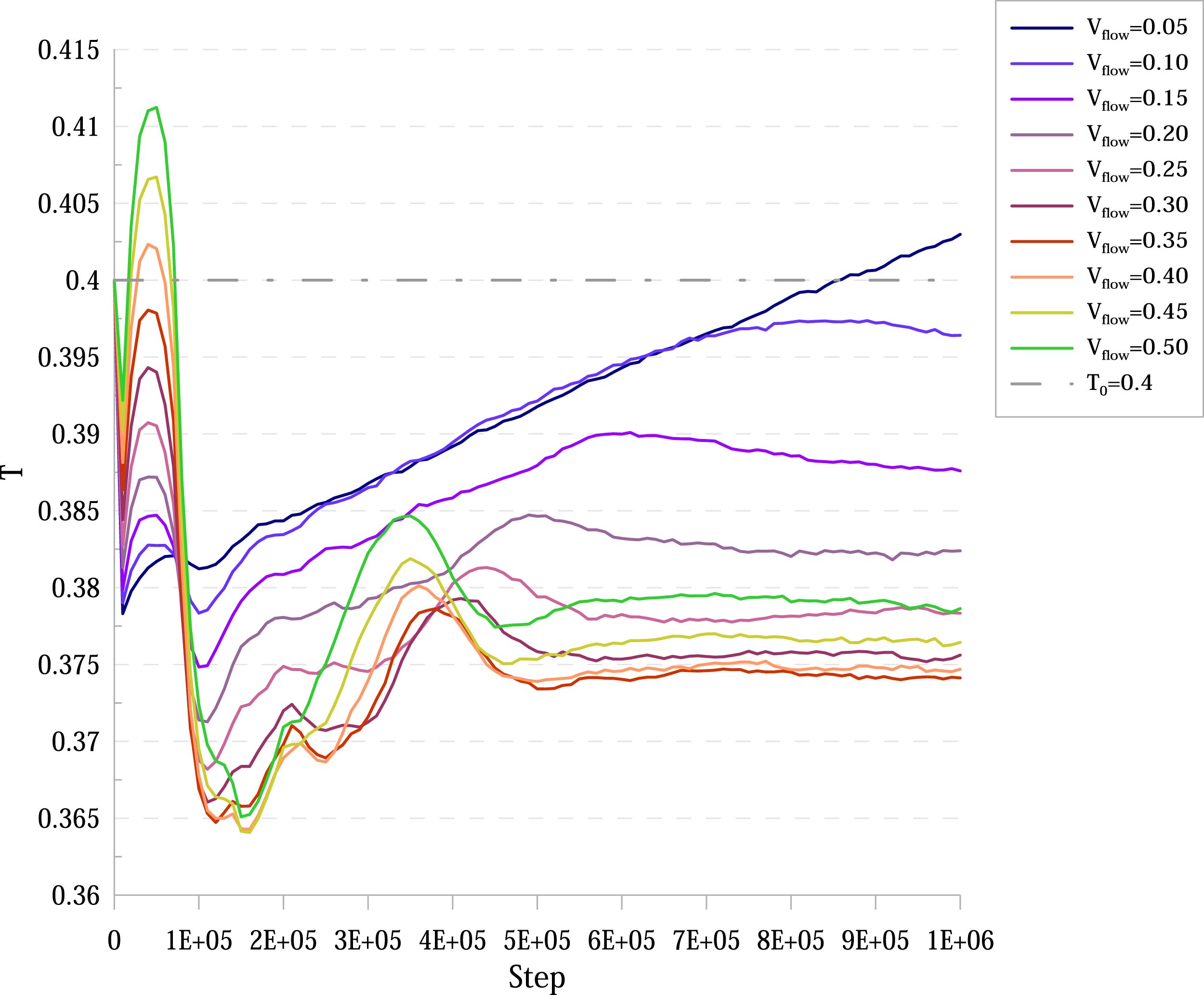
6.4. Pre-turbulent mode
As we are increasing the size of our simulation box, the nature of the flow is changing: at smaller scales it was laminar, at greater scales eddies began to appear as can be seen on Figure 9. We see that in the case of the Reynolds number Re ≈ 160 the Karman vortex street forms that is a typical feature of a pre-turbulent mode of the fluid flow. Density (left) and vorticity (right) plots for three different moments of time for a simulation with Natoms = 41 × 106 and vflow = 0.5. Karman vortex street can be seen forming. The Reynolds number in this case is Re ≈ 160. There are 4 additional FBC planes at the right side (five in total).
7. Discussion
Although the theoretical basis for our method is perfectly sound for ideal gases, as soon as we apply it to a non-ideal scenario, deviations from expected results such as vorticity leakages and density irregularities start to crop up. The likely cause of vorticity leakages is that particles on the opposite sides of a plane still interact with each other using short-range forces thereby “leaking” some information about vorticity on the other side.
The likely reason why density accumulates near the downstream boundary is that there is a higher-density region in front of the obstacle that repels particles that try to penetrate the boundary plane and many particles that do get through, get repelled right back into the plane, instantly losing all extra velocity. This results in a boundary being unable to “pump” the particles through in order to maintain a uniform and stationary flow. Luckily, this effect is apparent to the eye whenever it manifests, because a clear boundary between a lower-density and a higher-density region becomes visible on a density plot. Increasing the amount of planes or lowering the flow velocity helps alleviating this issue. For example, on Figure 9 the model dimensions are the same as on Figure 6 but there is no density transition because the flow velocity is lower.
One of the surprising performance effects that one of our implementations has is the progressive degradation of performance for the OpenMM implementation as the number of atoms increases. At first glance, it would seem that the neighbor list calculation that has
According to Smith (2015), it should be possible to observe turbulence at greater scales (at about Natoms ≈ 3 × 108, Re ≈ 400). Our LAMMPS/Kokkos implementation makes it possible to achieve these scales in the future if one were to dedicate sufficient GPU resources to such calculations. An interesting option for further development of the FBC method could be the adaptation for MD modelling of the turbulent inflow boundary conditions that are used in large eddy simulations Dhamankar et al. (2018). Our OpenMM implementation can be useful for studies of the vortex generation in non-Newtonian polymer fluids (e.g. see the recent papers of Handler et al. (2020) and Hopkins et al. (2020)) where the requirements for the number of atoms in MD models could be moderate but statistical averaging is important.
8. Conclusion
In this article, we have presented a novel type of flow boundary conditions for simulation of fluid flows on atomic level. We have described our GPU-based OpenMM implementation of this method, as well as the corresponding LAMMPS
We have compared the performance of the OpenMM implementation with the LAMMPS/Kokkos implementation and analysed the strong scaling of the LAMMPS/Kokkos implementation on two multi-GPU systems, showing our implementation to be sufficiently scalable for running multi-billion MD models on multi-GPU supercomputers.
We have traced a typical LAMMPS/Kokkos MD run and shown the time taken by our
Our LAMMPS/Kokkos
Footnotes
Acknowledgements
The authors are grateful to Yuri Grishichkin and Roman Chulkevich for the assistance with the system software tuning on the Desmos and cHARISMa supercomputers.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The development of the FBC algorithm and the performance analysis for the nodes of the Desmos supercomputer were supported by the Ministry of Science and Higher Education of the Russian Federation (agreement with the Joint Institute for High Temperatures of Russian Academy of Sciences). The implementation of the FBC algorithm in LAMMPS and the performance analysis for the nodes of the cHARISMa supercomputer were performed within the framework of the HSE University Basic Research Program.
Author biographies
Daniil Pavlov is a master’s student at the Moscow Institute of Physics and Technology and an intern researcher at the Joint Institute for High Temperatures of Russian Academy of Sciences, Moscow, Russia. His research interests include high-performance computing and GPU computing in materials science, their scalability and portability.
Vladislav Galigerov is master’s student at the Higher School of Economics and an intern researcher at the Joint Institute for High Temperatures of Russian Academy of Sciences, Moscow, Russia. His research interests include high-performance computing, general-purpose GPU computing and atomistic simulations.
Daniil Kolotinskii is a junior researcher at the Laboratory of Supercomputer Methods in Condensed Matter Physics of Moscow Institute of Physics and Technology (MIPT) and a junior researcher at the Joint Institute for High Temperatures Russian Academy of Sciences. He obtained a master’s degree in Applied Mathematics and Physics from MIPT in 2021. Now he is a Ph.D student at MIPT. His research interests include computational physics, numerical modelling and parallel computing.
Vsevolod Nikolskiy is a research fellow at the International Laboratory for Supercomputer Atomistic Modelling and Multiscale Analysis of Higher School of Economics, Moscow, Russian, and a junior researcher at the Joint Institute for High Temperatures Russian Academy of Sciences. He obtained a master’s degree in Applied Mathematics and Informatics from the Moscow Institute of Electronics and Mathematics of the Higher School of Economics in 2018. Now he is a Ph.D. student of the Doctoral School of Computer Science of HSE University. His research interests include high-performance and heterogeneous computing, efficiency of novel processor architectures, development of GPU-accelerated algorithms.
Vladimir Stegailov graduated from the Moscow Institute of Physics and Technology in 2004, received a PhD degree in 2005 and a DrSc degree in 2012. He is Head of Department at the Joint Institute for High Temperatures of Russian Academy of Sciences, Moscow, Russia. He is a professor of the Higher School of Economics and a leading researcher in the International Laboratory for Supercomputer Atomistic Modelling and Multi-scale Analysis. He is a professor in the Moscow Institute of Physics and Technology where he leads the Laboratory of Supercomputer Methods in Condensed Matter Physics. His research interests include high-performance computing and atomistic and multiscale modelling and simulation.