
Abstract
The non-orthogonal local submatrix method applied to electronic structure–based molecular dynamics simulations is shown to exceed 1.1 EFLOP/s in FP16/FP32-mixed floating-point arithmetic when using 4400 NVIDIA A100 GPUs of the Perlmutter system. This is enabled by a modification of the original method that pushes the sustained fraction of the peak performance to about 80%. Example calculations are performed for SARS-CoV-2 spike proteins with up to 83 million atoms.
Keywords
Introduction
Electronic structure–based ab-initio molecular dynamics (AIMD) simulations (Car et al. 1985; Payne et al., 1992; Kühne 2014) are an important tool in solid-state physics, chemistry, and material science. The explicit treatment of quantum-mechanical effects in the electronic structure is required in situations, where empirical model potentials used in classical molecular dynamics fail to describe the relevant physical or chemical phenomena.
To derive the forces acting on the atoms, the electronic structure problem has to be solved in every time step during the propagation of the atoms. To make this possible, linear-scaling methods have been developed, where the computational complexity scales only linearly with the number of atoms in the system (Goedecker, 1999; Yang, 1991; Galli et al., 1992; Richters and Kühne, 2014; Niklasson et al., 2016). We have proposed the non-orthogonal local submatrix method (NOLSM, Schade et al., 2022) as a massively parallel method to solve the electronic structure problem via an approximate solution of the required matrix functions. The local nature of the method avoids inter-node communication in the solution phase and has been shown to scale extremely well to more than one thousand GPUs, while efficiently using the mixed-precision tensor cores for linear algebra operations.
This article is building on the implementation described in Schade et al. (2022), but since we are focusing on improvements to increase the sustained peak performance, aspects like the compensation of noise from numerical approximations with an appropriately modified Langevin-type equation to obtain accurate thermodynamical expectation values are not touched here, but have been discussed in previous work (Richters and Kühne 2014; Rengaraj et al., 2020). Instead, Section II summarizes the tackled problem, whereas Section III puts the achievement in relation to the performance of related large-scale electronic structure–based structure relaxations or AIMD simulations. The innovations beyond those presented in Schade et al. (2022) are described in Section IV. In Section V-B, we discuss our evaluation and define the performance measurements. Finally, Section VI discusses the achieved performance.
Overview of the problem
Molecular dynamics calculations simulate the movement of atoms in molecules, surfaces, or solids by integrating Newton's equation of motion


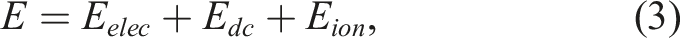

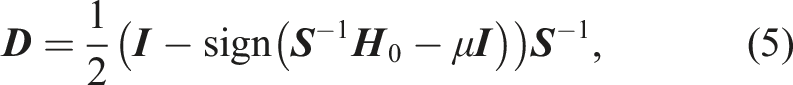
The evaluation of the matrix-sign function in equation (5) can be performed iteratively, for example, with the Newton–Schulz iteration (Schulz 1933)
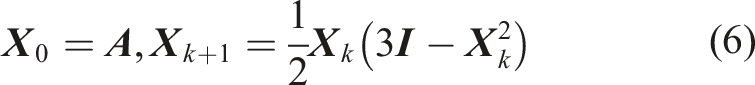
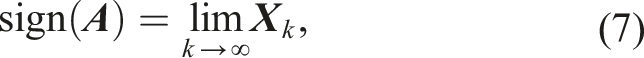
The submatrix method (Lass, Mohr, et al. 2018; Lass, Schade, et al. 2020) instead views the density matrix as a matrix function to be evaluated. Therein, the evaluation of a matrix function f( 1. In the first step, a submatrix is generated for column i of the input matrix 2. The matrix function is applied to the submatrix 3. The matrix elements of Schematic representation of the steps of the submatrix method for the approximate calculation of a matrix function f(
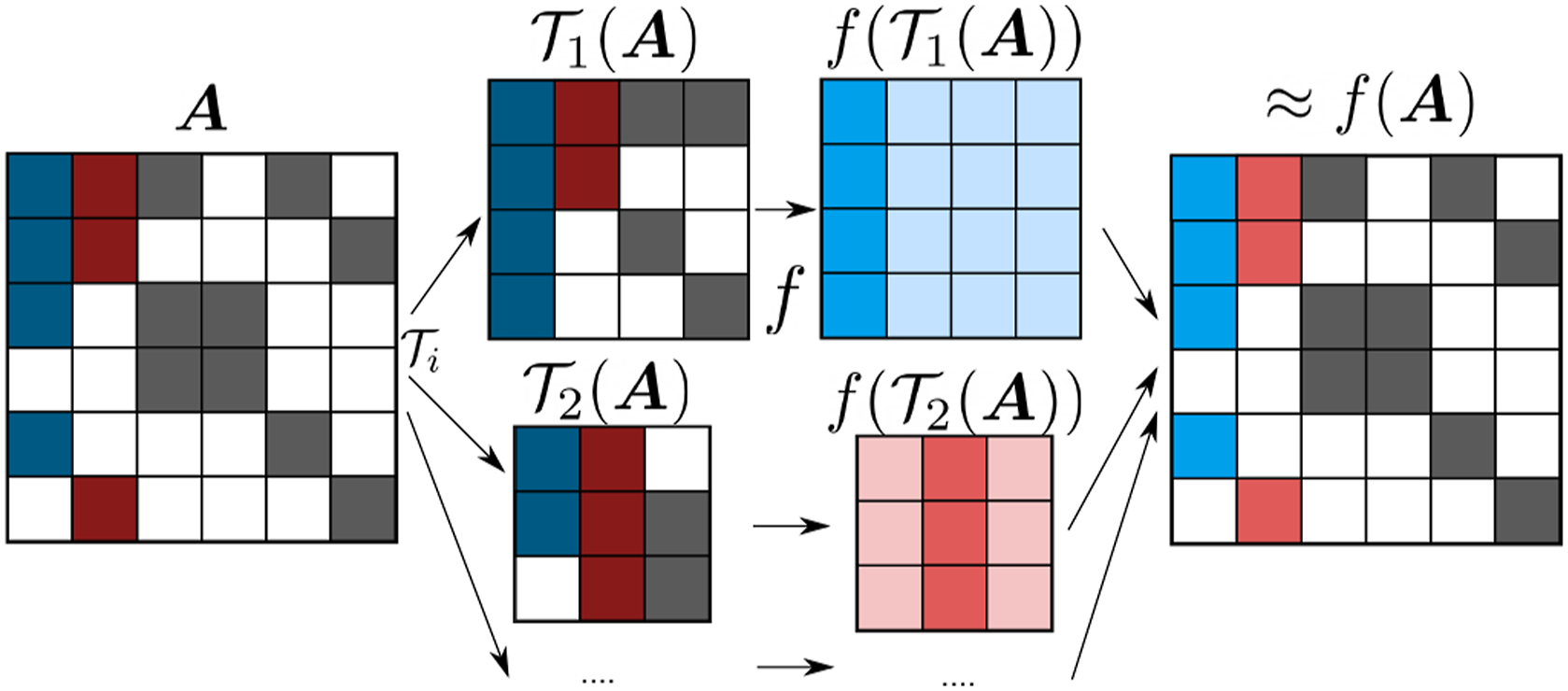
The resulting matrix
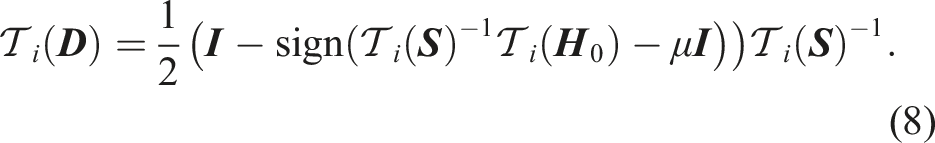
Note that the efficiency and accuracy of the submatrix method can be improved by generating one submatrix for multiple columns instead of just a single column as described in detail in Schade et al. (2022) together with the GPU implementation. Moreover, the use of the submatrix approximation and low-precision numerics can be compensated by an modified Langevin-type equation that replaces Newton's equation of motion so that exact ensemble-averaged expectation values can be obtained (Richters and Kühne 2014; Rengaraj et al., 2020).
Current state of the art
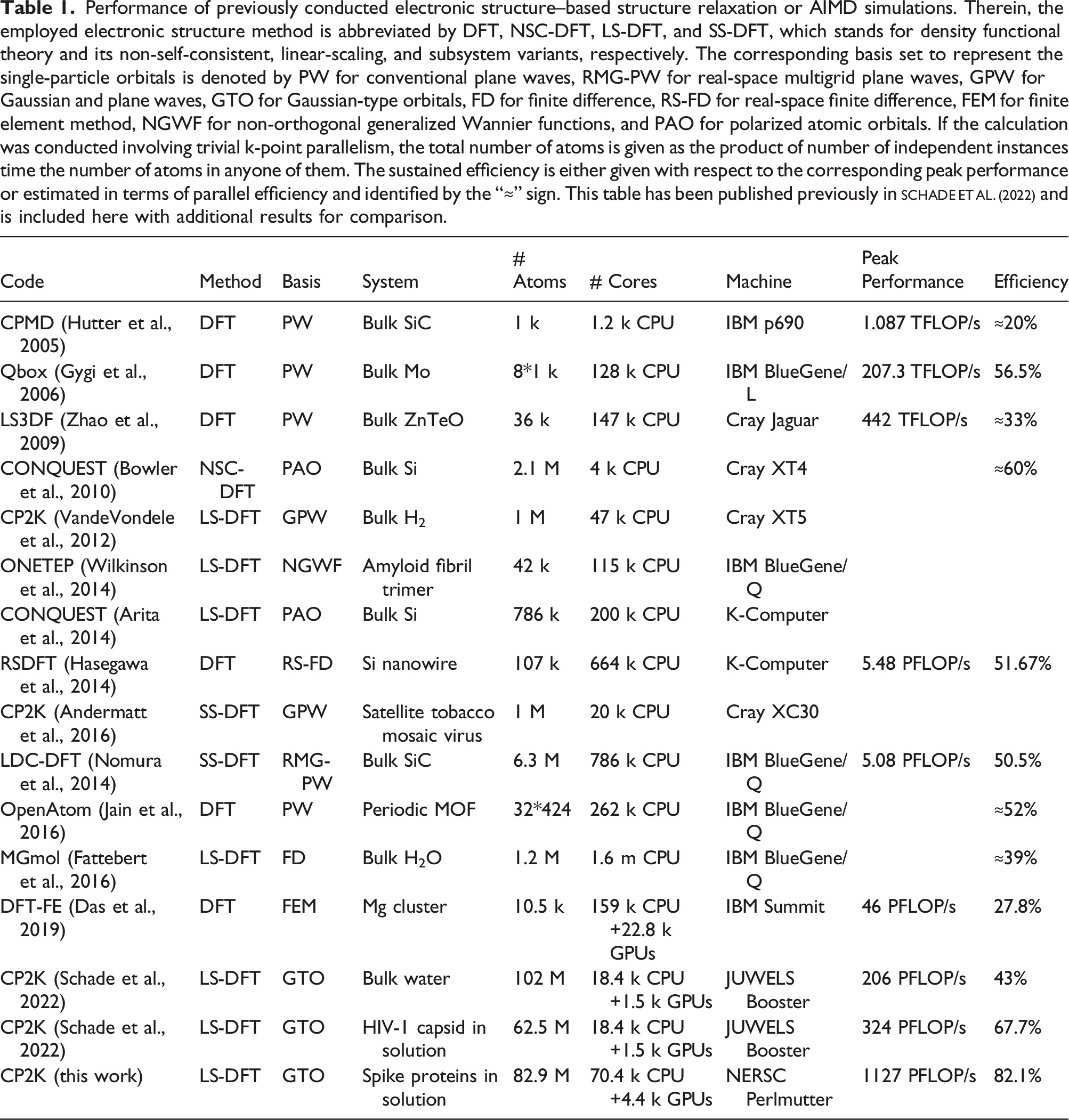
Performance of previously conducted electronic structure–based structure relaxation or AIMD simulations. Therein, the employed electronic structure method is abbreviated by DFT, NSC-DFT, LS-DFT, and SS-DFT, which stands for density functional theory and its non-self-consistent, linear-scaling, and subsystem variants, respectively. The corresponding basis set to represent the single-particle orbitals is denoted by PW for conventional plane waves, RMG-PW for real-space multigrid plane waves, GPW for Gaussian and plane waves, GTO for Gaussian-type orbitals, FD for finite difference, RS-FD for real-space finite difference, FEM for finite element method, NGWF for non-orthogonal generalized Wannier functions, and PAO for polarized atomic orbitals. If the calculation was conducted involving trivial k-point parallelism, the total number of atoms is given as the product of number of independent instances time the number of atoms in anyone of them. The sustained efficiency is either given with respect to the corresponding peak performance or estimated in terms of parallel efficiency and identified by the “≈” sign. This table has been published previously in Schade et al. (2022) and is included here with additional results for comparison.
Innovations realized
Summary of contributions
This work uses the previously reported algorithmic innovations like the use of approximate computing techniques, the non-orthogonal local submatrix method and its realization with GPUs, while minimizing the communication, as well as the heuristic combination of columns in the submatrix creation described in Schade et al. (2022). A new development beyond the implementation innovations already shown in Schade et al. (2022) like the efficient iterative evaluation of matrix functions for dense matrices on GPU tensor cores is introduced in Section IV-B: The matrix-size dependency of the GPU performance is now also considered for the combination of submatrices and yields an additional speedup.
Implementation innovations
1. Submatrix Combination Heuristics:
The combination of columns for the generation of submatrices introduced in Schade et al. (2022) used a cubic metric, that is, the combination of two columns yields an improvement if and only if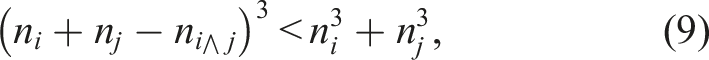
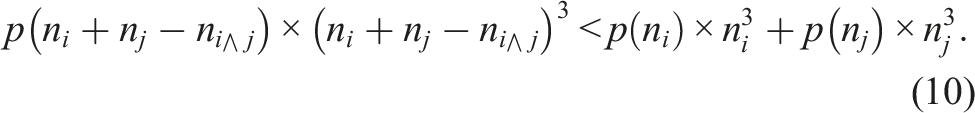
Influence of the two different submatrix combination criteria, equation (9) and equation (10), on the submatrix sizes, number of floating-point operations for one matrix multiplication of each submatrix, the estimated performance per NVIDIA A100 GPU, and the estimated speedup considering the matrix multiplication performance of an NVIDIA A100.
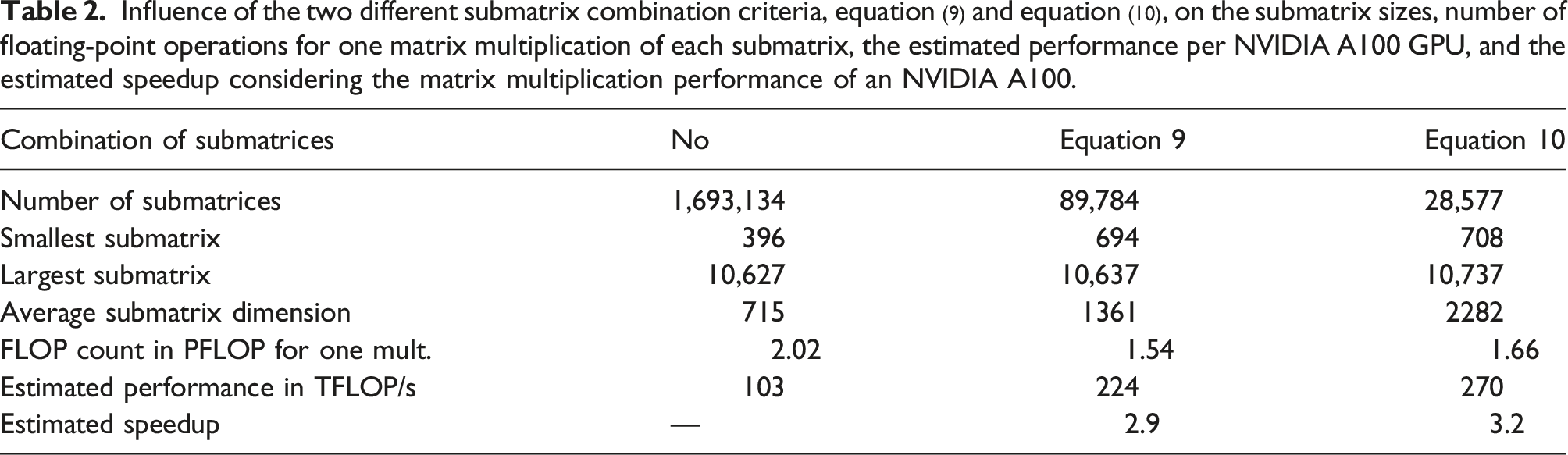
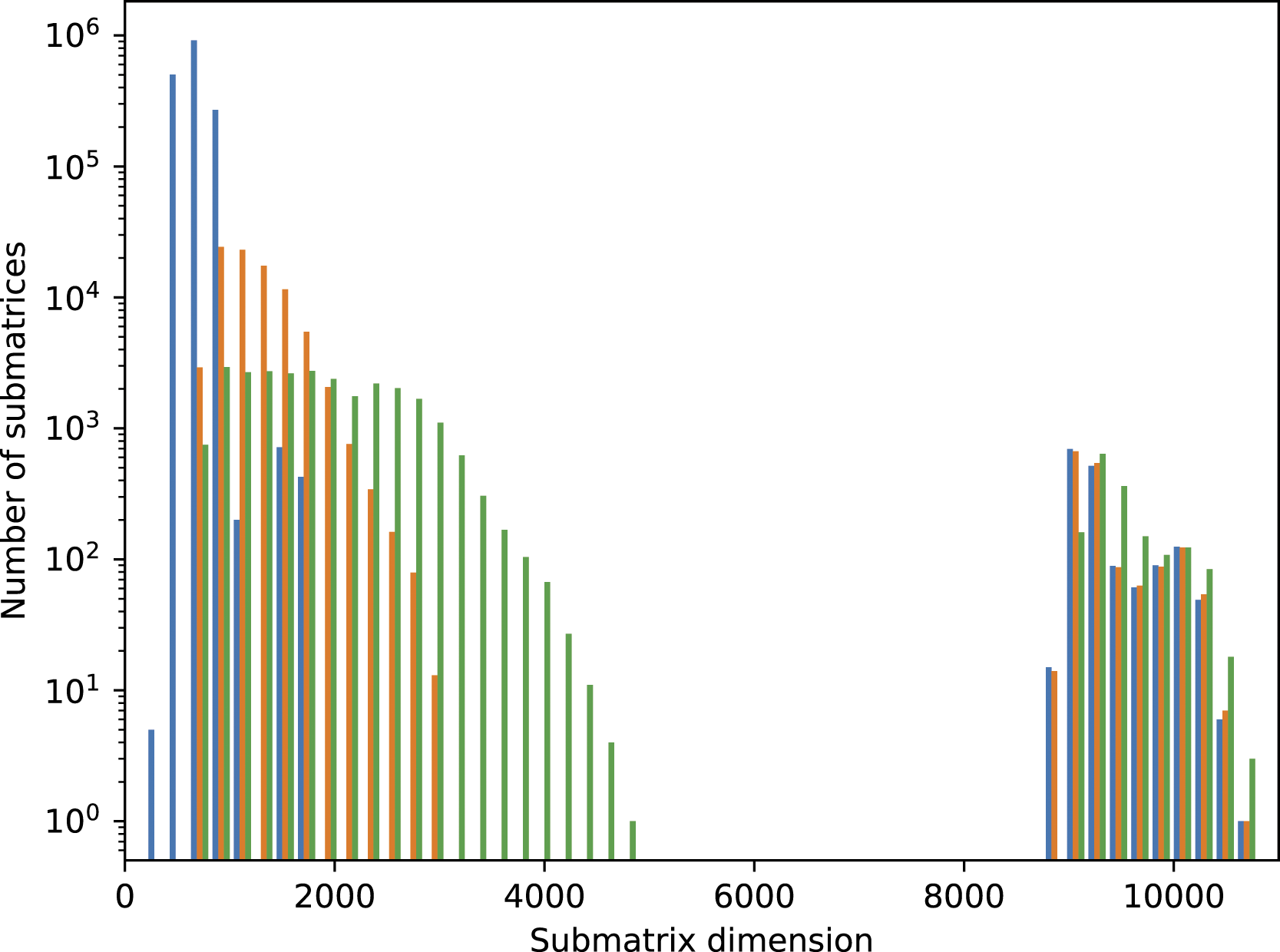
Histogram for the submatrix sizes in the case of the SARS-CoV-2 spike protein in aqueous solution with approx. 1.7 mio. atoms: without combining submatrices (blue), combination using criterion equation (9) (orange) and using criterion equation (10) (green). A discussion of the further structure can be found in Schade et al. (2022) and also applies to the spike protein system.
How performance was measured
Computational details
1. SARS-CoV-2 Spike Protein in Aqueous Solution:
As our benchmark system, we have used the full-length SARS-CoV-2 spike protein in the open state, anchored in a lipid bilayer (Reference PDB structure: 6VSB) and pre-equilibrated with all-atom MD using NAMD (Wrapp et al., 2020; Casalino et al., 2021). The system was solvated in aqueous solution in a simulation cell with dimensions 204.7 × 199.5 × 408.5 Å, and including 1,693,134 atoms. The single cell shown in Figure 3 can be easily repeated in a two-dimensional grid of spike proteins as a scalable benchmark system. 2. Simulation Details: SARS-CoV-2 spike protein in aqueous solution: full cell (left) and without hydrogen and oxygen atoms (right).
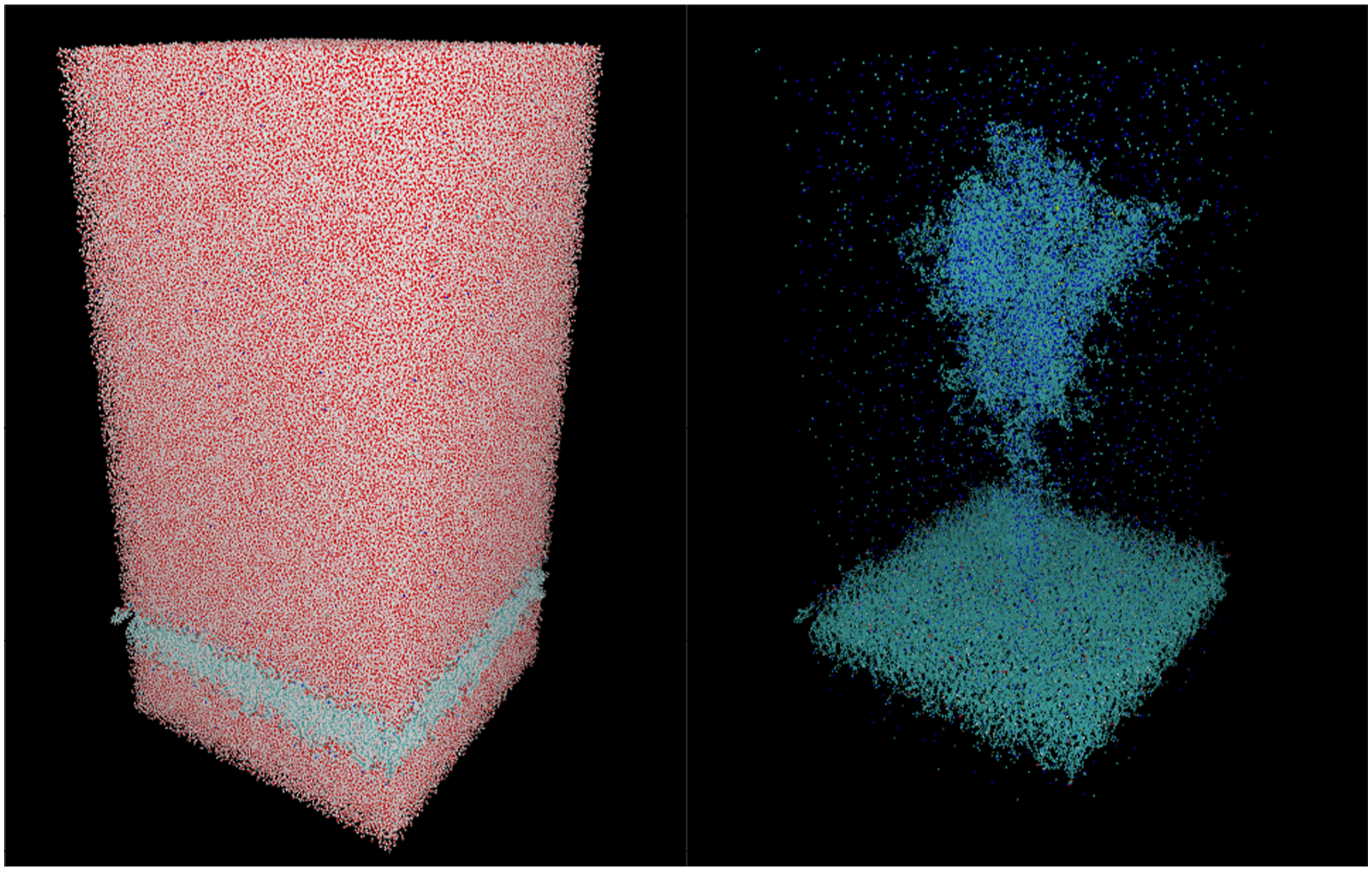
The electronic structure is simulated with the GFN-xTB approach in conjunction with a London dispersion correction based on the rational Becke–Johnson damping function (Grimme et al., 2011). Further details can be found in Schade et al. (2022).
For the sake of benchmark resources, we have restricted each simulation run to one SCF iteration in the spirit of the second-generation Car–Parrinello AIMD method (Kühne et al., 2007; Kühne and Prodan, 2018), but included the iterations for finding an appropriate chemical potential that produces a charge-neutral system.
Measurements
The main measurements presented here are as follows: 1. Wall clock time of the NOLSM method TNOLSM:
The wall clock time TNOLSM,i of the NOLSM method on node i is measured for each iteration of the chemical potential. Each iteration includes all transfers between host and GPU. The overall wall clock time TNOLSM is defined as the maximum over all node wall clock times. 2. FLOPs in the NOLSM method FLOPsNOLSM,i:
The per-node floating-point operations FLOPsNOLSM in the FP16/FP32-mixed-precision matrix iterations in the NOLSM method are estimated as 2n3 for a gemm-operation 3. Node performance of NOLSM method PNOLSM,i:
The node performances of the NOLSM method are defined as PNOLSM,i = FLOPsNOLSM,i/TNOLSM,i for each node i. 4. Performance of the NOLSM method PNOLSM:
The performance of the NOLSM method is defined as the sum of the node performances.
HPC system and environment
The benchmark runs presented here have been performed on the Perlmutter system at the National Energy Research Scientific Computing Center (NERSC). The Perlmutter system consists of 1536 GPU nodes with one AMD EPYC 7763 64-core CPU with 256 GB DDR4 memory and four NVIDIA A100 GPUs with 40 GB of HBM2 memory each. The peak performance of the tensor cores in one NVIDIA A100 GPU is 312 TFLOP/s in FP16 with FP32-based accumulate (NVIDIA Corporation, 2021). The system uses HPE Cray Slingshot as node interconnect.
The software environment used in this work consisted of GCC 11.2.0, Cray-MPICH 8.1.10, CUDA NVCC 11.5.119, and CUBLAS 11.5. One MPI rank per node and 64 CPU-threads per rank were used as well as four CUDA streams per GPU. Each stream was controlled by a single CPU-thread.
Performance measurements and results
Performance of the NOLSM method for the spike protein
We have performed calculations for three different grid sizes of spike proteins: 6 × 5 (51 mio. atoms), 6 × 6 (61 mio. atoms), and 7 × 7 (83 mio. atoms). All three example calculations have been performed with 1100 nodes of the Perlmutter system, that is, 4400 NVIDIA A100 GPUs.
The wall clock time of the NOLSM method TNOLSM is shown in Figure 4. The distribution of the performances of individual nodes is shown in Figure 5 for 7 × 7 spike proteins (83 mio. atoms) in relation to the peak performance of the GPUs. The performances of the nodes with 4 NVIDIA A100 GPUs mainly fall in the range between 1 PFLOP/s and 1.07 PFLOP/s with an average of 1.03 PFLOP/s. This represents about 80% of the peak performance of 1.248 PFLOP/s = 4 · 0.312 PFLOP/s per node. Wall time of the NOLSM method TNOLSM for a grid of SARS-CoV-2 spike proteins in aqueous solution on 1100 nodes of the Perlmutter system. Distribution of node performances for 83 mio. atoms (7 × 7 grid of SARS-CoV-2 spike proteins in aqueous solution) on 1100 nodes of the Perlmutter system.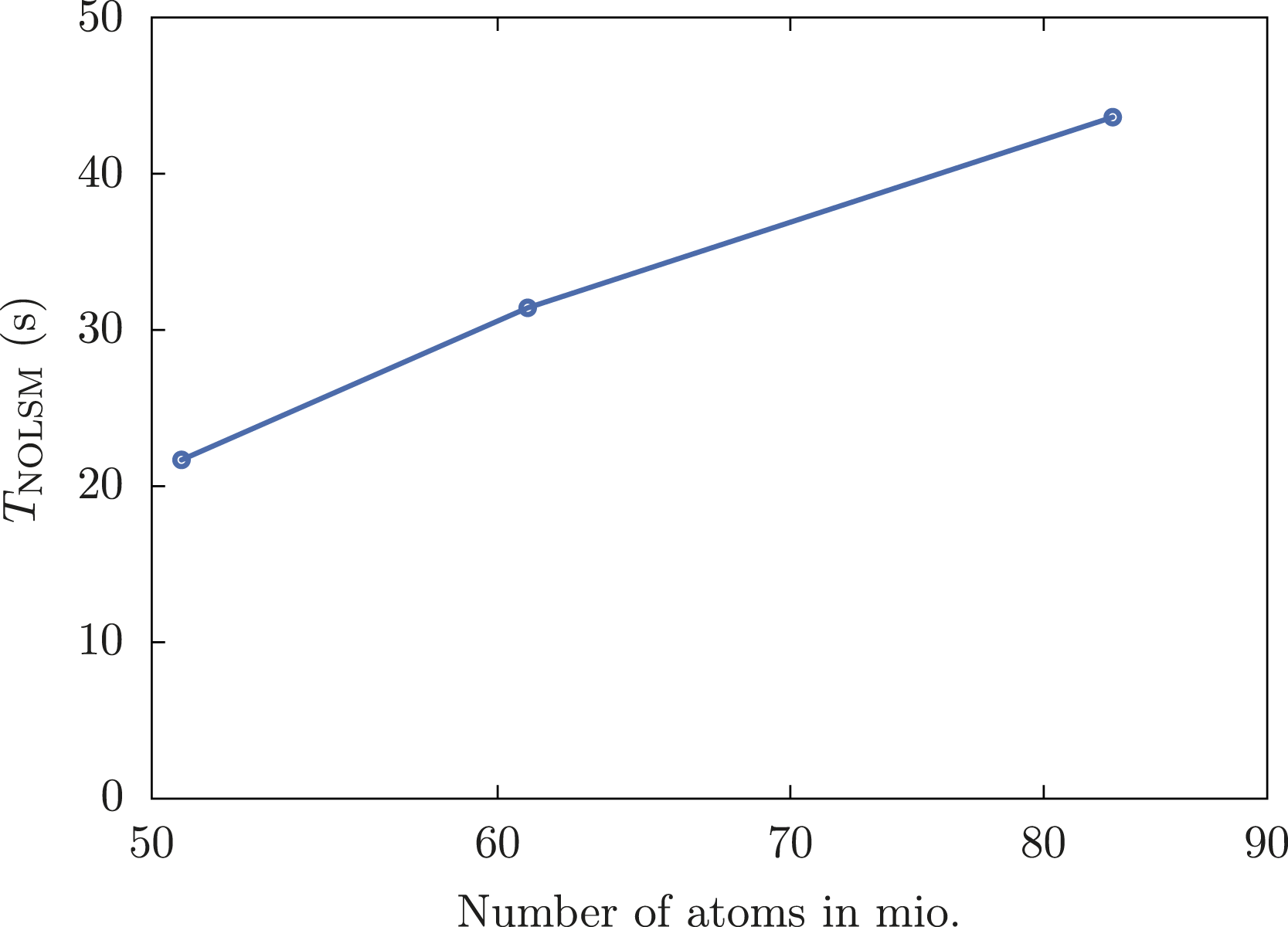
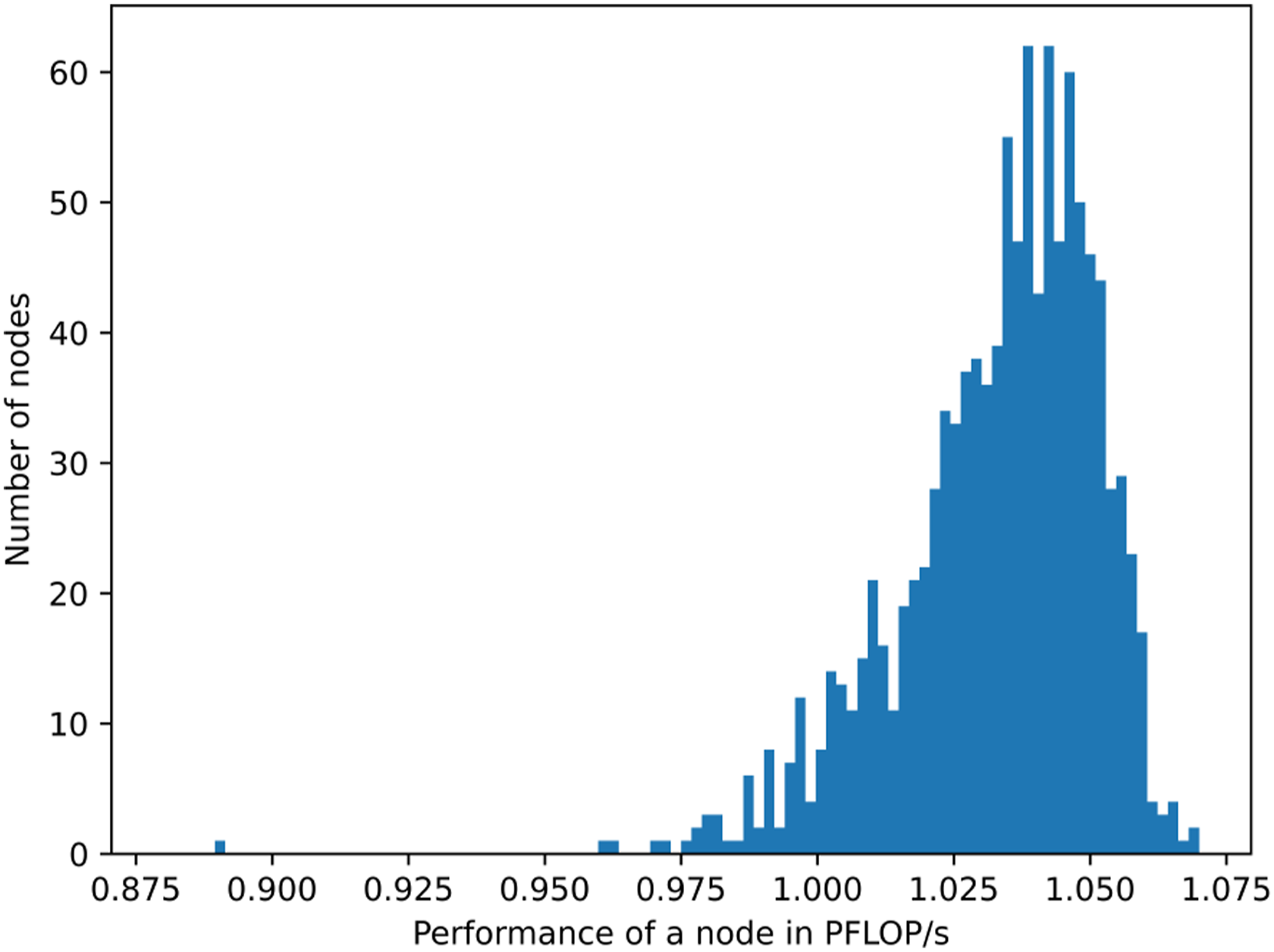
A floating-point throughput of 1.106 to 1.127 EFLOP/s with 4400 NVIDIA A100 GPUs is achieved representing about 80% of the theoretical peak performance of the tensor cores. Distribution of node performances for 83 mio. atoms (7 × 7 grid of SARS-CoV-2 spike proteins in aqueous solution) on 1100 nodes (4400 NVIDIA A100 GPUs) of the Perlmutter system.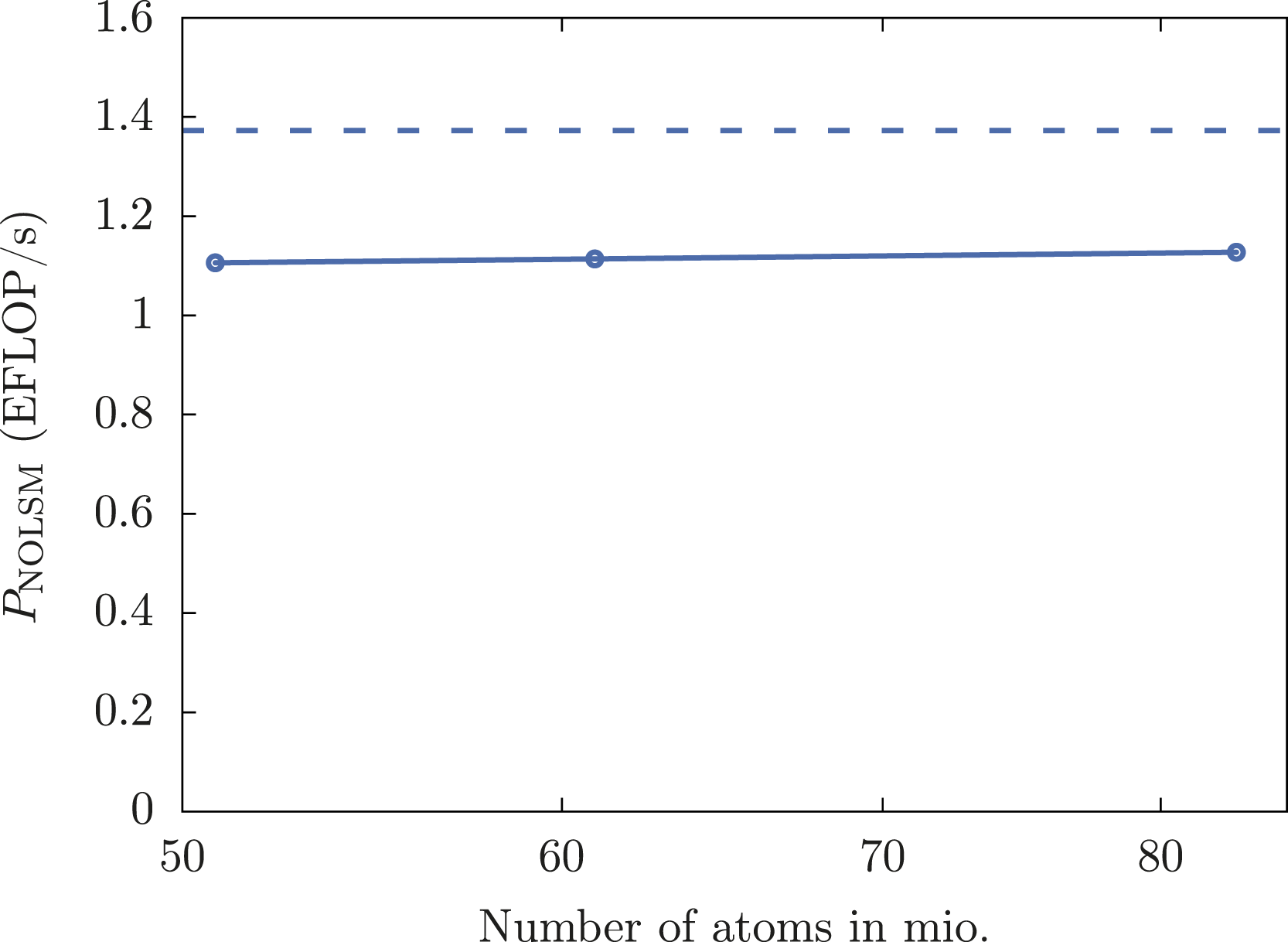
Conclusion
To the best of our knowledge, the achieved ∼1.1 EFLOP/s in FP16/FP32 floating-point arithmetic positions electronic structure–based molecular dynamics calculations with the non-orthogonal local submatrix method in CP2K (T. Kühne et al., 2020) among one of the first algorithms in computational natural science that has broken the exaflop barrier within a scientific application (Kurth et al., 2018; Joubert et al., 2018; Liu et al., 2021). The massively parallel nature of the method allows for an efficient use of many thousand GPUs. The method can not only be applied to electronic structure–based molecular dynamics, but also in other situations where a matrix function needs to be evaluated for a large sparse matrix or problems that can be transformed to such an operation.
Footnotes
Acknowledgements
This research used resources of the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility located at Lawrence Berkeley National Laboratory, operated under Contract No. DE-AC02-05CH11231 using NERSC award DDR-ERCAP0022240.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Additionally, we would like to thank for funding of this project by computing time provided by the Paderborn Center for Parallel Computing (PC2). This work is partially funded by Paderborn University’s research award for “GreenIT,” as well as the Federal Ministry of Education and Research (BMBF) and the state of North Rhine-Westphalia as part of the NHR Program. T.D.K. received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant Agreement No. 716142).
Author biographies
Robert Schade received his Ph.D. from Georg-August University Göttingen in 2019 for the development of new methods for the ab-initio simulation of materials with strong local electronic correlations. In his Ph.D. work, Robert Schade developed a novel method for the description of strong local electronic correlations in solids based on reduced density matrix functional theory (rDMFT). At PC2, Dr Schade is engaged in HPC-consulting for scientific users as well as general training of users to empower them to use the available computing resources in an efficient way. His main research focus is on the design and implementation of novel algorithms for enabling quantum chemistry on quantum computers as well as new highly parallel algorithms for quantum chemistry and their acceleration with FPGAs und GPUs.
Tobias Kenter received his PhD from Paderborn University in 2016 on the topic of productivity for FPGAs through overlays, compilation approaches, and tight coupling between FPGAs and CPUs. Since then, he focused on the acceleration of scientific applications on FPGAs using high-level synthesis–based development flows from Xilinx (now part of AMD) and Intel. As scientific advisor for FPGA acceleration at the Paderborn Center for Parallel Computing (PC2), he strives to bring more applications to this exciting technology and is involved in planning and operation of production systems with FPGAs.
Hossam Elgabarty graduated with a PhD in Chemistry in 2013 from the group of Daniel Sebastiani at the FU-Berlin. Later, he did two postdoctoral fellowships: At the University of Mainz with Prof. T. D. Kühne and at the University of Halle-Wittenberg with Prof. Daniel Sebastiani. In 2017, he moved to the University of Paderborn where he is currently a group leader at the Chair of Theoretical Chemistry. His group focuses on ab-initio MD simulations and theoretical condensed phase spectroscopy.
Michael Lass received his PhD from Paderborn University in 2022. In his dissertation, he dealt with the acceleration of a quantum chemistry code using accelerator devices, in particular GPUs and FPGAs, by exploiting algorithmic approximations and low-precision arithmetic. Since then, he works as a scientific advisor at the Paderborn Center for Parallel Computing (PC2), assisting users in the acceleration of their HPC codes and further developing the FPGA infrastructure.
Thomas D. Kühne studied computer science (B. Sc. ETH in 2003) and computational science and engineering (Dipl.-Rech. Wiss. ETH in 2005) with a focus in theoretical chemistry and computational astrophysics at ETH Zürich. Thereafter, he worked under the mentorship of Prof. Michele Parrinello in Lugano, where he obtained his Doctor of Science degree in theoretical physics in 2008, also from ETH Zürich. After postdoctoral research on multiscale simulation methods within the theoretical condensed matter group at Harvard University, he joined the University of Mainz as an assistant professor in theoretical chemistry in 2010. In 2014, he then moved to Paderborn University, as a tenured associate professor in Theoretical Interface Chemistry, where he was promoted to full professor in 2018 as the Chair of Theoretical Chemistry. Since May 2023, Prof. Kühne is the Founding Director of the Center for Advanced Systems Understanding (CASUS) at the Helmholtz-Zentrum Dresden-Rossendorf and Full Professor of Computational Systems Science at the TU Dresden. His research interests include the development of novel computational methods for ab-initio molecular dynamics and electronic structure theory, as well as the application of these techniques to study a large variety of different systems within Chemistry, Biophysics, and Materials Science.
Christian Plessl is professor for High-Performance Computing at the Department of Computer Science of Paderborn University. He is also the managing director of the Paderborn Center for Parallel Computing and member of the board of directors of the German National High-Performance Computing Alliance (NHR). He earned a PhD degree (2006) and MSc degree (2001), both from ETH Zurich. His research interests include architecture and tools for high-performance parallel and reconfigurable computing, hardware-software codesign, and adaptive computing systems.