
Abstract
Background/Objectives:
Neuroblastomas (NB) influenced by genetic alterations, which plays significant role in disease progression. Tumor suppressor genes (TSGs) are crucial in regulating cell growth, suppressing replication, and inducing apoptosis to prevent cancer formation. However, mutations in TSGs can lead to loss of normal activity, contributing to cancer development. This study aimed to identify TSG variants in NB patients and assess their clinical significance.
Methods:
One hundred two NB patients diagnosed and monitored according to the International Neuroblastoma Risk Group Staging System (INRGSS) protocol were included in this study. DNA was extracted from paraffin-embedded tissue samples, and Next-Generation Sequencing (NGS) was conducted using the Pillar ONCO/Reveal Multi-Cancer v4 panel.
Results:
The most frequently recurring TSG variant detected was RB1, p.P793S (n = 21; 25%) followed by ATM, p.D1853N (n = 20, 19.6%).
Stop-gain variants were identified in TP53 (p.R196*), FBXW7 (p.R367*), and PTEN (p.G129*).
Conclusions:
Our findings underscore the significance of specific TSG variants in NB, particularly in relation to disease progression and potential prognostic markers. Further research is needed to comprehensively assess the role of TSGs in NB, with an emphasis on germline variants and protein expression in larger patient cohorts.
Introduction
Neuroblastoma (NB) is a rare malignancy derived from the sympathetic nervous system of neural crest cells. The median age of NB diagnosis is 17 to 18 months, 1 and genetic alterations distinguish it. 2 Modifications in genetic expression play an important role in NB progression, influencing cellular processes such as growth. 3 Investigations of the genetic basis of NB, encompassing driver genes such as MYCN, RAS, and ALK, utilize clinical and biological variables, like MYCN amplification and chromosomal modifications, to categorize distinct risk groups in NB. 4 Children with high-risk NB frequently develop resistance to treatment because of tumor heterogeneity, resulting in recurrence and an unfavorable prognosis. The relevant therapeutic targets include anti-GD2 treatment and treatment for ALK, AURKA, and PHOX2B.5,6 The updated 2021 risk classification system was made by the Children’s Oncology Group (COG), NB has different risk categories (low, intermediate, and high-risk) that are assigned to based on tumor burden, biology, and prognosis. Risk assessment includes factors like patients age, tumor stage(L1, L2, M, and MS), surgical risk, imaging results, tumor differentiation grade, and biomarkers (MYCN oncogene amplification, DNA ploidy).7,8 Every year, approximately 3000 cases of pediatric malignancy are identified in the Turkish Republic, with roughly 500 being NB. 9 Fifty percent of these NB occurrences exhibit metastasis at the time of diagnosis. 10 TSGs play a crucial role in regulating cellular growth by inhibiting replication and promoting apoptosis, thereby preventing the development of cancer. These genes function essential of the genetic material, ensuring the stability of the genome and preventing the development of genetic alterations that may cause tumorigenesis. These genes operate through the preservation of genomic integrity, ingratiation of DNA repair mechanisms, and initiation of programmed cell death in instances of DNA damage.3,11 Mutations in TSGs may lead loss of function through somatic mutations (such as point mutations, missense mutations, and deletions), translocations, epigenetic changes, and chromosomal deletions. As a result, cells begin to proliferate uncontrollably and become resistant to apoptosis.12,13 Just as it is essential to investigate the mechanisms driving transformed oncogenes, studying the operation of adapted driver TSGs are also essential to understand the complex relationship between oncogenes and TSGs in cancer. Targeted therapies mostly focus on inhibiting increased oncogene activity. However, recent studies include new targets that regulate the activations of TSGs.14-17 NB is associated with alterations in TSGs, such as loss of heterozygosity on chromosome 1p, alterations on TP53, PHOX2B, and ATRX genes which is linked to poor prognosis in patients. 18 In contrast to adult cancers, TP53 mutations are infrequent in pediatric neuroblastoma, and its precise role in disease pathogenesis remains largely unexplored. However, beyond genetic alterations, TP53 may contribute to neuroblastoma development through alternative mechanisms. Some TP53 variants have been associated with increased neuroblastoma susceptibility, and abnormal TP53 protein accumulation has been frequently observed in neuroblastoma cells. Furthermore, MYCN, a key oncogenic driver in neuroblastoma, has been shown to directly regulate TP53 by binding to its promoter and enhancing its transcription. Additionally, TP53 dysfunction has been associated with resistance to radiotherapy in neuroblastoma, primarily through its influence on cellular metabolism. Moreover, MDM2, a principal negative regulator of TP53, plays a significant role in neuroblastoma progression. MDM2 is transcriptionally regulated by MYCN, and its overexpression contributes to MYCN stabilization and enhanced translation, thereby promoting tumorigenesis. The intricate relationship between TP53, MYCN, and MDM2 underscores critical molecular pathways in neuroblastoma pathophysiology, presenting potential targets for future therapeutic interventions, particularly in high-risk cases. 19
ATM is a TSG, with genomic alterations in chromosome 11q22-23 associated with tumorigenesis. NB cells with ATM zygosity and the ALT (Alternative Lengthening of Telomeres) mechanism of ATM kinase, which contributes to chemotherapy resistance. This resistance can potentially be reversed through the inhibition of ATM kinase. The function of ATM in NB is complex due to genetic diversity and MYCN gene amplification.20-22
The RB1 gene affects NB by potentially collaborating with other genetic elements such as MYCN and DDX1 in tumorigenesis. RB1 plays a role, in overseeing cell cycle advancement through the inhibition of the E2F transcription factor, causing arrest in the G1/S, and controlling cellular growth.23,24 The SMAD4 gene plays a notable role in NB by impacting diverse aspects of the condition. Moreover, SMAD4 establishes favorable transcriptional positive feedback cycle with MYCN, a gene correlated with high-risk NB, contributing to a distinct tumor reliance in MYCN-amplified NB. SMAD4 plays a role in boosting angiogenesis by stimulating proangiogenic factors like Vascular Endothelial Growth Factor (VEGF). These discoveries underscore the multifaceted function of SMAD4 in NB, influencing tumor aggressiveness, proliferation, and angiogenesis via diverse molecular mechanisms. 25 Mutations in TSGs can contribute to the development and progression of NB, emphasizing the importance of understanding their role in cancers. Therefore, gaining an understanding of TSGs functions in NB may pave the way for new therapeutic possibilities while also enabling the design of personalized treatment plans for patients with NB. Consequently, this may improve overall patient outcomes and lay the groundwork for the creation of new, low-side-effect treatment options for high-risk NB.26,27 Next-generation sequencing (NGS) is a high-capacity approach that enables the swift sequencing of DNA or RNA specimens, offering comprehensive insights into the hereditary composition of organisms. Within the sphere of NB investigations, NGS has played a key role in unveiling the genetic panorama of this pediatric cancer, which is imperative for the detection, prediction, and specialized targeted treatments. NGS is utilized to detect acquired variants, alterations in gene copy numbers, and additional genetic modifications that contribute to the origin and development of the ailment.28,29 Single nucleotide variants (SNVs) are alterations in the genome where a single nucleotide base is changed, inserted, or deleted. Studies have highlighted the significance of SNVs in NB pathogenesis, with genomic alterations like MYCN amplification, TERT rearrangements, and ATRX genomic alterations impacting telomere maintenance mechanisms and clinical outcomes in high-risk NB patients. 30 These discoveries cumulatively emphasize the crucial function of SNVs in oncology and the necessity for additional investigation to clarify their operational repercussions and treatment implications. The aim of this study was to identify rare variants in TSGs of NB patients.
Materials and Methods
The capture-based NGS analyses were carried out on 12 chosen TSGs, including ATM, TP53, RB1, SMAD4, APC, FBXW7, STK11, CDKN2A, SMARCB1, CDH1, PTEN, and MLH1 genes. DNA was isolated from tumor samples that were formalin-fixed paraffin-embedded (FFPE) from archival diagnostic tissue. Ethical approval was obtained from the Dokuz Eylül University Non-Invasive Research Ethics Committee (13.09.2023, No:2023/28-10), and was done by the Helsinki Declaration. The informed consent form was signed by patients’ parents or guardians.
Patients and Samples
This study included 102 NB cases. These cases were from 2300 NB samples diagnosed between 2012 and 2024. Cases were diagnosed according to the International Neuroblastoma Pathology Committee (INPC) classification, staged according to the International Neuroblastoma Risk Group Staging System (INRG-SS), and risk-stratified according to the Children’s Oncology Group (COG) risk classification system. These protocols were adapted to Turkish Pediatric Oncology Neuroblastoma Study Group. The primary diagnosis tumor samples were chosen for NGS analysis. NGS was planned for relapsed or refractory cases before but since 2022 NGS is planned at the diagnosis. NGS was used to identify potential options for targeted therapy based on clinical needs.
Tumor Tissue Selection and DNA Isolation
Tumor tissues 3mm in diameter each were selected from the most viable part of FFPE samples and DNA was isolated and measured.31
Targeted Next-Generation Sequencing (NGS)
Commercial Pillar ONCO/Reveal Multi-Cancer v4 kit Targeted NGS panel was performed. SNV were analysed Copy number variations were not assessed in this study.
Library Preparation and Sequencing
Genomic DNA targets were amplified by PCR. DNA was cut into 150 to 200 base pairs by exonclease. The DNA fragments of each case was indexed. Libraries were subsequently pooled, normalized. Sequencing was performed on the Illumina MiniSeq platforms described in our previous publication. 10
Data Analysis
Binary Alignment Map (BAM) files were generated compared with human reference genome. Variant calling was conducted with the Pillar Variant Analysis Toolkit (PiVAT). The annotation of variants was performed with reference data from COSMIC (v90), ClinVar (v201912), gnomAD (r2.1), and 1000 Genomes (Phase 3) databases.
Statistic
Variants causing significant changes in missense, frameshift, and stop-gained features were included in analysis. Clinical and laboratory information such as patient age, sex, risk group, EFS, OS, MYCN amplification, 11q deletion, and DNA ploidy condition were compared with the existence of variants utilizing non-parametric Chi-square analysis or Mann-Whitney U test, and Spearman Correlation analysis. The log rank test was used in Kaplan-Meier survival analysis to evaluate the impact of variants on survival. Statistical significance was attributed to P-values <.05. All statistical analyses were performed using SPSS version 29.0 (IBM Inc., Armonk, NY, USA).
Results
Patient Characteristics
The mean patient age at diagnosis was 48.57 ± 40.42 months (range: 2-204), 25 (24.5%) patients were less than 18 months old at diagnosis. The mean age of 2300 entire cohort is 38.55 ± 43.07. 57 (55.9%) patients were males, 45 (44.1%) patients were females. The 5-year OS rate was 27.0 ± 20.4 (min 4-max 113 months), and the 5-year EFS rate was 16.34 ± 15.76 (min 3-max 113 months). MYCN (2p24.3) amplification was observed in 39 (38.2%) of cases and 11q23.3 deletion in 31 (30.4%) while it is 21.4% for MYCN and 24.9% for 11q23.3 in the 2300 cases cohort. Furthermore, 73.6 % of patients were diploid or near-diploid upon initial diagnosis. According to Shimada classification 12 patients (11.8%) were classified as favorable histology, and 90 patients (88.2%) were classified under the unfavorable histology. For INRGSS, 9 patients (8.8%) L1 stage, 14 patients (13.7%) L2 stage, 79 patients (77.5%) M stage, and none of the patients MS stage. For the COG risk classification, 9 patients (8.8%) were low-risk, 9 (8.8%) intermediate-risk, and 84 (82.4%) high-risk. Detailed clinicopathologic information regarding this cohort of patients can be located in Table 1.
The Frequency of Amplification MYCN (2p24.3) Was Significantly Related to the Risk Group (P = .002) and INRGSS (P = .024).

The risk group the was significantly correlated with INRGSS (P < .001), Shimada classification (P < .001), and MYCN amplification (P = .02) in Spearman correlation analysis. No mutations were seen in the CDH1, CDKN2A, MLH1, or PTEN genes in the 11 patients with exitus, which may be associated with tumors with low variant burden being less responsive to treatment and having an inadequate immune response.
Profiles of Detected Variants
In this study, TSGs detected by NGS were analyzed for NB. Variants were identified in the exonic regions, including 724 missense variants, 31 stop-gained SNVs, and 4 frameshift variants. Variants located within intronic regions and synonymous variants were not documented.
After excluding synonymous SNVs, the gene with variants detected in the most patients was the ATM gene, affecting 37 patients (36.3%). RB1 variants were found in 35 patients (34.3%), TP53 variants in 30 patients (29.4%), SMAD4 variants in 23 patients (22.5%), STK11 variants in 20 patients (19.6%), FBXW7 variants in 20 patients (19.6%), SMARCB1 variants in 20 patients (19.6%), CDH1 variants in 18 patients (17.6%), APC variants in 18 patients (17.6%), CDKN2A variants in 16 patients (15.7%), and PTEN variants in 9 patients (8.8%). The gene with variants detected in the least number of patients was the MLH1 gene, with 6 (5.9%) patients.
The most frequently mutated gene was TP53 (n = 159). The other detected variants were SMAD4 (n = 107), ATM (n = 101), RB1 (n = 72), STK11 (n = 70), FBXW7 (n = 67), APC (n = 59), SMARCB1 (n = 42), CDH1 (n = 35), CDKN2A (n = 22), PTEN (n = 14), and MLH1 (n = 11).
The most frequently detected variants were p.P793S (n = 21, 20.58%) in the RB1 gene and p.D1853N (n = 20, 19.6%) in the ATM gene. When the clinical significance of the detected variants were examined, although mostly uncertain, 18 patients had pathogenic / likely pathogenic variants in TP53 gene. There were patients with multiple variants for the same gene. The variants detected more than once is given in Table 2.
Variants Detected Multiple Times.

Deletion.
Statistical Analysis of Detected Variants
In Spearman Correlation analysis, age, sex, survival, event, deletion of 11q23.3, MYCN amplification, Shimada classification, INRGSS, and COG risk classification was not correlated with any of 12 genes. In the Kaplan-Meier survival analysis with the Log Rank test, changes in any of the 12 genes were not statistically significantly linked to EFS or OS.
Discussion
Targeted therapies primarily focus on oncogenes. However, the role of TSGs in NB is rarely investigated. Studies and techniques developed on TSGs increase our knowledge to better understand tumorigenesis and provide better insight into the genome. This study performed NGS analysis on NB samples to determine the frequency of TSG variants in various risk categories and their potential clinical outcomes. Our study’s results revealed relevant TSG variants associated with NB. We found the TP53 gene variants to be the most frequently recurrent. In a study on NB cell lines, 32 it was shown that cell lines with the TSG TP53 undergo cell death by apoptosis. The findings show that NB cells with wild-type TP53 remain dormant in the G0/G1 phase of the normal cell cycle, whereas cells with mutated TP53 pause in the G2/M phase with increased DNA content.
These findings highlight the complex molecular mechanisms by which TP53 variants and related factors contribute to the development and progression of NB.
The most common hotspot mutations in the TP53 gene include p.R175, p.V157F, p.Y220C, p.G245, p.R248, p.R249, p.R273, and p.R282. These mutations are noteworthy because they are usually located in the DNA binding domain of the p53 protein. 33 The variants we identified in our study are R158H, R175H, R196*, R273H, R290H, R333H, D281G, E343G, E346G, G245S, G245V, I162T, L344P, M237V, F113S, F19S, S166P, S315P, T256A (Figure 1.).
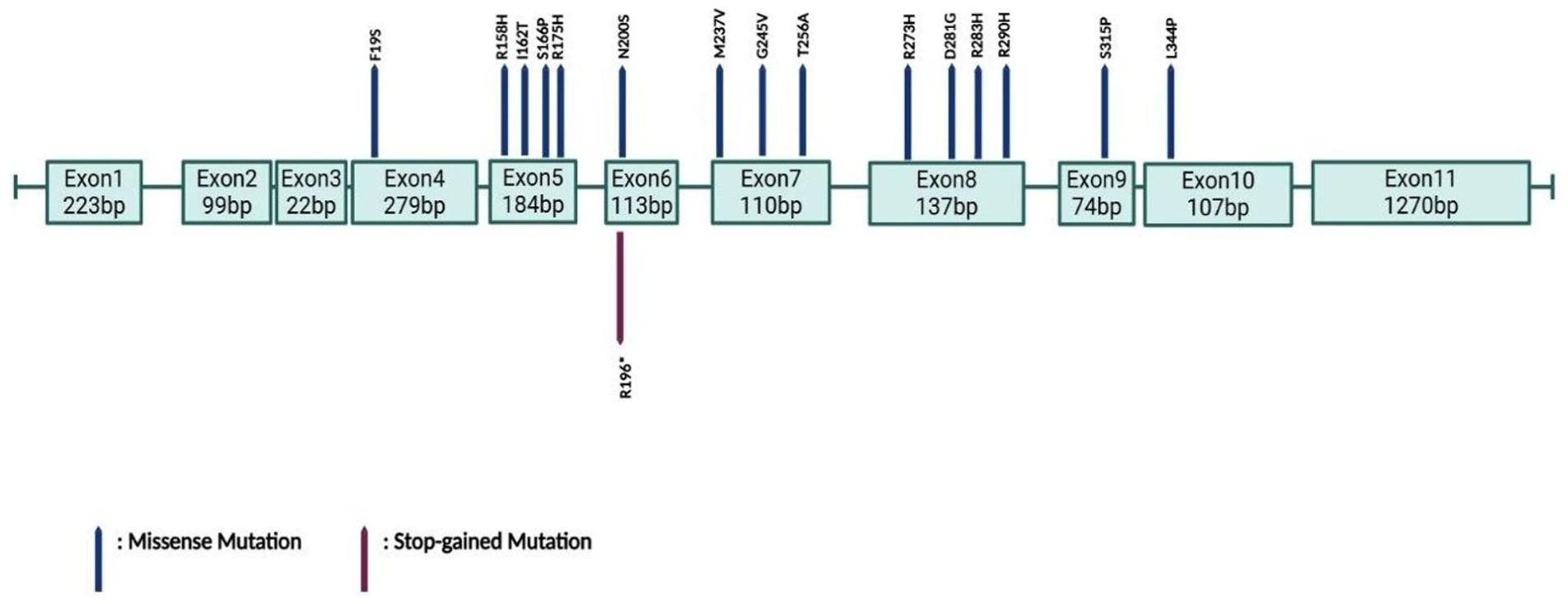
Clinically significant variants identified in the TP53 gene. Most of the variants. were found in the DNA binding domain (exon 5-8).
We also detected the p.R158H variant in the TP53 gene in 2 patients. This variant has been previously reported in the literature by Kharaziha et al. 34 Fischer et al 35 noted that missense mutations in TP53 frequently occur within the DNA binding domain (DBD), accounting for nearly 70% of TP53 alterations in cancer. It has been suggested that a historical sequencing bias between exons 5 and 8 may be contribute to this clustering.36-39
In a study 40 published in 2020, somatic mutations were detected in the TP53 gene at a rate of 0.9% in 685 patients. However, only 23 of the 685 patients in the study had relapsed refractory disease. In our series, 30 of 102 patients (29.4%) had various variants in the TP53 gene. The reason why this rate is higher compared to the literature is thought to be related to the fact that 47 of 102 patients had relapsed refractory disease. As reported in the literature, TP53 mutations are seen in 1% to 2% of primary tumors in NB and up to 15% of relapsed NB, and the rate increases dramatically in relapsed refractory disease.40-42 A study 43 using a genetically engineered mouse model investigated the role of TP53 in high-risk, treatment-resistant NB and reported that approximately 50% of relapsed tumors show a loss of TP53 function, typically after treatment. Unlike other MYC-driven cancers, p53 pathway mutations are rare at diagnosis, suggesting that they are selected for after treatment. In addition, Kaur et al 21 reported that from the current review and data from the COSMIC database, TP53 is the most frequently mutated gene in olfactory NB. Genomic instability that occurs during relapse may result in the accumulation of somatic variants in the TP53 gene. 44 The study 45 identified de novo mutations and genomic patterns in relapsed NBs, finding that the mutational burden in relapsed NB is significantly higher than in primary tumors. Unlike primary tumors, new mutational signatures were shown to emerge in relapsed genomes, with relapsed tumors exhibiting a greater number of SNVs. Most SNVs in relapsed tumors are either not detected in primary tumors or appear at low frequencies. Increased sequencing depth has been reported to reveal additional mutations in primary tumors. Additionally, local relapses show higher SNV numbers than metastatic relapses, suggesting distinct selection pressures. The study highlights that relapsed NBs display a notably higher mutational burden than primary tumors, with specific recurrent mutations likely contributing to tumor recurrence, and that clonal evolution in these tumors suggests a shift in tumor dynamics as the disease progresses.46 -49
We did not find a strong link between ATM variants and EFS or OS in our study.
However, another study 50 discovered a link in ATM haploinsufficiency and lower ATM expression between EFS, and OS in NB patients. Observations of ATM loss in high-stage NB without MYCN amplification suggest that ATM inactivation contributes to NB progression through a MYCN-independent mechanism. The ATM gene, located on chromosomes 11q22-q23, encodes a protein that is crucial for DNA damage response and cell cycle checkpoint regulation. In our study, 11q23.3 deletion was detected in 31 patients (30.4%). In NB, with highly heterogeneous biology, the hemizygous deletion of chromosome 11q is a well-established marker of poor prognosis. This region frequently deletes the ATM gene, and its loss significantly impacts the progression of NB.
In our study, we detected variants in the ATM gene in 37 patients (36.3%). The benign p.D1853N variant in the ATM gene was detected in 20 out of 102 patients, representing 19.6% of the sample. 51 In a 2010 study by Gao et al. 52 the relationship between the ATM gene p.D1853N polymorphism and cancer risk was examined through a meta-analysis. The ATM gene is essential for DNA damage repair and cell cycle control, and loss of heterozygosity has been observed in about 40% of sporadic breast tumors. Although the p.D1853N polymorphism is not seen as an independent risk factor, conflicting findings suggest that additional studies are needed to clarify its role. Notably, there is no research on its relevance to NB pathogenesis, highlighting the need for further investigation into a potential link. Our patients had a total of 21 p.P793S variants and 2 p.P793* variants in the RB1 gene. The literature does not specifically address this variant. Only 1 study 53 reported detecting a tolerable RB1 missense p.P793S variant within the C-terminal domain in the neuroendocrine prostate patient-derived xenograft (PDX) tumor model. In the literature, 54 a study examining the connection between the RB1 gene and MYCN in NB found that MYCN overexpression inhibits RB by increasing E2F3-responsive promoter activity, which leads to hyperphosphorylation of RB during the G1 phase. Amplifying MYCN makes RB indispensable for regulating the cell cycle of NB cells, suggesting a shift in reliance on cell cycle control mechanisms. The genetic interactions among amplified MYCN, wild-type RB1, and E2F3 reveal an unexpected molecular vulnerability. High MYCN and E2F3 expression correlated with poor prognosis in NB, independent of RB1 mRNA levels. This suggests a potential treatment target for NB patients with MYCN amplification and high RB1 level. The results demonstrate the potential of cyclin/CDK 4 to 6 complex inhibitors in treating this specific group of NB patients, highlighting the importance of personalized therapeutic approaches.
Furthermore, recent studies have highlighted the role of telomere maintenance mechanisms in neuroblastoma biology. The Alternative Lengthening of Telomeres (ALT) phenotype has been shown to associate with TP53 and RB1 dysfunction, leading to telomere instability, genomic stress, and adverse clinical outcomes. These findings suggest that alterations in TP53 and RB1 may indirectly promote tumor progression through deregulated telomere maintenance.55,56
While homozygous CDKN2A deletions occur in approximately 3% of primary NB, Schubert et al 57 identified this copy number loss in 13% of recurrent tumors. This
group found that the loss of CDKN2A in NB cells was associated with a phenotypic shift toward a more progenitor-like state and increased sensitivity to EGFR inhibitors; suggesting potential therapeutic effects. Bassi et al 58 explained that homozygous deletion mutations are not the main mechanism of inactivation for the CDKN2B/p15 and CDKN2A/p16 genes in NB, loss of heterozygosity in the 9p region is associated with advanced disease stages and a poor prognosis, regardless of MYCN amplification.
The p.R173H variant was detected in 3 patients (2.94%) in the PTEN gene. In a study, 59 it was concluded that the p.R173H variant detected in MeWo cells, a cutaneous melanoma cell line, causes resistance to 5′-Aza-2 deoxycytidine (Aza-C), an immunoregulatory agent used clinically in the treatment of myelodysplastic syndrome and acute myeloid leukemia.
In our study, we detected variants in the SMAD4 gene that stop-gain: p.G176*, p.W398*, and p.Q388*. Isidori et al 60 found a strong association between SMAD4 loss and recurrence in esophageal adenocarcinoma. Their study, involving 38 individuals, detected p.G176* variant in 1 patient who had 100% loss in the SMAD gene and mutations in the TP53/cyclin-dependent kinase inhibitor 2A (CDKN2A; TP53 pathway). In our study, multiple variants were detected in the TP53 and CDKN2A genes in all 3 patients carrying the p.G176* p.W398*, and p.Q388* variant, a finding consistent with existing literature. Isidori et al 58 reported a significant correlation between SMAD4 loss and cancer recurrence, showing that 29% of patients experienced recurrence when SMAD4 immunoreactivity was intact, compared to 75% when SMAD4 was lost. Notably, the 3 patients in our study with variants in the SMAD4, TP53, and CDKN2A genes presented with relapsed, refractory disease.
We detected variants in the STK11 gene in 19.6% of 102 patients, with p.F354L as the most frequent variant. One study identified a single heterozygous germline variant, STK11, p.F354L, in a child who developed an encapsulated follicular variant of papillary thyroid carcinoma (FVPTC) within 6 months of completing the NB treatment. The STK11 variant was located in chromosome band 19p13.3 by NGS, which revealed 3 identical heterozygous missense variants in both tumors. The STK11, p.F354L variant was confirmed as a germline variant in the patient’s peripheral blood mononuclear cells. 61
This study has several limitations. Firstly, we did not perform additional sequencing methods to further validate the identified variants. Secondly, the sample size of 102 patients is relatively small, limiting the generalizability of our findings. Additionally, 82.4% of the patient cohort consisted of high-risk patients, which may introduce a selection bias. Further large-scale studies are needed, particularly to assess the impact of rare variants with low allele frequencies on neuroblastoma (NB) oncogenesis.
The elevated mutational burden and increased SNV frequency in relapsed tumors highlight the genomic instability characteristic of advanced NB stages. Clonal evolution in relapsed tumors, characterized by new mutational signatures and SNVs, suggests distinct selective pressures shaping tumor cells throughout disease progression. Further investigation into the timing and functional impact of tumor suppressor gene (TSG) mutations in NB could enhance our understanding of its molecular mechanisms and contribute to the development of targeted therapies.
Future research should include paired tumor-normal sequencing and germline analyses to explore hereditary predisposition. Additionally, RNA and protein expression analyses in larger patient cohorts are essential to determine the functional consequences of these variants. Such studies will provide deeper insights into the genetic landscape of NB and facilitate the development of more effective therapeutic strategies.
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.