
Abstract
Liver involvement by histiocytic and dendritic cell neoplasms signals high-risk disease, often necessitating closer monitoring and aggressive management. Severe cases may progress to liver failure, requiring transplantation. Liver involvement occurs in about one-third of patients with systemic juvenile xanthogranuloma (JXG) and 20% to 60% of pediatric patients with Langerhans cell histiocytosis (LCH), particularly in multiorgan disease. Tyrosine kinase inhibitors show promise in LCH treatment, but optimal timing for treatment cessation remains uncertain. We present 2 pediatric cases, 1 with LCH, and the other with disseminated JXG, along with a literature review emphasizing liver histopathology and transplant considerations. These cases highlight distinct histological patterns. In LCH, progressive bile duct destruction led to ductopenic cholestatic cirrhosis and secondary sclerosing cholangitis. In contrast, in the case of JXG, bile ducts remained intact despite being surrounded by histiocytes. In both, disease localization to larger, segmental portal tracts may reduce liver biopsy sensitivity. In LCH, BRAF inhibitor therapy triggered a granulomatous reaction that could mimic disease recurrence in the liver graft. Other histiocytoses typically spare the bile ducts and do not cause biliary cirrhosis. Recognizing these distinct infiltration patterns can aid diagnosis and management.
Background
Histiocytic and dendritic cell neoplasms are rare, complex disorders with variable and often non-specific symptoms, depending on whether disease is localized or systemic.
Systemic forms fall under the “L (Langerhans) group” per the 2016 revised classification by the Histiocyte Society. 1 This group includes Langerhans cell histiocytosis (LCH), Erdheim-Chester disease (ECD), and extracutaneous and disseminated juvenile xanthogranuloma (JXG), all marked by frequent activating mutations in genes of the MAPK pathway, resulting in ERK overexpression. 2 The 5th edition of the WHO classification of hematolymphoid tumors 3 divides these neoplasms into 3 categories: (1) Plasmacytoid dendritic cell neoplasms, (2) Langerhans cell and other dendritic cell neoplasms, and (3) Histiocyte/macrophage neoplasms. The latter category groups ECD and JXG together with Rosai-Dorfman disease, ALK-positive histiocytosis, and histiocytic sarcoma. Diagnosis often relies on immunophenotyping, though overlapping features can complicate classification.
Liver involvement occurs in approximately one-third (31.4%) of patients with systemic JXG 4 and in 20% to 60% of pediatric LCH patients 5 , especially in multiorgan disease, sometimes requiring liver transplantation (LT).
We present 2 pediatric cases, 1 with LCH, and the other with disseminated JXG, highlighting distinct hepatic histology. We also review literature on liver pathology and molecular features in histiocytoses, and the potential role of LT.
Methods
We reviewed 2 pediatric cases of histiocytosis with liver involvement requiring transplantation. Written informed consent was obtained from the parents. As this study fell outside the scope of Swiss legislation on human research, ethics committee approval was waived.
Histology, Immunohistochemistry, and In Situ Hybridization
The liver biopsies, explants, and skin biopsies from both patients were processed following standard protocol. For routine histology, 3-μm sections were stained with hematoxylin-eosin (H&E). Representative liver samples were additionally stained with Masson’s trichrome, reticulin, Perl’s, and PAS-diastase. Immunohistochemistry was performed on 4-μm sections formalin-fixed, paraffin-embedded (FFPE) sections using the Ultraview or Optiview detection systems (Roche) for the following antibodies: BRAF V600E, CD1a, CD163, CD68, CK7, Factor XIIIa, Fascin, CD207/Langerin, and S100 protein. Epstein-Barr virus detection was assessed by in situ hybridization using EBER probes. Antibodies and probes are detailed in Table 1.
Reagents for Immunohistochemistry and In Situ Hybridization.
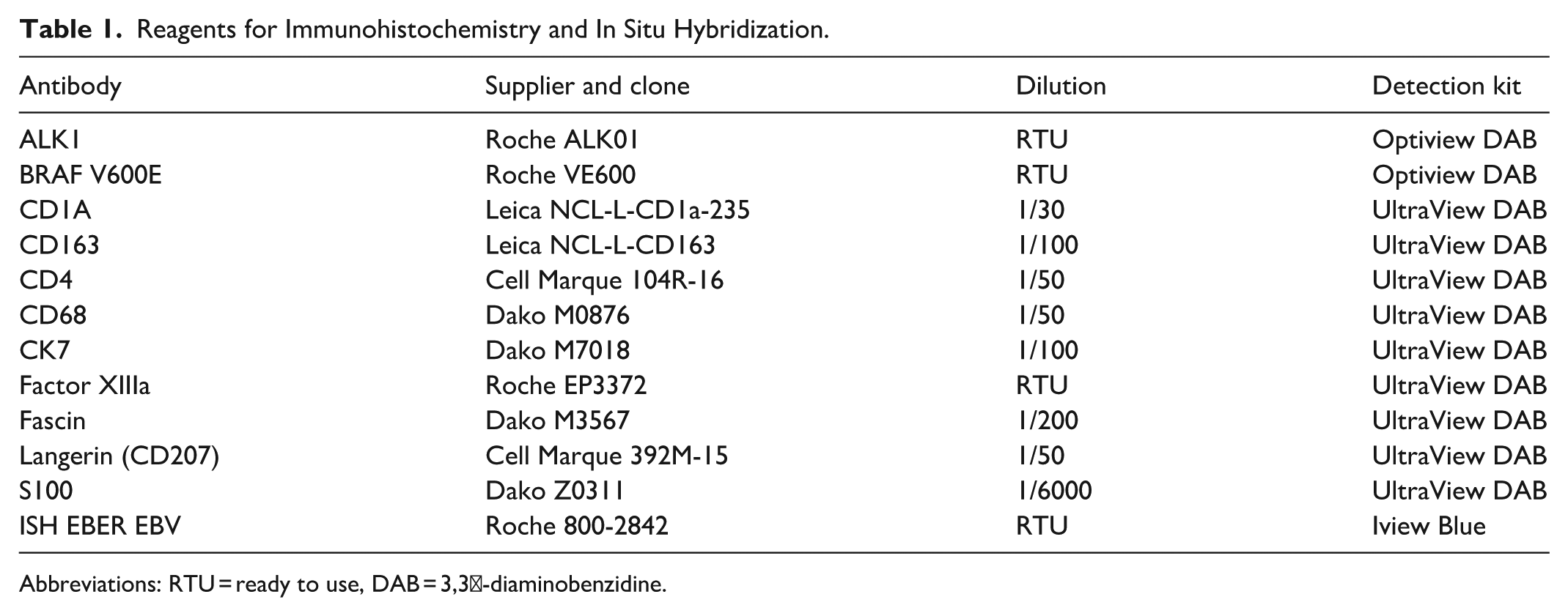
Abbreviations: RTU = ready to use, DAB = 3,3′-diaminobenzidine.
Molecular Analysis
Tumor cell content was assessed on H&E slides. Genomic DNA was extracted from FFPE tumor tissue using the QIAamp DNA FFPE Tissue Kit (Qiagen). For targeted DNA sequencing, libraries were prepared with a custom 462-gene panel (SureSelect-HS, Agilent) and sequenced on a NextSeq500 (Illumina). Reads were aligned to the hg19 reference genome using BWA-MEM, and variants were called with Mutect2 and Strelka2, using matched normal tissue to filter germline variants.
For RNA sequencing, RNA extracted from FFPE tissue was processed with the TruSight RNA Fusion Panel (Illumina), targeting 507 fusion-associated genes. Libraries were sequenced on a NextSeq, and fusion transcripts were identified using MapSplice 2.1.5.
BRAF V600E mutation was excluded by SYBR Green-based qRT-PCR. RNA was isolated (NucleoSpin RNA Kit, Macherey-Nagel), reverse-transcribed (Applied Biosystems, ref. 43698813), and amplified on a LightCycler 480 (Roche), with Ct values normalized to TBP.
Review of the Literature
A systematic search of the MEDLINE (PubMed) database was conducted through June 2025 to identify pediatric cases of liver transplantation due to histiocytic or dendritic cell neoplasms. Search terms included: pediatric OR children AND liver transplantation AND histiocytosis, OR Langerhans cell, OR juvenile xanthogranulomatosis, OR ALK-positive histiocytosis.
Liver involvement in LCH was defined by hepatomegaly (>3 cm below the costal margin, midclavicular line) and liver dysfunction (hypoproteinemia <55 g/L and hypoalbuminemia <25 g/L), excluding other causes. 6 As histological confirmation is not required for diagnosis, we included all reported pediatric liver transplant cases for LCH, JXG, or ALK-positive histiocytosis that provided histological descriptions.
Results
Case 1: 5-Year-Old Female at Liver Transplantation—LCH
A 1-year-old female presented with cutaneous lesions diagnosed as LCH), initially managed with topical therapy. Over the following year, she experienced multiple cutaneous flares, which remained responsive to topical treatment. At age 2 years, she developed diabetes insipidus, controlled with desmopressin.
A focal liver lesion was identified during disease work-up and monitored closely. At 2.5 years, she was started on LCH-III protocol treatment (Group 1, multisystem risk). By age 4 years, she developed signs of chronic liver disease, including hepatosplenomegaly, jaundice, and telangiectasias. Liver biopsy revealed sclerosing cholangitis, attributed to LCH.
Soon after, a new skin lesion appeared. Histology showed dermal infiltration by histiocytes with grooved, reniform nuclei and eosinophilic cytoplasm. Cells were positive for CD68, S100, CD207/Langerin, and CD1a. BRAF V600E (VE1) immunostaining was focally positive; mutation analysis confirmed BRAF V600E mutation with a variant allele frequency (VAF) of 3.96%, also subsequently detected in blood and bone marrow. Vemurafenib treatment was initiated.
After 4 months, total-body MRI revealed a lytic lesion in the right humerus, suggesting refractory disease. Treatment was discontinued, leading to rapid worsening of cholestasis and necessitating urgent liver transplantation at age 5 years.
The explant liver showed cholestatic cirrhosis with ductopenia, concentric periductal fibrosis around remaining bile ducts, and segmental hypertrophy (segments I and IV) with regenerative macronodules. Cytokeratin 7 highlighted ductopenia, ductular reaction, and aberrant expression in hepatocytes consistent with chronic cholestasis. Langerhans cells (CD68, protein S100, CD207/Langerin, and CD1a positive) were focally present in a large portal tract and gallbladder. NGS confirmed BRAF V600E mutation (VAF 0.5%), supporting active liver disease at the time of transplantation, warranting resumption of vemurafenib post-transplant.
The patient has remained on vemurafenib for 8 years post-LT 8 years ago and is disease-free. She undergoes yearly follow up for her LCH including serial liver biopsies. Three years post-LT, protocol biopsies revealed persistent portal and lobular granulomas. However, no recurrence of LCH was seen by immunohistochemistry (S100 and CD1a), and BRAF V600E testing has remained negative. Special stains ruled out fungal or mycobacterial infection.
Figures 1 and 2 illustrate the main pathological findings and vemurafenib-associated granulomatous changes.
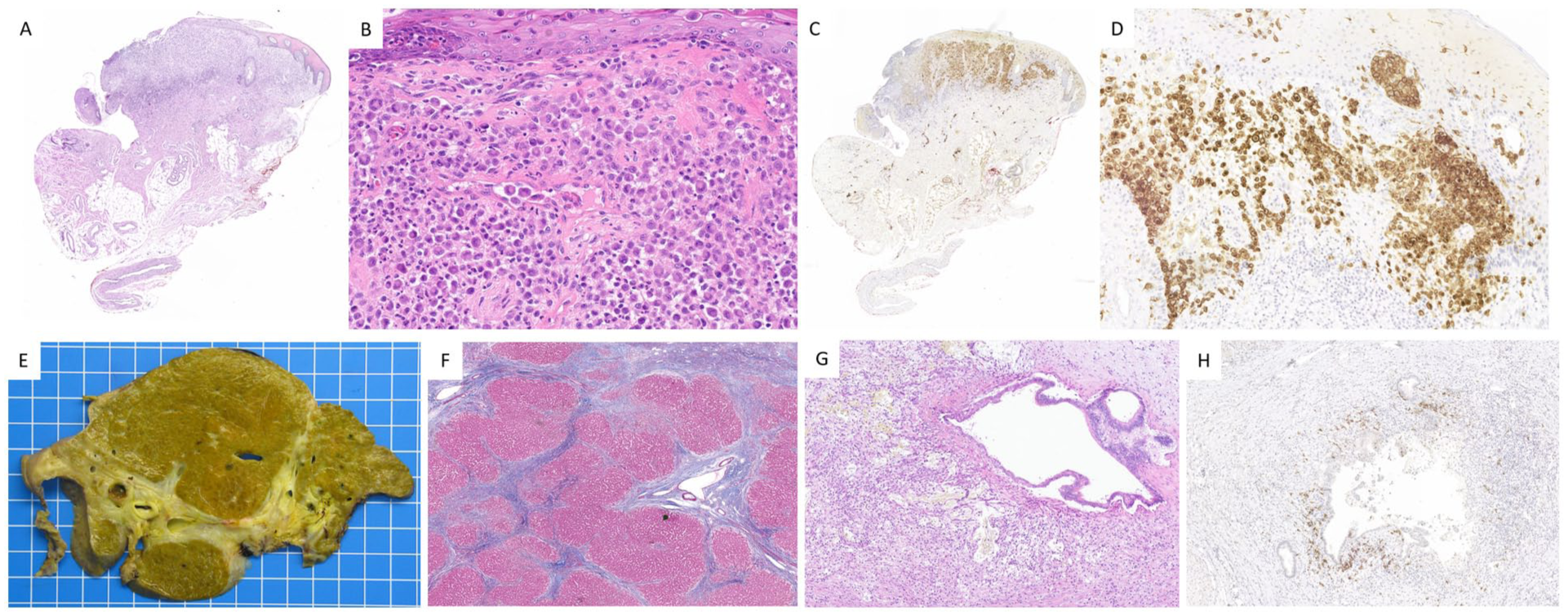
Case 1, LCH. A-D. Skin lesion. The dermis shows infiltration by histiocytes characterized by large vesicular, grooved, and often reniform nucleus, along with moderately abundant eosinophilic cytoplasm (A, B, Hematoxylin and Eosin, H&E). These cells exhibit immunoreactivity for S100 protein (C) and CD1a (D). EH. Liver explant. Cut section shows liver cirrhosis (E), confirmed by Masson’s trichrome (F). A dystrophic, partially disrupted, segmental bile duct is surrounded by a xanthomatous inflammatory reaction (G, H&E), comprising histiocytes reactive to the BRAF V600E (VE1) antibody (H).
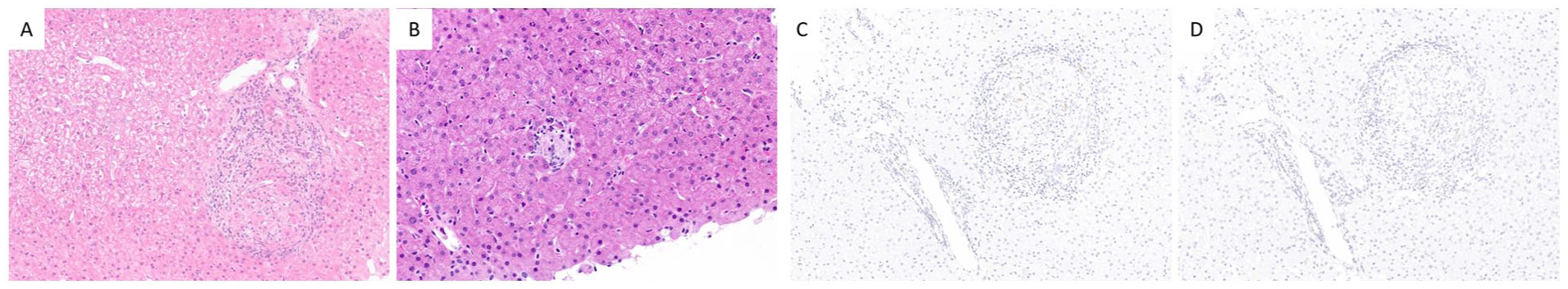
Case 1, LCH. A-D. Vemurafenib-induced granulomatous reaction in the liver transplant. Nonnecrotizing granulomas are seen within both the portal tracts (A) and hepatic lobules (B, H&E). There is no reactivity to S100 protein (C) or to CD1a (D), arguing against recurrent LCH.
Main Histological Findings in the Liver: LCH

Case 2: Infant Male Aged 4 months at Liver Transplantation—Disseminated JXG
A male infant was born at 36 weeks via induction for fetal ascites detected at 33 weeks. Postnatally, he presented with refractory ascites, thrombocytopenia, and an umbilico-iliac porto-systemic shunt on Doppler ultrasound. Portal hypertension raised suspicion for intrahepatic disease, but biopsy was contraindicated due to severe thrombocytopenia.
At 2 months, non-confluent, macular pigmented skin lesions (≤0.4 cm) appeared on the right hemithorax and shoulder, initially attributed to iron overload from transfusions. Despite supportive therapy, he underwent LT at 4 months for cholestatic liver failure and refractory ascites of unknown origin. Pre-transplant liver biopsy was not performed. The post-operative course was mostly uneventful aside from 1 intra-abdominal infection requiring surgical intervention.
At transplantation, hepatomegaly was confirmed (liver weight: 462 g; expected: 160 g). Histology of the explant showed canalicular and hepatocytic cholestasis, bridging necrosis, perisinusoidal fibrosis, and dense histiocytic infiltration of the liver hilum, segmental portal tracts, and round ligament. No Touton cells or xanthomatous giant cells were seen. Infiltrating cells were mononuclear with oval/spindle nuclei, and reactive to histiocytic markers CD68, CD163, and CD4, further expressing Fascin, and Factor XIIIA. They remained negative for CD1a, S100, CD207/Langerin, ALK1, and BRAF V600E; EBER in situ hybridization was negative. Findings were diagnostic of disseminated JXG with liver involvement.
Molecular analysis revealed a class 5 hotspot mutation in PTPN11 (p.E76V, VAF 34%), and a class 4 CSF1R deletion (p.P566_N572del, VAF 20%). Fusion gene screening was negative, including ALK1 and NTRK rearrangements.
Post-LT imaging identified multifocal bone lesions, bilateral renal and pituitary involvement, CNS lesions, and pulmonary infiltration. At 6 months of age, new skin lesions appeared on the face and groin. Biopsies of skin and kidney confirmed histiocytic infiltration with foamy mononuclear cells, sharing the liver immunoprofile, without Touton cells.
Treatment following the multisystem LCH arm of the LCH-IV protocol was initiated at 5 months (1-month post-LT; see Supplemental Figure 1). One year after LT, the patient showed partial systemic remission. Persistent lesions remained in mediastinal vessels, lungs, liver capsule, peritoneum, IVC, meninges, and coronary arteries, but treatment continued without modification.
At 6 years post-LT (and 4 years after completing chemotherapy), the patient remains free of active disease. Imaging shows residual pulmonary and vascular calcifications. Brain MRI at 4 years post-LT revealed white matter rarefaction, periventricular T2 signal changes, and a thin corpus callosum. Clinically, he has normal liver function, few infections, but significant neurocognitive delay and failure to thrive, largely due to oral aversion.
Key pathological findings are illustrated in Figure 3.
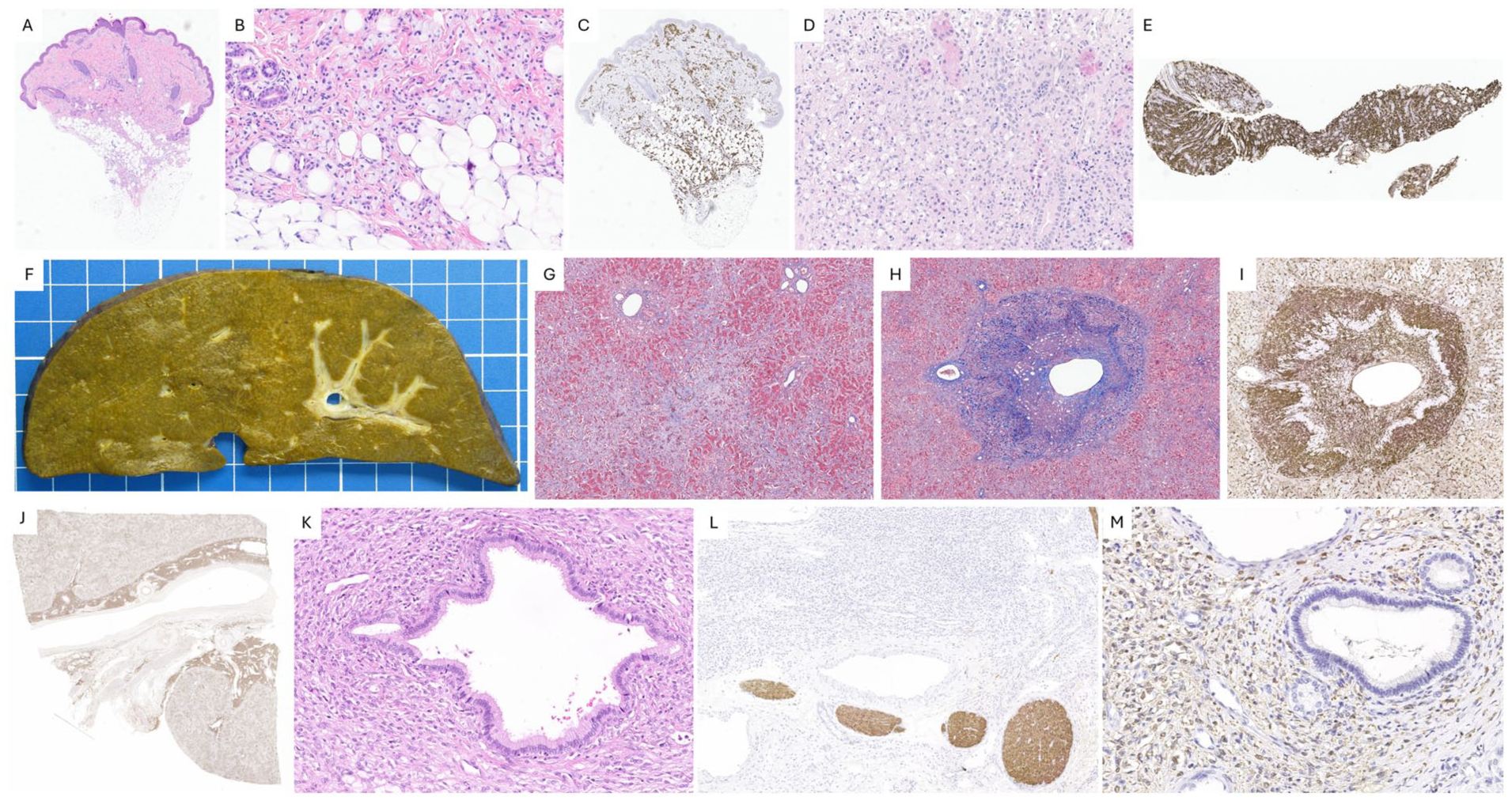
Case 2, JXG. A-C. Skin lesion. The dermis and hypodermis show infiltration by numerous xanthomatous mononuclear foam cells (A, B. H&E), which are positive for CD68 (C). D-E. Kidney infiltration. Renal biopsy reveals extensive infiltration (D, H&E) by similar CD68-positive histiocytes (E). F-M. Liver explant. Nodularity is seen on gross examination (F), while Masson’s trichrome highlights the perisinusoidal fibrosis (G). Centrilobular endotheliitis: histiocytes infiltrate and disrupt the wall of a centrilobular vein (H, Masson’s trichrome, I, Fascin). In the liver hilum, CD68-positive histiocytes form a dense infiltrate (J), surrounding but sparing a large bile duct with intact epithelial lining (K, H&E). These histiocytes are negative for S100 protein, which highlights nerves (L), but show immunoreactivity for Factor XIIIa (M).
Main Histological Findings in the Liver: JXG
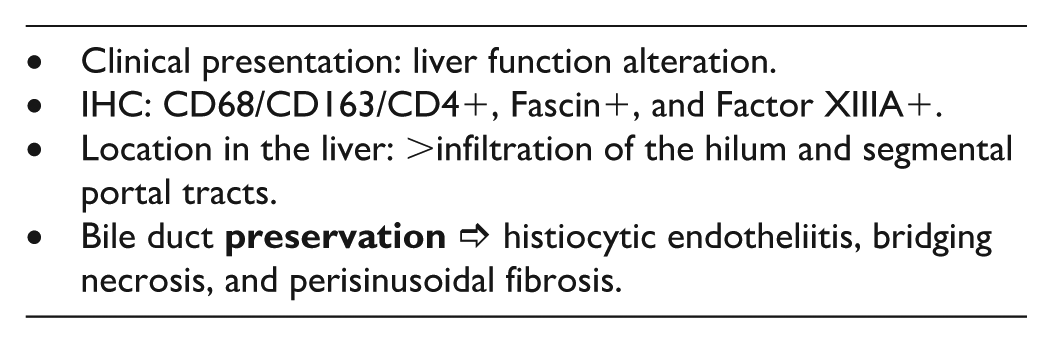
The main findings from these 2 cases are presented in Table 2. It is noteworthy that both cases were severe and required potentially lifesaving liver transplantation. While they serve as illustrative situations, they do not capture the full spectrum of liver infiltration in LCH and JXG. In particular, JXG often follows an indolent course. Our findings are compared with previously reported histological findings from pediatric liver transplants in LCH and JXG.
Main Clinical and Histopathological Features of Pediatric Liver Explants in LCH and JXG.
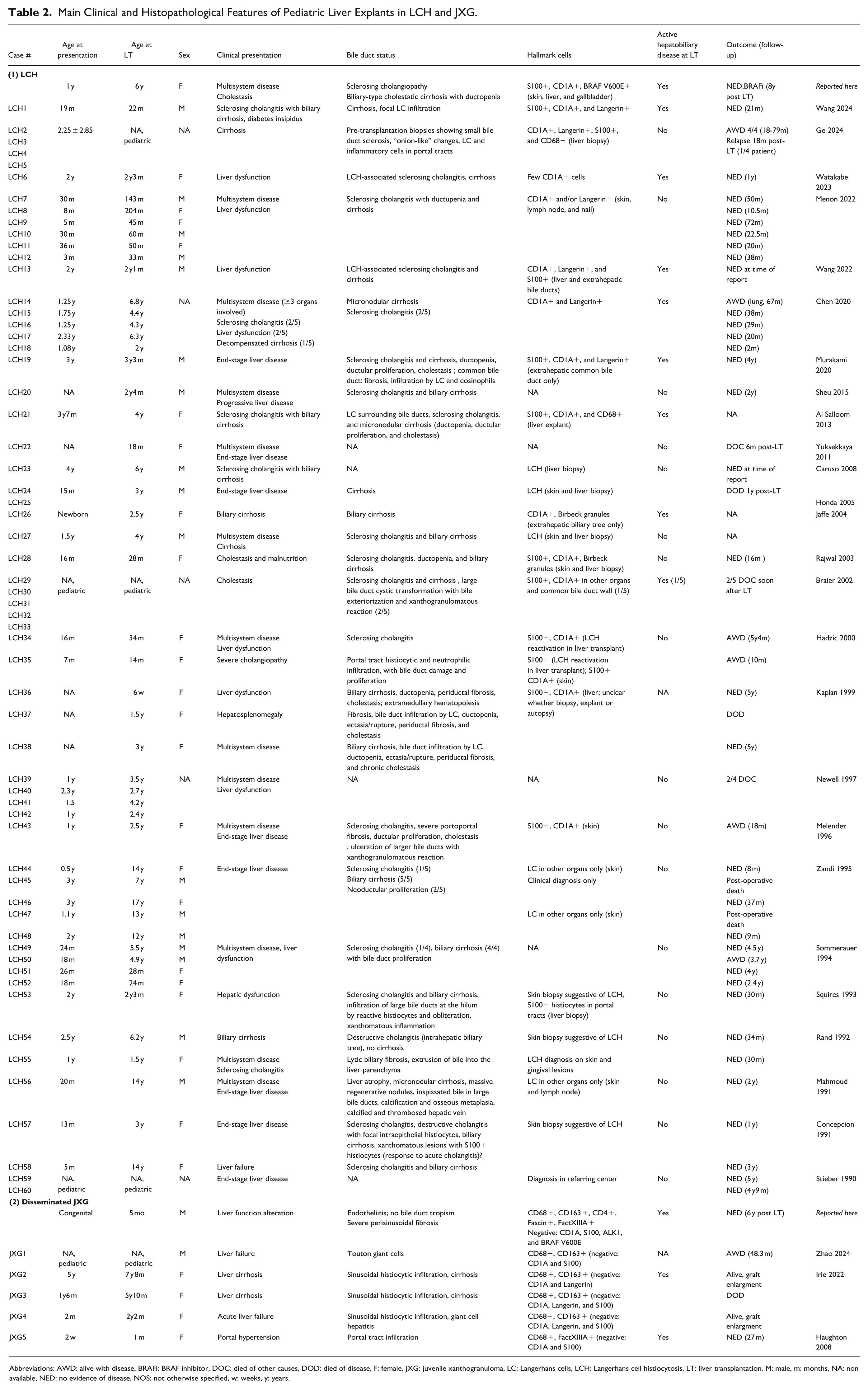
Abbreviations: AWD: alive with disease, BRAFi: BRAF inhibitor, DOC: died of other causes, DOD: died of disease, F: female, JXG: juvenile xanthogranuloma, LC: Langerhans cells, LCH: Langerhans cell histiocytosis, LT: liver transplantation, M: male, m: months, NA: non available, NED: no evidence of disease, NOS: not otherwise specified, w: weeks, y: years.
Discussion
Histiocytic neoplasms range from localized, self-limiting lesions to life-threatening disseminated disease, with potential liver involvement at any stage. Diagnosis is often complicated by overlapping histological features, but infiltration patterns can provide important clues. When involvement is limited to large portal tracts or the hepatic hilum, liver biopsy may be insufficient for accurate diagnosis.
Liver Involvement by Histiocytic Disorders
Liver involvement in histiocytic disorders can be differentiated histologically by the behavior of histiocytic infiltrates around bile ducts. In LCH, infiltrates cause progressive bile duct destruction, leading to sclerosing cholangitis, whereas in JXG, bile ducts are surrounded but remain intact. Both conditions may ultimately cause biliary-type cirrhosis. Other histiocytoses typically do not target bile ducts or cause biliary cirrhosis.
The clinical presentation and histological patterns of liver involvement in LCH, JXG, and ALK-positive histiocytosis are summarized in Table 3.
Modes of Presentation and Histological Patterns of Liver Involvement in LCH, JXG, and in ALK-Positive Histiocytosis.
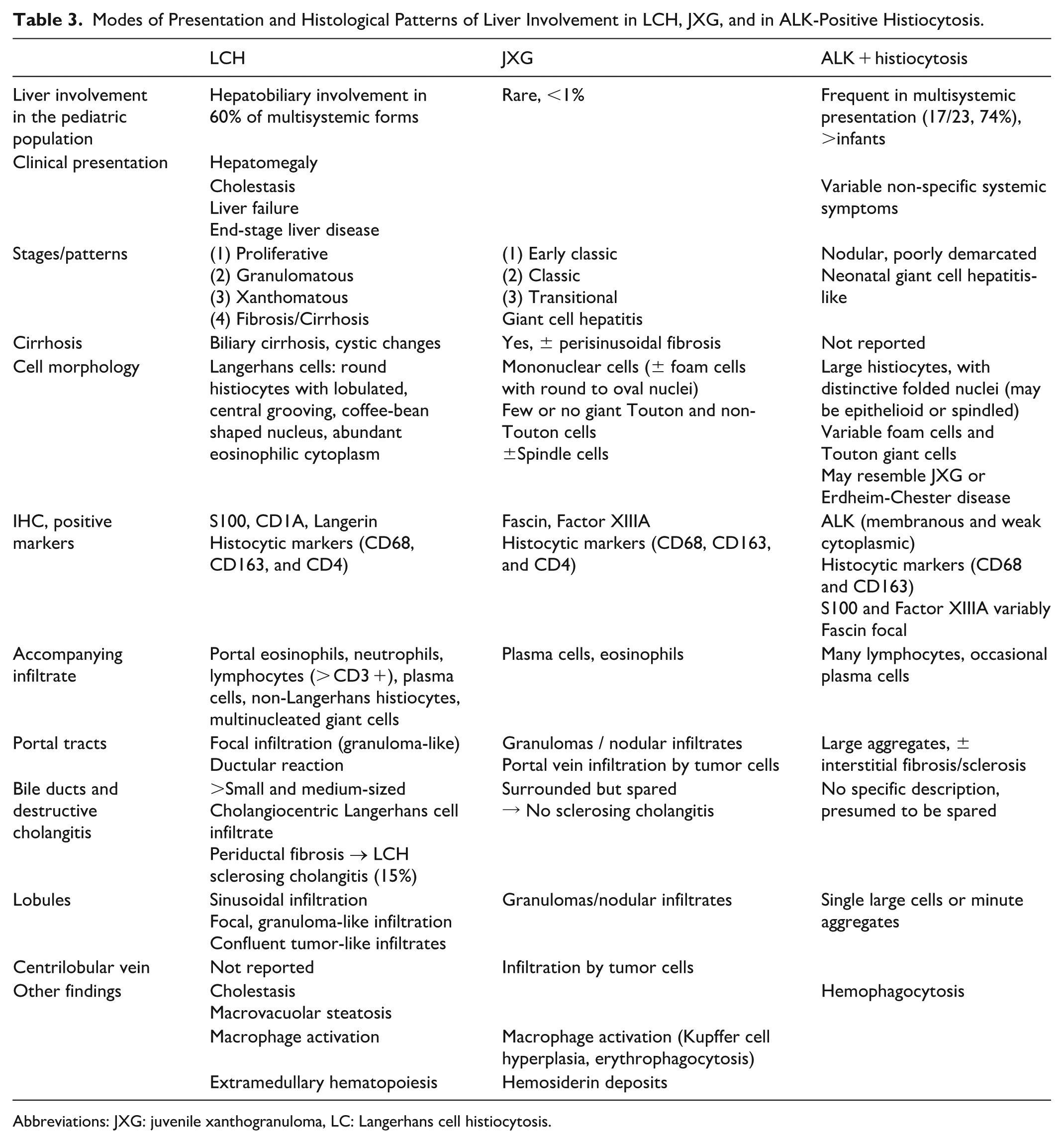
Abbreviations: JXG: juvenile xanthogranuloma, LC: Langerhans cell histiocytosis.
LCH: A Histiocytosis With Selective Bile Duct Affinity
LCH primarily affects children aged 1 to 3 years, with an incidence of 1 to 4 cases per million children annually. 6 Multisystem involvement is classified as risk organ (RO) positive, or high-risk LCH, when liver, spleen, or bone marrow are affected. 2 Hepatobiliary involvement occurs in up to 60% of pediatric multisystem cases,6,7 whereas isolated liver LCH is rare8,9 and often only diagnosed at liver explant histology. 9 Liver involvement triples mortality risk, accounting for about 20% of deaths in LCH,2,10 and cholangitis is associated with significantly worse 2-year overall and progression-free survival. 10
Clinically, patients may present with jaundice and hypoalbuminemia though, hepatomegaly without cholestasis can reflect macrophage activation syndrome without Langerhans cell infiltration. 11 Portal tract infiltration by Langerhans cells causes progressive bile duct destruction, advancing from larger to peripheral ducts, leading to sclerosing cholangitis and biliary cirrhosis, 12 as seen in Case 1. Biliary cirrhosis may develop rapidly within 1 to 3 years, faster than in primary sclerosing cholangitis or that associated with inflammatory bowel disease. 6 LCH can also cause cystic dilatation of bile duct dilatation due to destructive cholangitis of the larger bile ducts, and present as granulomatous lesions or liver masses. 12
Four histological stages are recognized: (1) an initial, early proliferative phase, (2) a granulomatous stage, (3) a xanthomatous phase, and (4) a later fibrotic stage.7,13 The liver explant from Patient 1 showed late, stage 4 fibrosis with focal xanthomatous activity consistent with stage 3 disease. In the liver, these 4 histological LCH stages correspond to characteristic imaging patterns. During the early stages, periportal inflammation and edema produce bandlike or nodular hypoechogenicity on ultrasound, hypoattenuation on CT, and moderate to high signal intensity on T2-weighted MRI. In the xanthomatous phase, lipid-laden nodules appear hyperechoic on ultrasound, hypoattenuating on CT, and hyperintense on contrast-enhanced T1-weighted MRI without fat suppression. In the later fibrotic stage, typical features of sclerosing cholangitis, such as segmental bile duct narrowing or dilatation with a bead-like appearance, may become evident. 7 Chemotherapy aimed at inducing remission is most effective during the 3 first stages of active infiltration, whereas the late fibrotic stage typically reflects irreversible disease, with limited treatment options beyond liver transplantation. 14 Established bile duct injury may persist even if the primary disease becomes “burnt out,” explaining why pretreatment biopsies can be non-diagnostic and liver explants may lack active disease. 11 Biliary injury can continue to progress despite absence of active disease. 15
JXG: A Histiocytosis Sparing the Bile Ducts
Liver involvement in JXG is exceedingly rare. In a large retrospective series of 525 patients, only 4 cases (<1%) demonstrated hepatic disease, all in individuals under 20 years of age (0.8%). 16 Similarly, a meta-analysis of 2949 pediatric patients with cutaneous JXG reported systemic manifestations in only 0.75% of cases. Within their own cohort of 338 patients, Samuelov et al. described a 6-week-old infant with multiple hepatic nodules that spontaneously regressed by 7 months of age. 17 Their literature review identified 10 additional pediatric cases with hepatic involvement,17 -20 including 1 patient presenting with hepatosplenomegaly but lacking histological confirmation. 19 Overall, liver infiltration was documented in just 11 of 2949 cases (0.37%).
Notably, a recurrent NTRK1 fusion has been documented in solitary lesions within the JXG spectrum, giving rise to a provisional new entity of NTRK-histiocytosis.2,21,22 This underscores the importance of ongoing molecular refinement in the classification of histiocytic disorders.
ALK-Positive Histiocytosis
Liver involvement was first described in 2008 in 3 infants with multisystemic disease. 23 Patients are classified into 3 groups: Group 1A includes infants with multisystemic disease involving the liver and hematopoietic system; Group 1B comprises patients with multisystemic disease without liver involvement; and Group 2 includes those with single-system disease, where liver involvement is typically absent. 23 Histologically, the liver shows ALK-positive histiocytic infiltration, with immunohistochemistry enabling the detection of individual infiltrating cells, especially within the lobules.23,24 Over time, increasing numbers of Touton and foam cells may appear, potentially mimicking JXG. 24 Prominent interstitial fibrosis has also been reported, notably in an adult case. 25
Other Histiocytoses and Histiocytic Neoplasms
Indeterminate cell histiocytosis/indeterminate dendritic cell tumor, mainly a cutaneous disorder affecting adults, has rare reports of liver involvement by CD1a-positive histiocytes lacking Birbeck granules ultrastructurally. 26 This neoplasm is composed of a clonal proliferation of histiocytes expressing the dendritic cells markers CD1a and S100 protein, but not CD207 (Langerin). 3 Rosai-Dorfman disease 27 and Erdheim-Chester disease infrequently involve the liver, with the latter rarely diagnosed in children. 28 Finally, histiocytic sarcoma, a rare malignant histiocytosis involves the liver in approximately 16% of the cases, characterized by sinusoidal infiltration of atypical tumor cells with large, pleomorphic nuclei. 29
Molecular Findings and Targeted Therapy, With Focus on LCH and JXG
Histiocytoses are now understood as a spectrum of clonal hematopoietic disorders primarily driven by aberrant activation of the mitogen-activated protein kinase (MAPK) signaling pathway. 21 Lesions may show mixed phenotypes, combining 2 or more histiocytic disorders, and distinct histiocytoses may co-occur and vary according by anatomical localization. 30
Recurrent gain-of-function BRAFV600E mutations are frequently found in LCH, 31 while MAP2K1 mutations typically occur in BRAF wild-type lesions, consistent with broad ERK pathway activation in LCH.30,32 Other alterations affecting the Ras/Raf/MEK/ERK (MAPK) pathway are identified less frequently in patients lacking BRAF or MAP2K1 alterations. Importantly, LCH cases harboring BRAFV600E mutations are linked to higher relapse rates and increased involvement of risk organs such as the liver.33,34 Using sensitive techniques like digital droplet PCR (ddPCR), BRAFV600E mutations can be detected in liver tissue even without clear histological evidence of active Langerhans cell infiltration, suggesting persistence of clonal mutation-bearing cells in “burnt-out” lesions or circulating precursors”. 34
This mutation status informs therapeutic approaches, with the BRAF inhibitor vemurafenib demonstrating efficacy in pediatric patients when combined with chemotherapy. However, cessation of vemurafenib often leads to rapid disease relapse, 35 and sustained remission after treatment cessation remains uncommon, with median remission duration of around 17 months. 36 The long-term safety and effects of vemurafenib treatment in children still require further evaluation.
In JXG, the MAPK and PI3K pathways are also implicated, though no consistent molecular findings have been identified.32,37 Instead of point mutations, BRAF fusions are notably enriched in JXG lesions. 38 Moreover, mutations in the colony stimulating factor-1 receptor (CSF1R) gene, which encodes a receptor tyrosine kinase regulating monocyte and macrophage functions, are found in about 10% of JXG cases. 21 Activation of CSF1R leads to MAPK/ERK pathway signaling. 39 The p.P566_N572del mutation in CSF1R, as observed in Patient 2, affects intracellular domains and results in enhanced kinase activity with cytokine-independent cellular proliferation. 21 Since CSF-1R signaling is also essential for Langerhans cell development from precursor monocytes, 40 inhibiting the CSF-1/CSF-1R axis offers a promising therapeutic target for both JXG and LCH.21,39,40
The PTPN11 gene, a key regulator of the RAS/MAPK-pathway, is classically associated with juvenile myelomonocytic leukemia (JMML). 41 There are documented cases of JXG co-occurring with JMML. For example, 1 male infant diagnosed with JXG at 3 months later developed JMML, and sequencing revealed the same p.E76K PTPN11 mutation in both diseases. 42 A hotspot PTPN11 p.E76V mutation was also detected in Patient 2, who has not exhibited signs of JMML. Furthermore, JXG can occur in the context of Noonan syndrome, a RASopathy caused by germline mutations in genes of the Ras/MAPK pathway, illustrated by a case of a 3-month-old female with a germline p.Y62D PTPN11 mutation presenting multiple JXG skin lesions. 43 Coexistence of neurofibromatosis type 1, JXG, and JMML has also been reported. 44
To our knowledge, co-occurrence of CSF1R and PTPN11 mutations has not been specifically documented in JXG. In Patient 2, the variant allele frequencies were 20% and 34%, respectively, making it unlikely that these alterations arose in separate subclones and were mutually exclusive. This raises the possibility that their simultaneous occurrence exerted a synergistic effect, converging toward more robust and durable downstream MAPK pathway activation, thereby contributing to the increased disease severity observed.
Indication for Liver Transplantation
In LCH, LT is indicated mainly when disease progresses to sclerosing cholangitis, liver failure, or end-stage liver disease, especially if chemotherapy fails or is not tolerated. 45 Most LT cases are pediatric (82%), with a median age of 3 years. 9 At transplantation, over half of patients (56.4%) have active extrahepatic disease, which correlates with a risk of liver recurrence; however, recurrence in the transplanted liver has been reported in only 8% of patients and is generally responsive to salvage therapy. 9 Thus, LT can be considered in selected patients with decompensated liver disease and active LCH when chemotherapy is ineffective or contraindicated. Maintenance therapy with vemurafenib post-LT, as in Patient 1, has shown durable disease control, although optimal treatment duration remains unclear.
LT has been reported in 59 pediatric LCH patients,10 -15,46 -64 with active liver disease at the time of transplantation in 12 cases,11,46,49,54,61 -63 including Patient 1. Recent cases show favorable outcomes, with follow-up up to 5.5 years49,54,61 -63 Notably, the case we report remains disease-free 8 years post-LT while continuing BRAF inhibitor therapy, reflecting an unusually prolonged favorable evolution.
For disseminated JXG, only 6 patients including the case discussed here have undergone LT, with the youngest at 1 month old.65 -67 Although Patient 2 met LT criteria at age 1 month, transplant was deferred until age 5 months due to risk considerations.
No reports of LT exist for ALK-positive histiocytosis.
Potential Mimics for Recurrence in Liver Transplants
Pathologists should be aware of 2 potential diagnostic pitfalls when evaluating suspected recurrence: (1) granulomatous reactions (GRs), and (2) presence of CD1a+, Langerin+, Langerans cells in portal tracts in association with cholangiopathy.
(1) Adverse cutaneous events to BRAF inhibitors (BRAFi) occur in more than 75% of the patients, with GRs increasingly recognized. These GRs may resemble multiorgan sarcoidosis-like disease but are more commonly limited to the skin. 68 GRs occur mainly after dabrafenib (59%) and vemurafenib (37%) use, with 1 reported pediatric case involving skin-only lesions. 69 A review by Pham et al. 68 identified 55 BRAFi-related GR cases, with only 1 pediatric patient. Time to onset varies widely (1 month-5 years, mean = 10 months). Liver involvement as granulomatous hepatitis is rare. 70 GRs affect 4.2% to 6% of histiocytosis patients on BRAFi. 68 In the context of known histiocytic neoplasms, granulomas may signal recurrence. Patient 1 remains on vemurafenib, currently tolerating treatment despite GR, raising questions about treatment duration and long-term side effects versus recurrence risk.
(2) Entenmann et al., 71 reported CD1a+, Langerin+ Langerhans cells infiltrating bile duct epithelium or portal connective tissue during acute cellular rejection (ACR) without evidence of LCH. These Langerhans cells represent an inflammatory response associated with ACR-related cholangiopathy and should not be mistaken for LCH recurrence.
In both scenarios, immunohistochemical and molecular testing for known mutations is essential to avoid misdiagnosis of recurrence, allowing accurate assessment within the graft.
In conclusion, hepatic involvement in histiocytosis may lead to end-stage liver disease requiring transplantation. Histiocytoses display distinct hepatic infiltration patterns that aid diagnosis. Targeted therapies, particularly tyrosine kinase inhibitors, have transformed management, but determining when and whether to discontinue treatment remains a critical and unresolved challenge.
Supplemental Material
sj-pptx-1-pdp-10.1177_10935266251385405 – Supplemental material for Bile Duct Targeting or Preservation: Contrasting Liver Histology in Langerhans Cell Histiocytosis and Disseminated Juvenile Xanthogranuloma
Supplemental material, sj-pptx-1-pdp-10.1177_10935266251385405 for Bile Duct Targeting or Preservation: Contrasting Liver Histology in Langerhans Cell Histiocytosis and Disseminated Juvenile Xanthogranuloma by Margaux Däniker, Frédéric Baleydier, Nathalie M. Rock, Sébastien Menzinger, Barbara E. Wildhaber, Valérie A. McLin and Anne-Laure Rougemont in Pediatric and Developmental Pathology
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.