
Abstract
Congenital diaphragmatic hernia is a genetically heterogeneous condition with a developmental defect in the diaphragm. The GATA4 gene is essential for fetal heart development, and pathogenic GATA4 variants are a known cause of structural congenital heart diseases. Haploinsufficiency of GATA4 is also associated with diaphragmatic hernia. Pathogenic GATA4 sequence variants with isolated diaphragmatic hernia in the absence of congenital heart defects are extremely rare. Our report expands the phenotypic spectrum related to GATA4.
We report a fetus with a prenatal isolated diaphragmatic hernia detected during a routine screening ultrasound. An autopsy of the fetus confirmed a large isolated posterolateral hernia, which affected the left lung volume significantly. Clinical exome sequencing revealed a novel heterozygous nonsense variant c.826C>T,p.(Gln276*) in the GATA4 gene, which was predicted to cause haploinsufficiency. The variant occurred de novo and was classified as pathogenic.
The report presents a detailed clinical description of the fetus with ultrasound, MRI, and post-mortem pictures of a rare prenatal isolated diaphragmatic hernia related to a novel pathogenic GATA4 sequence variant. Prenatal ultrasound screening with further investigation by MRI and a comprehensive gene panel holds a key role in determining the prognosis of a fetus with a diaphragmatic hernia.
Introduction
Congenital diaphragmatic hernia (CDH) is estimated to occur in 2–4/10 000 pregnancies. 1 It is a genetically heterogeneous condition, with even the most common underlying causes accounting for only a few percent each of all CDH incidences. 2 CDH may be associated with chromosomal aneuploidies, copy number variants, or monogenic disorders,2,3 and 40–60% of cases are part of syndromic manifestations. 2 The defining feature of CDH is a developmental defect in the diaphragm, most commonly located in the left posterolateral region (Bochdalek hernia).
While ultrasound is the primary diagnostic modality in prenatal settings, fetal MRI is used for further assessment of a hernia, screening for additional anomalies, and lung volumetric measurements. 3 Prenatal detection allows for parental counselling, planning of delivery and postnatal care, and even prenatal interventions such as tracheal occlusion.
Clinical severity of CDH is variable, with mortality after birth mostly dependent on the degree of lung hypoplasia. Lung hypoplasia is caused by the abdominal organs herniating into the thoracic cavity and can be quantified by lung area to head circumference ratio with ultrasound or lung volumetry with MRI. These measurements can be used to evaluate the probability of postnatal survival and extracorporeal membrane oxygenation (ECMO) treatment requirement. 4
GATA4 (GATA-binding protein 4) encodes a zinc finger transcription factor, which binds GATA motifs of several genes (OMIM:*600576). The GATA gene family is involved in multiple embryogenic events. 5 GATA4, GATA5 and GATA6 cooperate in cardiogenesis and are critical in the regulation of fetal cardiac cell specification and heart formation. 6 Diseases linked to GATA4 include atrial septal defect (ASD), atrioventricular septal defect (AVSD), ventricular septal defect (VSD) and tetralogy of Fallot (OMIM:*600576).
Notably, GATA4 has a crucial role in the developing diaphragm, and its reduced expression has been linked to CDH. 7 The chromosome 8p23.1 microdeletion encompassing GATA4 is a well-recognised cause of congenital heart defects and diaphragmatic hernia, contributing to 3-5% of CDH cases.2,8 GATA4 is considered the candidate gene for CDH in recurrent 8p23.1 microdeletions.2,8-11 In contrast, GATA4 sequence variants are a rare cause of CDH.2,12
Case Report
A 29-year-old healthy G2P0 was referred to the Turku University Hospital Fetal Medicine unit due to dextroposition of the fetal heart detected in the morphological ultrasound screening at 20 + 3 weeks of gestation. The first-trimester ultrasound screening and non-invasive prenatal testing (NIPT) had been normal at 12 + 1 weeks. At 21 + 1 weeks of gestation, the heart was situated to the right in the thorax, with the heart axis pointed to the left. Four-chamber and three-vessel views were detected normally. VSD was suspected in the outflow area. The aortic and ductal arches appeared normal. The diaphragm was identified, and the stomach was located within the abdominal cavity. There was a hyperechogenic, non-cystic, space-occupying process in the thorax’s left side, compressing the heart to the right (Figure 1).
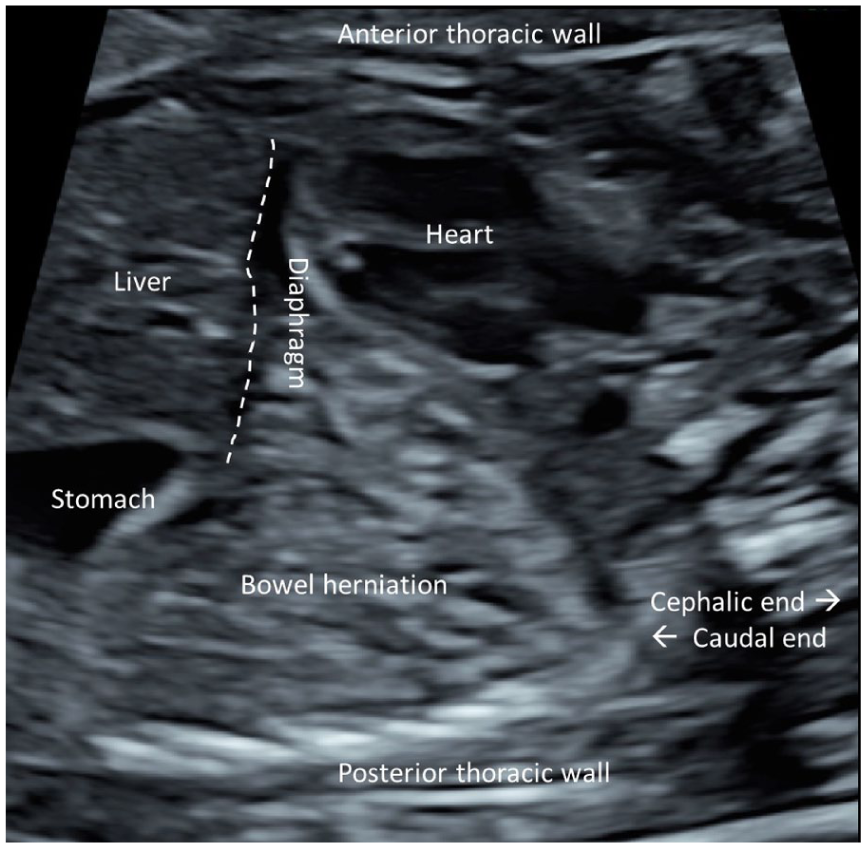
Parasagittal view of the fetus showing the heart dislocated anteriorly to the right side of the thorax due to diaphragmatic herniation of the small bowel to the thoracic cavity posteriorly in the morphological ultrasound image of a 21-week-old fetus. The localisation of the diaphragm is illustrated with a dashed line.
Diaphragmatic hernia was suspected. The patient was sent for an MRI, and an amniotic fluid sample was extracted for exome sequencing.
MRI was conducted at 21 weeks of gestation and revealed a left posterior diaphragmatic defect, with bowel herniating into the left thoracic cavity (Figure 2). Total fetal lung volume was estimated to be 10.9 ml (−1.3SD for gestational age 13 ). The heart and mediastinum were displaced rightwards. No additional abnormalities were identified.
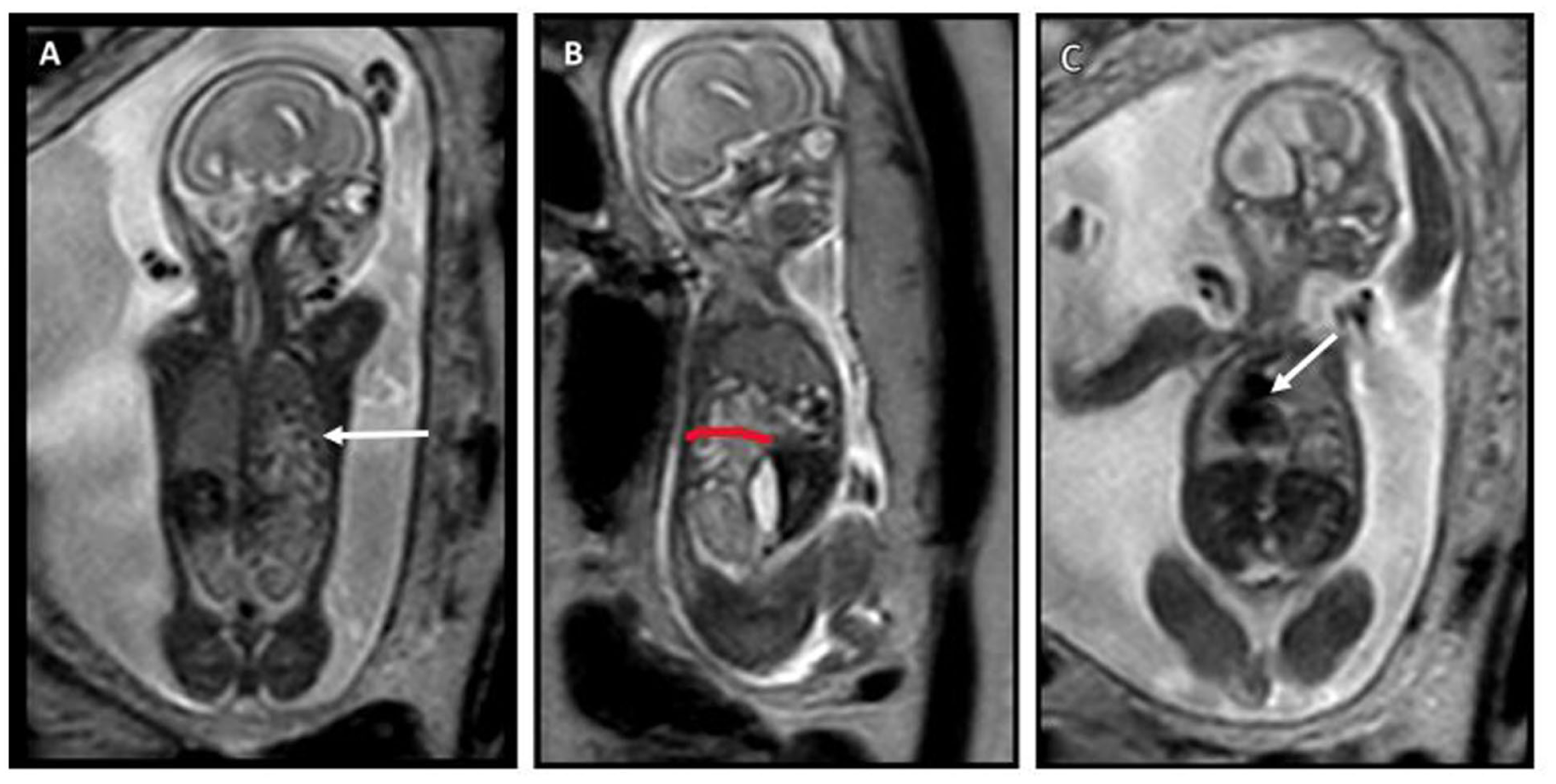
T2-weighted MRI images. (A) A coronal image at the posterior level near the vertebra, showing the left diaphragmatic defect and fluid-filled structures in the thorax (arrow), consistent with bowel loops. (B) A left sagittal image, more clearly depicting the posterior diaphragmatic defect (red line) and bowel in the thoracic cavity. (C) A more anterior coronal image, displaying the rightward shift of the heart and mediastinum (arrow).
The pregnancy was terminated at 22 + 2 weeks of gestation. Autopsy revealed slight coarseness of facial features and a large posterolateral hernia covering half of the diaphragm’s left side. Most of the intestines and spleen were herniated into the thoracic cavity, displacing the heart to the right and withholding space from the developing lungs. The anatomy and axis of the heart were normal. There were no signs of heart defects. The positioning of the great arteries and veins was normal. (Figure 3). The left lung was significantly smaller than the right lung (right: 7.7 g; left: 3.5 g) (Table S1). Histology revealed slight developmental delay of lung parenchyma: they were in early canalicular phase, in contrast to the expected late canalicular phase. A more detailed description of the macroscopic and microscopic autopsy, with fetal measurements and organ weights, is presented in the Supplemental data.
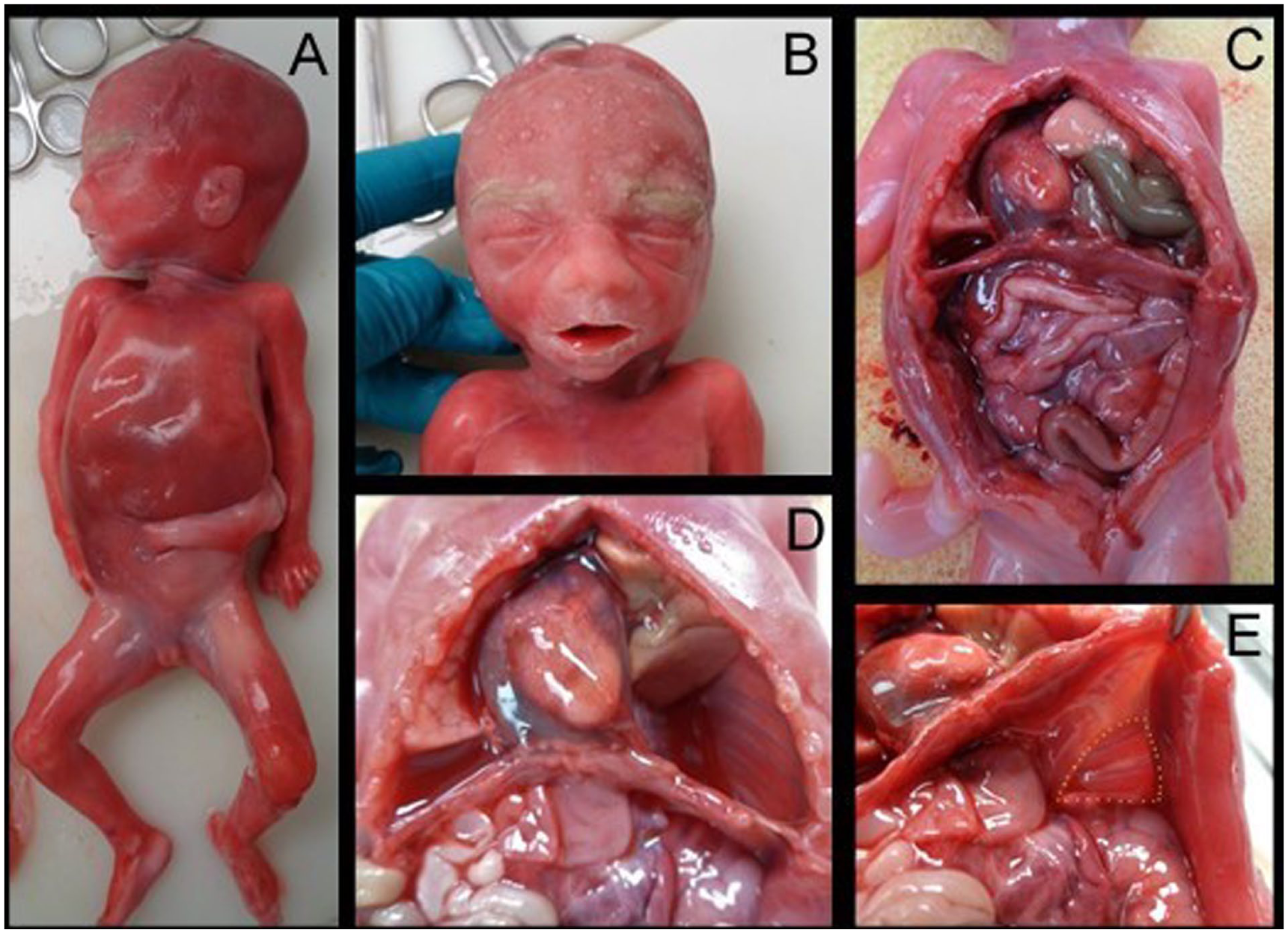
Autopsy photographs. Whole fetus (A) with mild coarseness of facial features (B). The prolapsed intestines and the displacement of the heart to the right side of the thoracic cavity are seen (C). After removing the prolapsed intestines and spleen from the thoracic cavity, the space they were occupying, as well as the displacement of the left lung and heart, could be seen clearly (D). A view of the diaphragm seen from the abdominal cavity (E). The hernia defect is highlighted with yellow dashed lines.
The Genomics laboratory of Turku University Hospital performed clinical exome sequencing from fetal DNA. A heterozygous nonsense variant c.826C>T,p.(Gln276*) in the GATA4 gene was discovered. The novel variant occurred de novo and was classified as pathogenic according to American College of Medical Genetics and Genomics (ACMG) classification criteria.14,15 Detailed description of data analysis and variant interpretation is presented in Supplemental Data.
Discussion
Screening ultrasound at 20 + 3 weeks of gestation revealed rightward displacement of the fetal heart and a possible VSD. In addition, the left hemithorax showed abnormally echogenic, unidentifiable tissue, which raised suspicion of a diaphragmatic hernia. MRI of the fetus revealed a significant left posterior diaphragmatic hernia, explaining the dextroposition of the heart, which was further confirmed in the autopsy. The internal structures and the chirality of the heart visualised normally in the ultrasound and autopsy. There were no signs of GATA4-related congenital heart defects. Given the findings, the prognosis was deemed poor, leading to termination of the pregnancy following consultation with a paediatrician.
Genetic testing revealed a pathogenic de novo variant c.826C>T,p.(Gln276*) in the GATA4 gene. The novel nonsense variant has not been previously reported in the literature or variant databases (ClinVar, HGMD). The variant was classified as pathogenic and undoubtedly explains the fetal phenotype. Pathogenic GATA4 sequence variants typically cause congenital heart defects. 6 GATA4 sequence variants have been reported only in rare instances with diaphragmatic hernia, supporting the notion that diaphragmatic hernia is not a common presentation.2,12,16
The ClinVar database lists 32 sequence variants as pathogenic or likely pathogenic in the GATA4 gene, including nonsense, frameshift and missense variants. 17 Most of these variants are related to structural congenital heart diseases, and a minority are related to testicular anomalies with or without congenital heart disease, whereas none are directly related to diaphragmatic hernia. However, one pathogenic GATA4 missense variant has been observed in a patient with congenital left-sided diaphragmatic hernia and septal defects. 16 Interestingly, this variant is located near the variant c.826C>T reported in our case.
Prenatal CDH cases with GATA4 variants are rare. Kammoun et al. 18 reported GATA4 missense variants causing severe one-sided isolated CDH in two fetuses, with adverse outcomes. 18 In one family, a small, isolated left-sided hernia was reported in two seemingly asymptomatic GATA4 carriers. The index patient in the family presented prenatally with isolated CDH, although postnatally with syndromic features including bilateral hydronephrosis, undescended testis, and patent foramen ovale. 12 In addition, 8p23.1 microdeletions encompassing GATA4 have been reported prenatally with CDH, congenital heart defects, and growth restriction.19,20 Awareness of the whole phenotypic spectrum is needed and can only be improved through accumulating published cases; thus, it is important to report novel findings like our case.
Diaphragmatic hernia has been associated with reduced expression of GATA4, 7 and CDH related to pathogenic GATA4 variants is predicted to result from haploinsufficiency through a loss-of-function mechanism. 21 Functional studies in mice have demonstrated that GATA4 deficiency, especially in mesenchymal-derived muscle connective tissue fibroblasts, causes CDH. Interestingly, both humans and mice with GATA4 haploinsufficiency present CDH with reduced penetrance. 22 The incomplete penetrance and variable expressivity reported with GATA4 variants may explain the relatively low incidence of GATA4-related CDH. The complex interplay between genetic, epigenetic and environmental factors may account for the different phenotypic presentations between reported cases. Environmental factors such as maternal vitamin A insufficiency during the first trimester, a known risk factor for CDH, may act as a sensitising factor for the development of CDH following GATA4 haploinsufficiency.2,22
Future research should focus on functional studies of GATA4 variants to better understand its crucial role during embryogenesis and to understand the complete phenotypic spectrum of GATA4 variants. Larger cohorts will help in determining the prevalence of GATA4 variants in CDH cases. This will result in a more comprehensive understanding of the genetic landscape of CDH and its association with GATA4. It is recommended that the GATA4 gene be added to the list of genes of interest for isolated or syndromic prenatal CDH.
Supplemental Material
sj-docx-1-pdp-10.1177_10935266251381440 – Supplemental material for Prenatal Isolated Congenital Diaphragmatic Hernia: A Rare Clinical Presentation of a GATA4 Pathogenic Variant
Supplemental material, sj-docx-1-pdp-10.1177_10935266251381440 for Prenatal Isolated Congenital Diaphragmatic Hernia: A Rare Clinical Presentation of a GATA4 Pathogenic Variant by Nea Tulonen, Jussi Tallus, Heidi Kaprio, Jukka Laine, Mirjami Mattila, Maria Haanpää and Sini Keskinen in Pediatric and Developmental Pathology
Footnotes
Acknowledgements
We thank the family for their participation in this study and their consent to publication.
Abbreviations
ACMG; American College of Medical Genetics and Genomics
ASD; atrial septal defect
AVSD; atrioventricular septal defect
CDH; congenital diaphragmatic hernia
ECMO; extracorporeal membrane oxygenation
NIPT; non-invasive prenatal screening
VSD; ventricular septal defect
Ethical Considerations
All participants provided written informed consent prior to enrolment in the study. Our institution does not require ethical approval for reporting individual cases or case series.
Consent to Participate
A written consent was obtained from the family.
Author Contributions
The authors declare that all authors contributed to this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.