
Abstract
Background:
Maple syrup urine disease (MSUD) is an autosomal recessive inherited disorder characterized by deficiency of branched-chain α-keto acid dehydrogenase complex. The affected patients can experience severe metabolic intoxication and encephalopathy in the first few years of life. Liver transplantation is an effective long-term treatment. There has been a lack of histologic description of explanted livers from MSUD patients in the literature.
Methods:
A search of the medical record system was performed for cases carrying a diagnosis of MSUD between January 2003 and May 2024. Eight patients who underwent liver transplantation were identified. Their explanted livers were evaluated and their medical records were extensively reviewed.
Results:
The weights of explanted livers were within normal range for patients’ age. Histologic examination demonstrated features of nodular regenerative hyperplasia (NRH) in 5 (62.5%) liver explants. Other histologic findings included minimal to mild lymphocytic portal inflammation seen in 6 cases and mild steatosis in 2 cases. A detailed review of clinical histories revealed no signs of portal hypertension or specific underlying conditions conducive to NRH development.
Conclusion:
NRH is a frequent histologic finding in explanted livers from MSUD patients, although the underlying etiopathogenesis and clinical implication remain to be elucidated.
Keywords
Introduction
Maple syrup urine disease (MSUD) is an autosomal recessive neurometabolic disorder resulting from severe deficiency of the branched-chain α-keto acid dehydrogenase complex (BCKDH). This enzyme complex is crucial for the oxidative decarboxylation of branched-chain α-keto acids (BCKA). The metabolic block leads to blood, tissue, and urine accumulation of branched-chain amino acids (BCAA) leucine, isoleucine, and valine, along with alloisoleucine and their corresponding BCKAs α-ketoisocaproic, α-ketoisovaleric, and α-keto-β-methylvaleric acids. In the first few weeks of life, patients typically experience severe episodes of encephalopathy, characterized by seizure, coma, and potentially fatal cerebral edema. This initial phase is followed by a progressive decline in neurological functions, marked by motor delay, ataxia, intellectual disability, and psychological issues. 1 Suggestive biochemical findings in newborn screening for MSUD include whole-blood concentration ratios of (leucine + isoleucine) to (alanine + phenylalanine) that exceed the threshold values for the screening laboratory. Follow-up plasma amino acid analysis typically reveals increased concentrations of BCAAs and alloisoleucine. The diagnosis of MSUD is confirmed by identifying biallelic pathogenic mutations in BCKDHA, BCKDHB, or DBT genes. 2
Increased blood BCAA levels impair the functions of the immune system, skeletal muscles, and central nervous system. They also cause tissue swelling, disrupt glutamate homeostasis, and result in a relative lack of large neutral amino acids, which reduces the synthesis of neurotransmitters such as dopamine and serotonin.3,4 Significant findings from autopsies of MSUD patients are mostly limited to the central nervous system, which include global status spongiosis of the white matter, reduced number of myelin sheaths, increased cerebral water content, and decreased total brain lipid content. 5 MSUD is also linked to anomalies in muscle fibers, where fiber sizes vary significantly, characterized by localized or diffuse destruction of myofibrils. 6
To avoid neurological symptoms, patients with MSUD are treated with protein-restricted diet and supplementation with a specialized formula including necessary amino acids (excluding BCAAs) and minerals. 7 However, brain damage, chronic psychological burden, and even death may still occur in older patients, even with appropriate diet and medical treatment. Orthotopic liver transplantation (OLT) provides an enzymatic reprieve and improves cognitive functioning without worsening neuropsychiatric conditions, while allowing for an unrestricted diet. 8 At the same time, the livers explanted from MSUD patients have been used for domino liver transplant (DLT), 8 which involves transplanting the liver from a MSUD patient into a patient with end-stage liver disease with the expectation that the recipient will not develop MSUD, while the MSUD patient receives a different liver from a deceased donor or a liver segment from a living donor. 9 To our knowledge, there has been a lack of histopathologic description of explanted livers from MSUD patients in the literature.
Materials and Methods
After approval by the Internal Review Board of our institution, a retrospective search of the pathology database within the electronic medical record system was conducted for cases carrying a clinical diagnosis of MSUD between January 2003 to May 2024. A total of 9 cases was identified. This included 8 liver transplantations and 1 autopsy. The autopsy case was a 6-year-old boy who died of multi-microbial pneumonia leading to acute respiratory distress syndrome. Postmortem examination of the liver showed ductular cholestasis (cholangitis lenta) resulting from septicemia. This case was excluded from the study because of limited tissue sampling and complicated clinical course. Only the 8 explanted native livers were included for the study.
Hematoxylin and eosin (H&E)-stained slides, along with special stains for trichrome, reticulin, periodic acid-Schiff (PAS) with and without diastase, and iron, were available for detailed histologic evaluation, which was initially conducted by 2 resident pathologists (YS and YG) followed by independent review and verification by 2 liver pathologists (BVN and HLW). The electronic medical records were extensively reviewed and pertinent clinical data were collected.
Results
As shown in Table 1, 7 patients were infants and toddlers ranging in age from 11 to 42 months at the time of liver transplantation. One patient was a 27-year-old male. All patients were diagnosed between 2 and 15 days of life, including 3 patients detected by neonatal screening. Two patients were siblings. Seizures were documented in 5 patients, which were generally treated with Carbamazepine (Tegretol), Levetiracetam (Keppra), and/or phenobarbital. Gastroenteritis, bronchiolitis, and urinary tract infection were documented in 5 patients. One patient (#1) has an episode of atrial arrhythmia of unclear etiology, which was successfully treated with amiodarone and digoxin. Plasma leucine concentrations before transplantation were available in 7 patients, which were all markedly elevated. Liver enzymes and function tests (including serum albumin) were all normal except for 1 patient (#3) who had transient mild elevation of serum alanine aminotransferase and aspartate aminotransferase levels. Genetic data were available in 4 patients, including homozygous mutations in the BCKDHB gene in 2, compound heterozygous mutations for 3 variants in the BCKDHB gene in 1, and compound heterozygous mutations for 2 variants in the BCKDHA gene in 1.
Clinical Data of Patients With Maple Syrup Urine Disease.
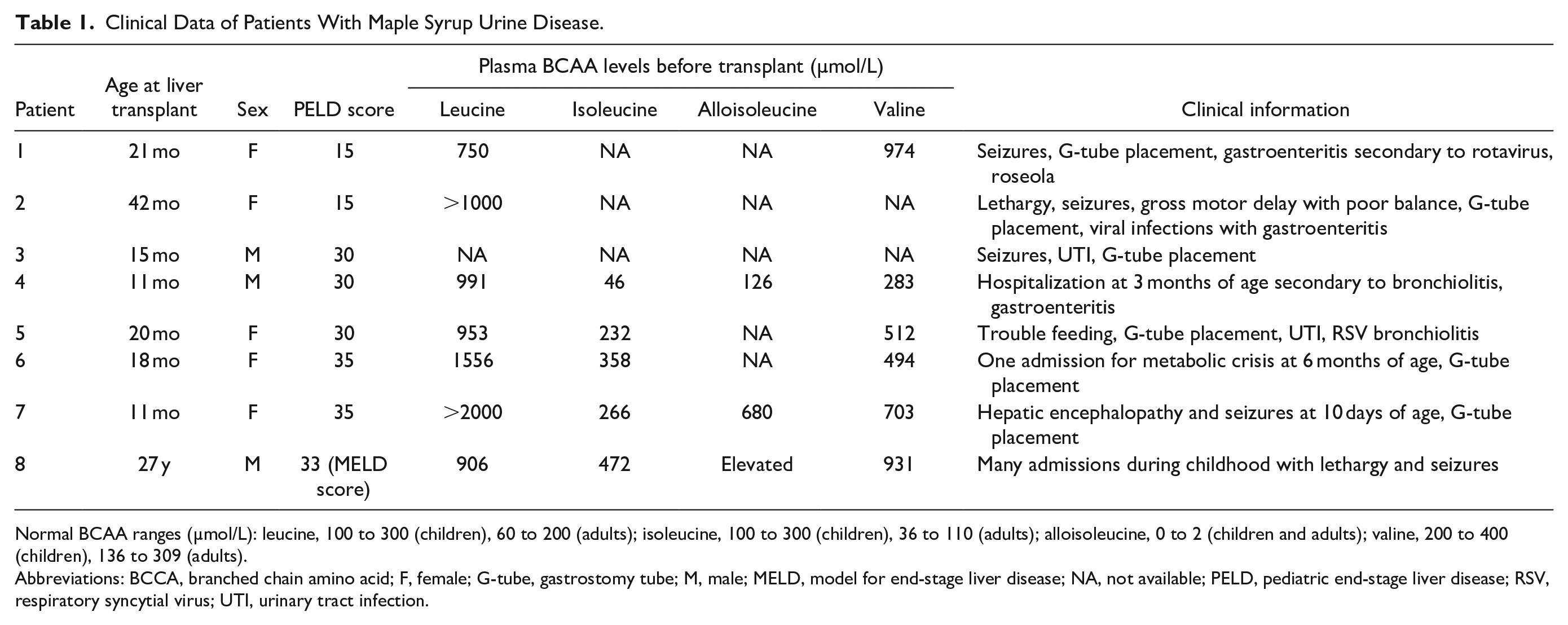
Normal BCAA ranges (µmol/L): leucine, 100 to 300 (children), 60 to 200 (adults); isoleucine, 100 to 300 (children), 36 to 110 (adults); alloisoleucine, 0 to 2 (children and adults); valine, 200 to 400 (children), 136 to 309 (adults).
Abbreviations: BCCA, branched chain amino acid; F, female; G-tube, gastrostomy tube; M, male; MELD, model for end-stage liver disease; NA, not available; PELD, pediatric end-stage liver disease; RSV, respiratory syncytial virus; UTI, urinary tract infection.
All patients were on protein-restricted formula/diet with nutritional supplements before liver transplantation. Gastrostomy tube placement was documented in 6 patients. No developmental delay was documented in these patients, although 1 patient (#1) had been on growth hormone treatment. None of the patients had a documented history of portal hypertension.
The weights of explanted livers were all within normal ranges for patients’ age (Table 2). Gross examinations were unremarkable, and showed no nodularity, fibrosis, focal lesion, or discoloration (Figure 1). On histologic examination, diffuse nodularity was observed in 5 (62.5%) cases. Nodularity was prominent on H&E stain at low-power view in 3 cases (Figure 2(a)) but was vaguely seen in 2 cases (Figure 2(b)). The nodules were small (1–2 mm) and relatively uniform, with compressed intervening hepatocytes, as highlighted by reticulin stain (Figure 2(c) and (d)). The findings were consistent with nodular regenerative hyperplasia (NRH). Of the 4 patients with available genetic data, NRH was observed in 3. We were unable to establish a meaningful correlation between specific mutations and NRH due to the small number of cases.
Pathologic Findings in Explanted Livers of Patients With Maple Syrup Urine Disease.
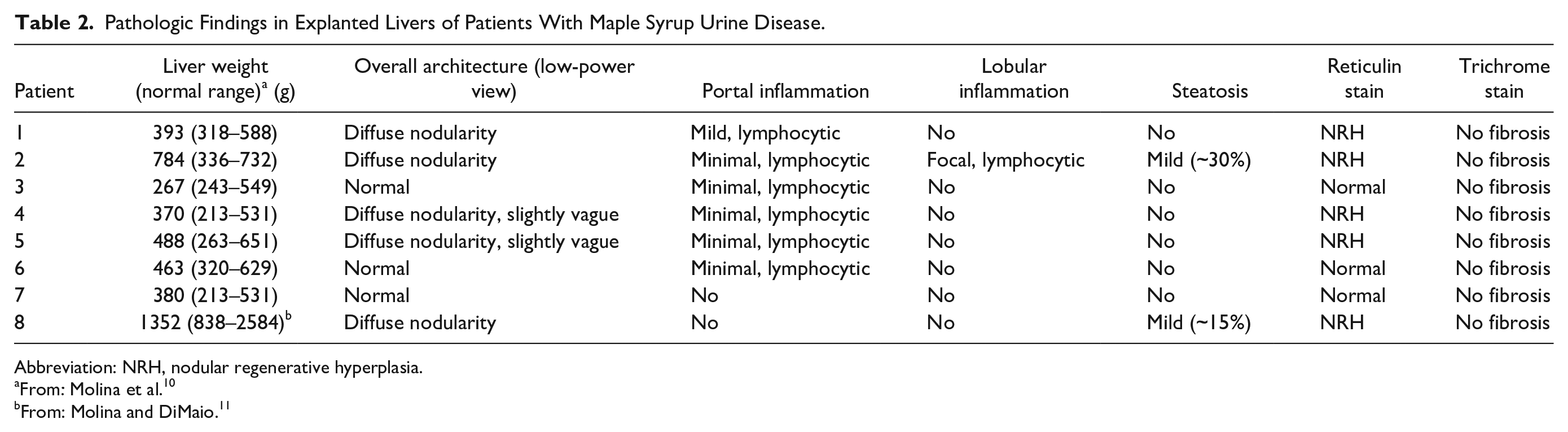
Abbreviation: NRH, nodular regenerative hyperplasia.
From: Molina et al. 10
From: Molina and DiMaio. 11
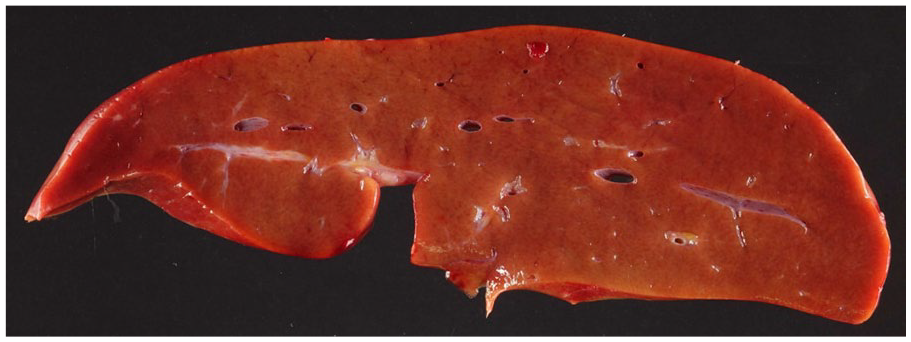
Normal gross examination of an explanted liver (case 5). There were no nodularity, fibrosis, focal lesion, or discoloration.
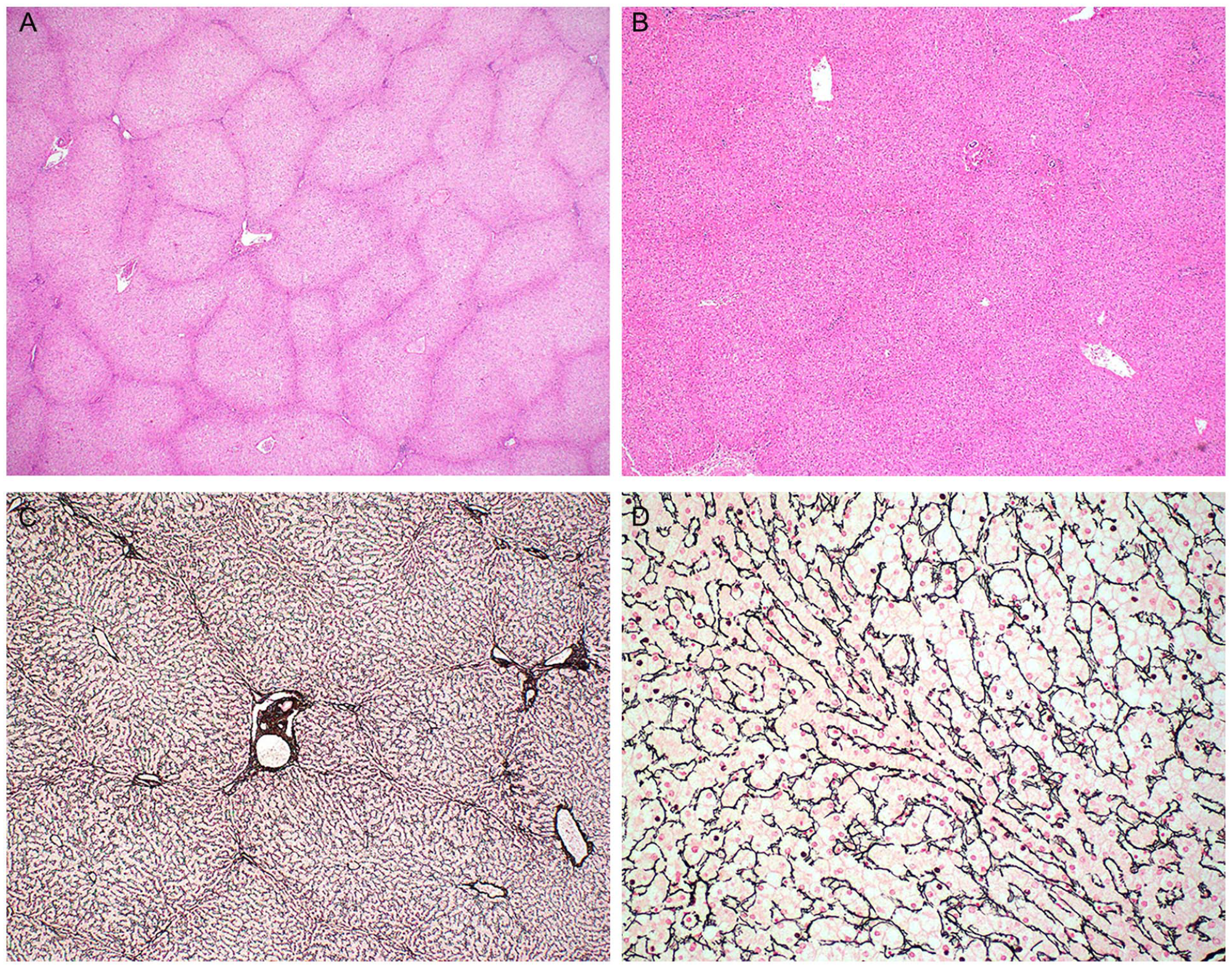
Nodular regenerative hyperplasia. Diffuse nodularity was evident for case 2 (A; H&E, ×20) but slightly vague for case 4 (B; H&E, ×40) at low-power view. Reticulin stain highlighted nodularity at low-power view (C; case 5; ×40) and compressed hepatocytes between regenerative nodules at higher-power view (D; case 5; ×200).
Other histologic findings were quite minimal. As shown in Table 2, 6 cases showed minimal to mild nonspecific lymphocytic infiltrates in scattered portal tracts (Figure 3(a)). Mild steatosis was seen in 2 cases (#2 and #8), but no ballooned hepatocytes or Mallory-Denk bodies were identified. Three cases (#3, #4, and #7) showed patchy mild hepatocyte swelling with somewhat clear cytoplasm (Figure 3(b)). This finding was interpreted as potential drug-induced injury. One case (#3) showed glycogen depletion on PAS stain. No features of glycogen overload were appreciated. No abnormal findings were observed in bile ducts and vasculature. There was no cholestasis, iron deposition, ductular reaction, or fibrosis.
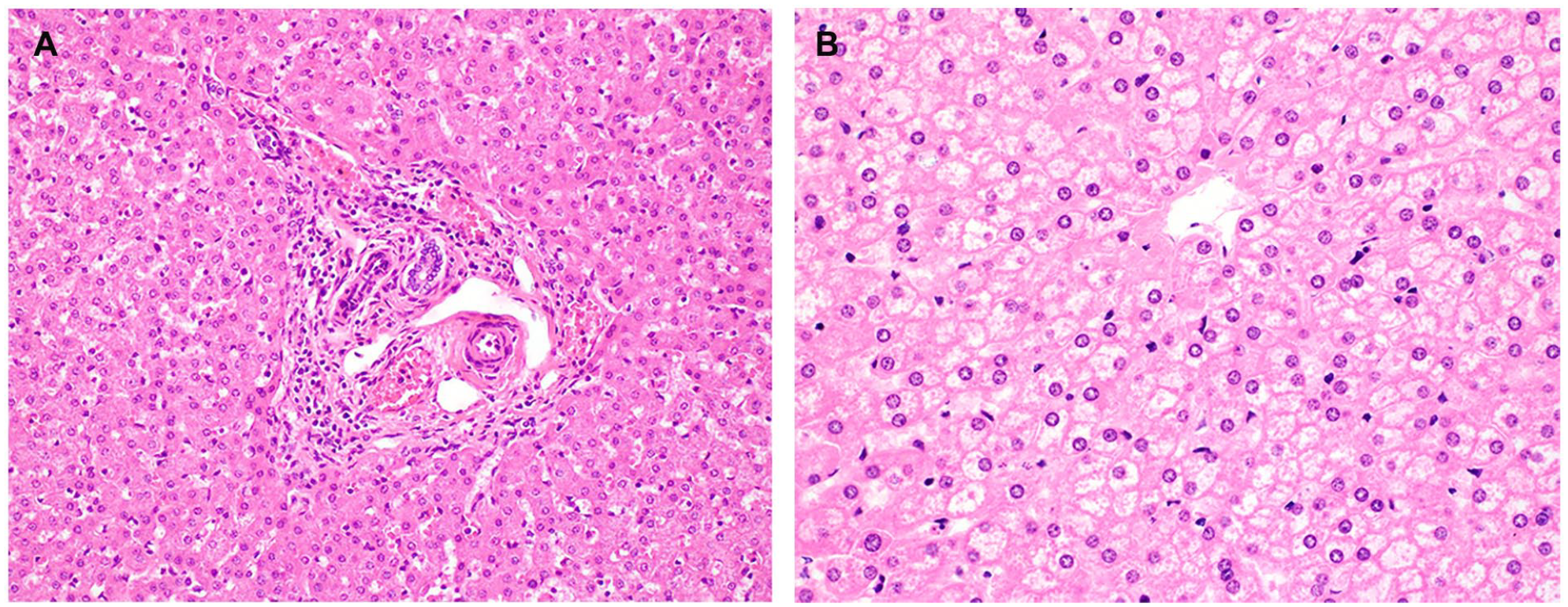
(A) Minimal portal lymphocytic infiltrates noted in case 6 (H&E, ×200). (B) Patchy mild hepatocyte swelling with somewhat clear cytoplasm noted in case 3 (H&E, ×400).
Discussion
MSUD is a type of organic acidemia resulting from BCKDH deficiency that leads to BCAA accumulation to cause life-threatening ketoacidosis and neurotoxicity. Given that BCKDH activity is predominantly found in skeletal muscle (60%), liver (9−13%), brain (10–20%), and kidney (8–13%), elective OLT has emerged as a therapeutic option for MSUD, which provides an enzymatic reprieve to improve cognitive functions without causing neuropsychiatric deterioration, even on an unrestricted diet.8,12,13 The distribution of BCKDH activity in the body also supports the use of livers from MSUD patients as a promising source for DLT. 8 This sequential organ transplantation, or “domino” transplantation, was first performed with heart transplantation in the late 1980s and early 1990s. 14 DLT was first performed in the early 1990s, allowing the liver from a recipient of liver transplantation to be used as a marginally qualified donor graft for another transplantation. 15 The key principle of DLT is that certain inherited and metabolic disorders can be treated with liver transplantation, producing an explanted liver that is structurally sound and can preserve hepatic function when implanted into carefully selected recipients.16,17 In the United States, a total of 181,976 liver transplantations were performed between October 1987 and December 2020, of which 185 (0.1%) were DLTs. Livers from MSUD patients accounted for 22% of these domino transplants. 18 However, there is a lack of histopathologic description of explanted livers from MSUD patients in the literature. Only 1 autopsy report published in 1954 described hepatomegaly with large amounts of glycogen and minimal periportal infiltrates with lymphocytes, mononuclear cells, and occasionally eosinophils in an infant with presumed MSUD who died of respiratory failure at 11 days of life. 19
In this study, we evaluated 8 explanted native livers from MSUD patients and found NRH in 5 (62.5%) cases. Minimal to mild lymphocytic infiltration in scattered portal tracts was a common observation. However, we did not find hepatomegaly or increased glycogen storage in these livers.
NRH is an uncommon hepatic vascular disorder characterized by diffuse transformation of the hepatic parenchyma into small regenerative nodules with no significant fibrosis. This condition is thought to be an adaptive hyperplastic response of hepatocytes to mechanical or functional abnormalities in hepatic microvasculature, caused by imbalances between arterial and venous blood flow.20-22 NRH falls under the category of noncirrhotic portal hypertension, 23 and is recently proposed as part of the histologic spectrum of porto-sinusoidal vascular disorder.24,25 Clinical presentations can range from asymptomatic, abnormal liver function tests, to portal hypertension with complications such as esophageal varices, splenomegaly, and ascites. Management of NRH focuses on treating the underlying conditions, if identified, and addressing the complications of portal hypertension. 21 The overall outcomes of patients with NRH are largely determined by underlying illness rather than liver-related complications. 26
The occurrence of NRH is associated with a wide spectrum of systemic diseases. Examples include vascular and prothrombotic disorders such as portal vein thrombosis and venous outflow obstruction, connective tissue and autoimmune diseases such as rheumatoid arthritis and lupus erythematosus, hematological malignancies such as leukemia and lymphoma, medications such as azathioprine and 6-thioguanine, post-OLT status, and other miscellaneous diseases such as common variable immunodeficiency and primary biliary cholangitis. 22 However, many patients with NRH lack identifiable risk factors. In a study of 167 patients with NRH, a potentially associated causal condition was identified in only 73 (44%) patients, including hematological diseases (44%), rheumatological diseases (26%), drug exposure (23%), other/miscellaneous etiologies (3.6%), and multiple risk factors (3.4%). 27 The risk factors associated with NRH development in MSUD patients observed in our study remain elusive, but we speculate that drugs are the most likely culprit. These patients were constantly treated with anticonvulsants for seizures and antiviral/antibacterial agents for recurrent infections, although we are unable to identify the exact drugs that have been well-documented to cause NRH. 28 It is interesting to note that the MSUD patients with NRH are generally older than those without, but we have no reason to hypothesize that the disease process itself is a potential risk factor.
The clinical significance of the finding of NRH in MSUD patients remains unclear. The fact that none of the patients had portal hypertension argues against an important clinical implication. Regarding the long-term prognosis of DLT using liver allografts from MSUD patients, there has been no report of MSUD development in DLT recipients. This is not surprising because the liver accounts for only 9–13% of total BCKDH activity in the body.8,29-31 The postoperative technical complications, such as portal vein or hepatic artery stenosis and biliary stricture, are similar to those observed in non-DLT recipients. Only 1 patient was reported to have portal vein thrombosis 3 years post-DLT. 8 Other common posttransplant complications, such as acute T cell-mediated rejection, are also similarly described in DLT. However, none of the follow-up studies has documented the presence of NRH in MSUD allografts nor portal hypertension in recipients. The possibility exists that the diagnosis of NRH might be missed because the histopathologic features of NRH can be subtle and easily overlooked, 32 or the diagnostic features of NRH are reversed because the allografts are no longer exposed to the same medications. 33 Nonetheless, the presence of NRH in the livers of MSUD patients does not appear to have an adverse impact on DLT recipients.
In conclusion, NRH is a frequent histologic finding in explanted livers from MSUD patients. The underlying etiopathogenesis and clinical implication remain unclear. Other nonspecific findings, such as minimal to mild portal lymphocytic infiltrates and mild steatosis, can also be seen in these livers.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.