
Abstract
Tbx4 protein, expressed in mesenchyme of the developing lung, contributes to airway branching and distal lung growth. An association between pediatric onset of pulmonary arterial hypertension (PAH) and genetic variations coding for the T-box transcription factor 4 gene (TBX4) has been increasingly recognized. Tbx4-related PAH onset has a bimodal age distribution, including severe to lethal PAH in newborns and later onset PAH. We present an autopsy study of a 24-year-old male with a heterozygous TBX4 variant, who developed pulmonary arterial hypertension at age 12 years. This unique case highlights the complex pulmonary histopathology leading to lethal cardiopulmonary failure in the setting of TBX4 mutation.
Introduction
Despite advances in pulmonary hypertension-targeted drug therapies, pulmonary arterial hypertension (PAH) contributes significantly to high morbidity and mortality in diverse settings, including genetic causes of PAH.1,2 Advances in molecular medicine have led to the recognition of multiple genetic mechanisms underlying PAH in both pediatric and adult populations.3-6 Past work has shown that genetic variations coding for the T-box transcription factor 4 gene (TBX4), including complete deletion, are strongly associated with pediatric and adult onset of PAH.3-5,7-10 Tbx4 protein, expressed embryologically in developing lung mesenchyme and lower extremity limb buds, contributes to distal lung development and hind-limb formation.11-20 Past studies have shown a bimodal age distribution of the onset of clinical disease in TBX4-related PAH: 1) neonatal patients with severe and often lethal pulmonary hypertension with respiratory failure; and 2) PAH associated with variable severity of chronic lung disease, which can present late in childhood and in adults.7,8,9,21-24 Structural features of TBX4-related PAH and associated lung disease, especially in subjects presenting beyond the immediate postnatal period, have been incompletely characterized. While PAH is the most common presentation for TBX4-associated pulmonary disease, additional features seen in TBX4-associated lethal lung development may present in neonates as acinar dysplasia or congenital alveolar dysplasia. 25 In this autopsy study, we explore the long-term clinical implications and histopathologic features of childhood-onset PAH in the setting of a heterozygous TBX4 variant.
Clinical history
A 12-year-old male with unremarkable perinatal, birth and family history presented for surgical correction of bilateral genu valgum (knock-knees). During the postoperative period, he was observed to have persistent nocturnal oxygen desaturations. Further diagnostic evaluations were performed, including an echocardiogram that led to the diagnosis of PAH. Subsequent right heart catheterization revealed near systemic levels of PAH that was not responsive to acute vasoreactivity testing. Despite aggressive PAH-targeted drug therapy, he demonstrated a continuous decline in exercise tolerance and worsening right ventricular function with pericardial effusion and the development of recurrent hematemesis due to portal hypertension with esophageal varices and gastropathy. He experienced worsening biventricular congestive heart failure and was hospitalized for recurrent pneumonia with increasing oxygen requirements. The patient and his family declined lung transplantation. At 24 years of age, while suffering from worsening hypoxic respiratory failure and anasarca, he was transitioned to palliative-focused care and died 4 weeks later. Permission for a lung-restricted autopsy was granted.
In addition to PAH, the patient’s clinical history is significant for infantile renal tubal acidosis, autism spectrum disorder, keratoconus (conical cornea), angiofibromas, and mild growth restriction, reaching a final adult height of 64 inches (163 cm). Karyotype analysis, conducted at 2 years of age revealed a de novo chromosome 3 deficiency (46, XY, del(3)(q26.2q26.32)) of unknown significance. Targeted genetic sequencing, performed in his late teenage years, revealed a heterozygous germline point-mutation in the TBX4 gene (p.Gly106Ser) resulting in a missense transcription error for the resulting Tbx4 protein. This particular single-nucleotide variant has not been described previously in another individual suffering from childhood-onset PAH. However, variations at differing loci of the TBX4 gene have been associated with PAH and coxopodopatellar syndrome.7,25-26 Additionally, population studies have suggested that this variant is likely to result in a pathologic loss-of-function behavior for the TBX4 gene. 25 Parental cytogenetic studies were normal and molecular sequencing of the TBX4 gene was not performed in the parents.
Postmortem findings
Gross findings were consistent with the sequelae associated with TBX4 mutation, including dense, heavy lungs with incomplete horizontal fissure of right lung (Figure 1), cyanosis of the lips and fingers, an enlarged thoracic cavity, and valgus rotation of the lower extremity joints and evidence of surgical correction. Findings related to PAH included cardiomegaly with massive hemopericardium that significantly reduced the left pleural space, marked lower extremity edema, pleural effusions, and superficial skin breakdown along the spine from prolonged bed rest. Lung tissue palpation, prior to sectioning, demonstrated dense parenchymal foci associated with pleural tethering and puckering.
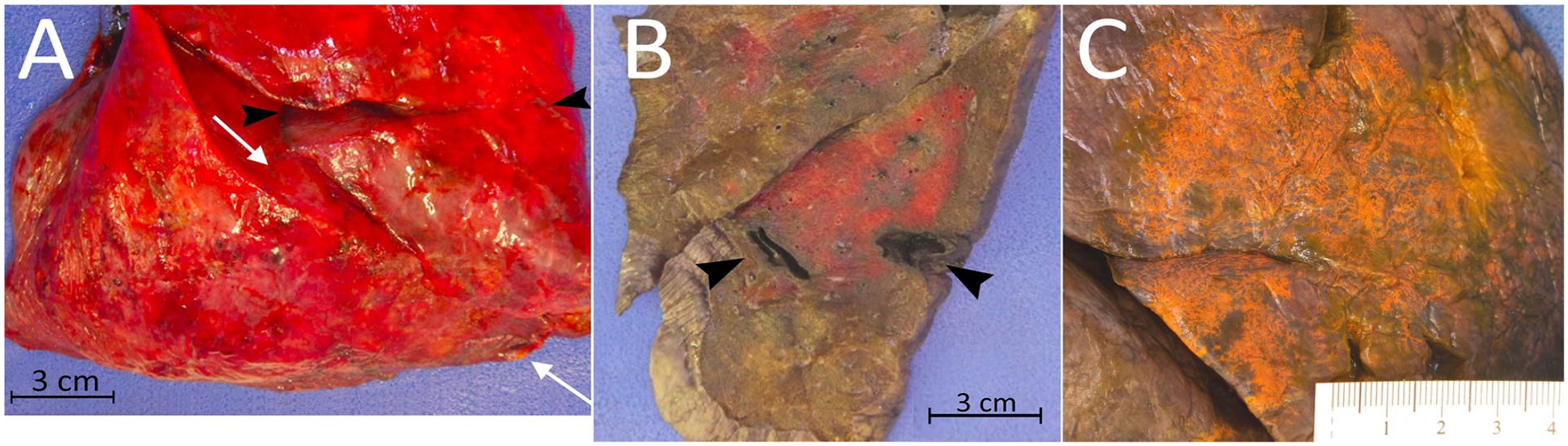
(A) Incomplete fissures of the right lung: horizontal (between black arrowheads) and oblique (between white arrows). (B) Dense heterogeneous parenchyma and infarct cavities (arrowheads). (C) Intravascular dye injection highlighting atypical pleural vascular patterns.
Following overnight fixation with infused formalin, the large pulmonary vessels were injected with inks of contrasting colors, including black ink in the pulmonary arteries and orange ink into the pulmonary veins (Figure 1). Serial sectioning revealed necrotized infarct cavities, as well as heterogeneous regions of overly firm parenchyma. Twelve tissue blocks of lung parenchyma were formalin-fixed and embedded in paraffin for histologic evaluation. Routine hematoxylin and eosin (H&E) stain was applied. Select blocks were chosen for serial sectioning and trichrome special staining prior to imaging by synchrotron-based phase contrast micro-CT, and 3-dimensional image reconstruction of the vascular microenvironment was done using the Amira software (Thermo Fischer Scientific) as described previously.27,28
Histologic evaluation revealed maldevelopment of multiple lung compartments: simplified, enlarged alveoli and back-to-back bronchiolar profiles; bony and fibrotic mesenchymal elements regionally replacing pulmonary parenchyma; pleural and lymphatic vessel muscularization; vascular plexiform lesions; and prominent intrapulmonary bronchopulmonary vascular anastomoses (IBA) (Figure 2). Additional findings included features associated with long-standing vascular remodeling, which included smooth muscle hypertrophy, thrombosis and parenchymal infarcts, vessel wall hyalinization, obliteration of vascular lumens associated with prominent IBA channels. The postmortem ink infusions demonstrated extensive recruitment of bronchial vessels via the pulmonary vein (orange ink) and impeded infusion within pulmonary arteries, based on the reduced spread of black ink. Intermingling of the 2 inks suggests the presence of anastomoses due to vascular remodeling with recruitment of IBAs between the pulmonary and bronchial arteries that bypass the terminal capillary networks essential for effective gas exchange (Figure 3). The synchrotron-based three-dimensional image reconstruction of the vasculature highlights these extensive anastomotic networks between bronchial circulation and remodeled pulmonary arteries within the diseased lung (Figure 4). Control lung tissue does not show shunt vessels, plexiform lesions, or recruited bronchial vessels.27,29-32
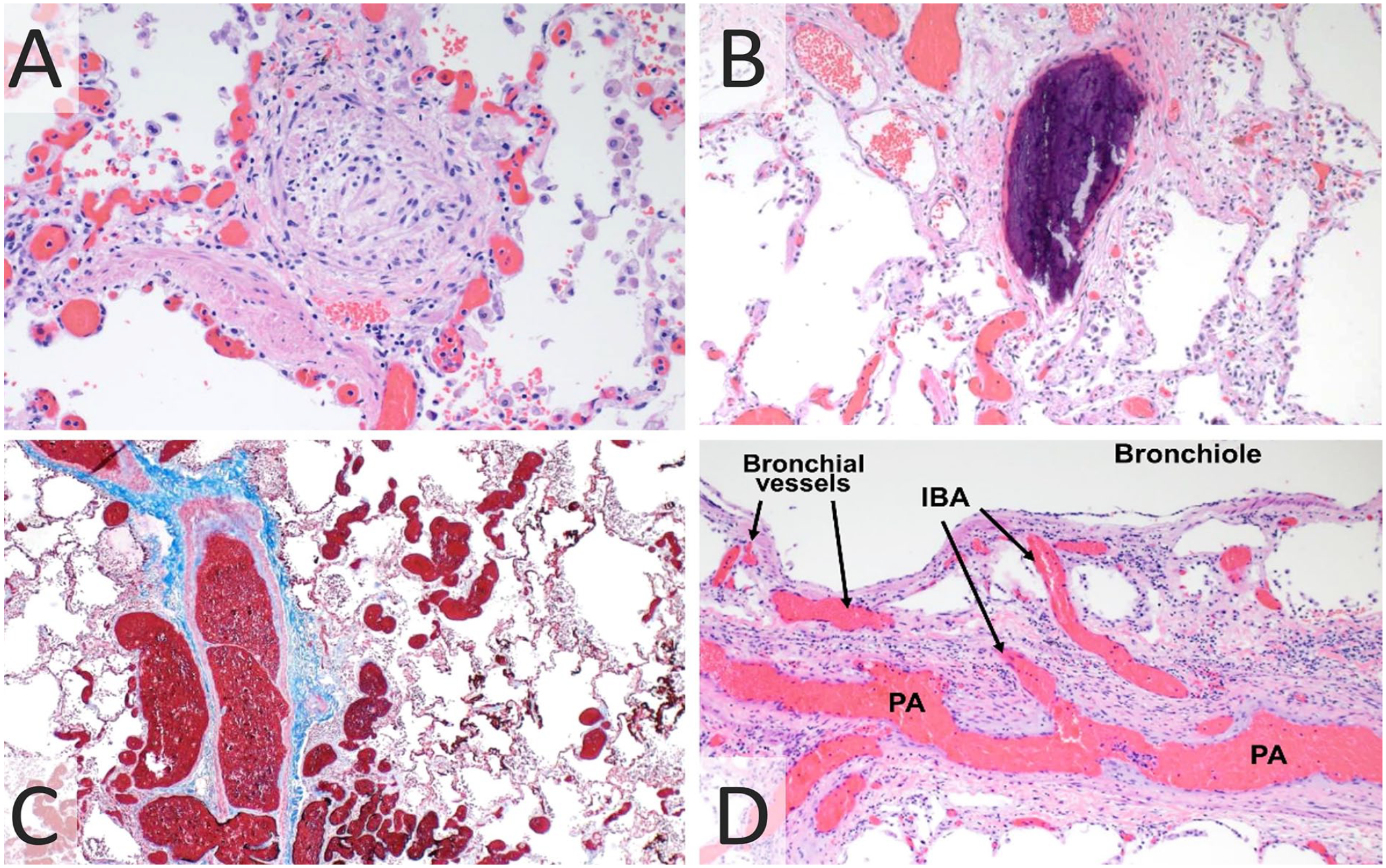
(A) Pulmonary artery lumen obliteration and wall muscularization (H&E, 200×). (B) Ectopic bone formation and stromal vessel dilatation with congestion (H&E, 100×). (C) Plexiform lesion characterized by many dilated thin-walled vessels surrounding the bronchoarterial bundle (Trichrome, 40×). (D) Intrapulmonary bronchopulmonary anastomoses (IBA) allow blood flow between bronchial vessels and pulmonary artery (PA) branches (H&E, 100×).
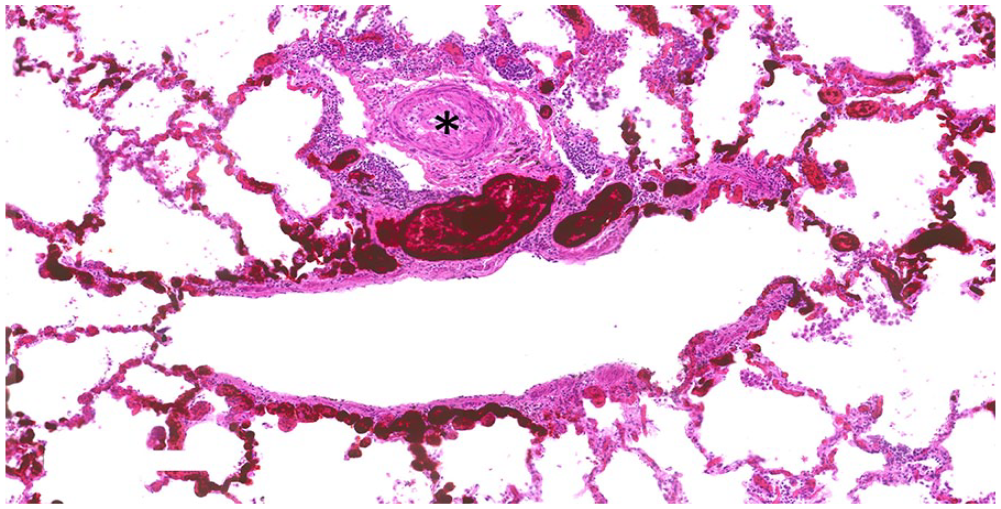
Occluded pulmonary artery (*) surrounded by vascular remodeling demonstrating intermingling of inks, orange (venous flow) and black (arterial flow), suggestive of intrapulmonary bronchopulmonary anastomoses (IBA) bypassing the terminal capillary network.
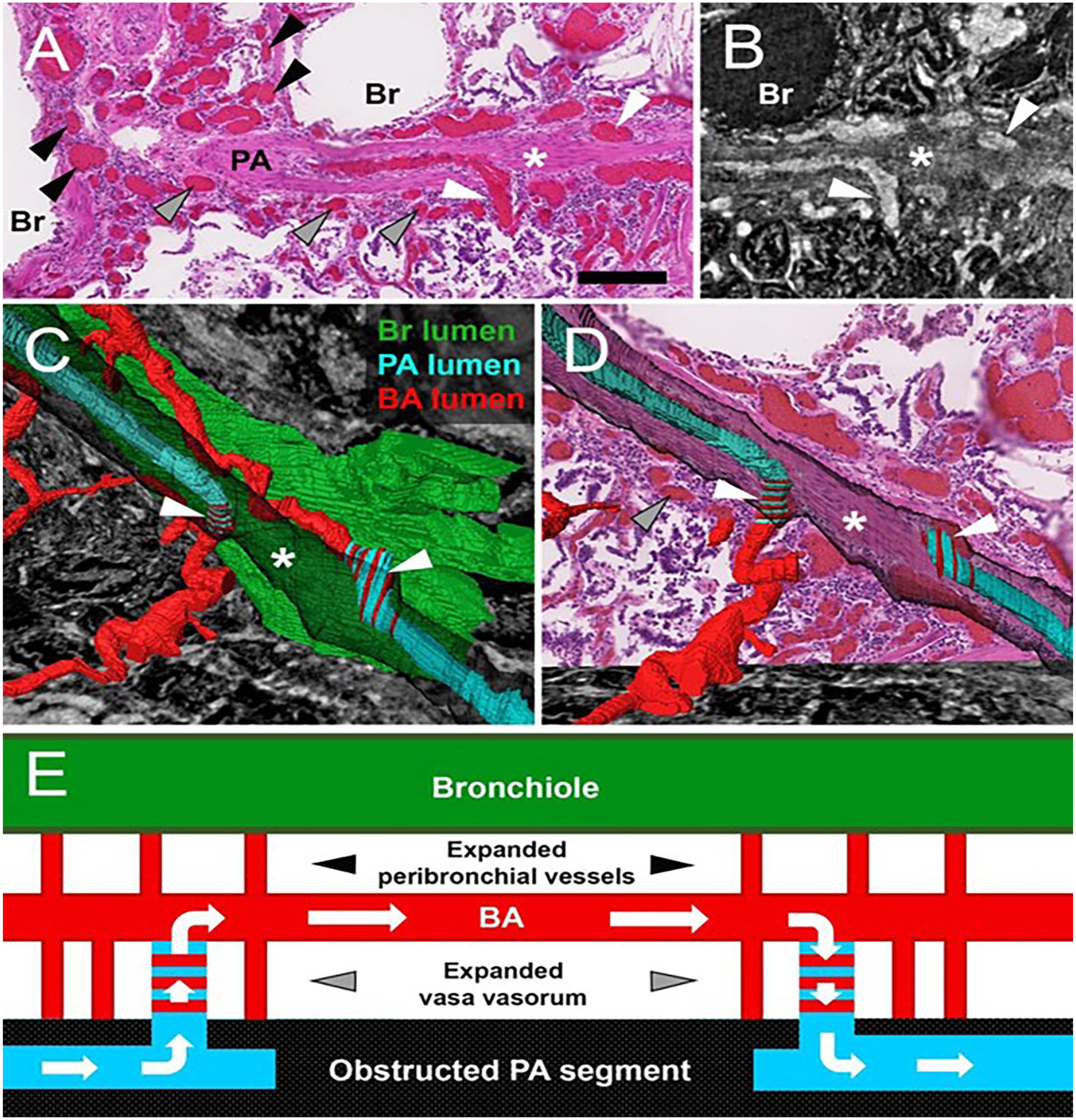
3D reconstruction of microscopic computed tomography (CT) images confirms the presence of recruited intrapulmonary bronchopulmonary anastomoses (IBA) in TBX4-associated pulmonary arterial hypertension (PAH). (A) A partially obstructed (*) pulmonary artery (PA) associated with recruited IBA proximal and distal to obstruction (white arrowheads), expanded vasa vasorum (grey arrowheads) and peribronchial vessels (black arrowheads) (H&E, scalebar = 200 um). (B) CT slice of area shown in A. (C and D) 3-D reconstruction of the PA lumen (light blue) shows connection with the vasa vasorum and peribronchial vessels (red) via a recruited IBA (white arrowheads, left) proximal to the obstructed PA segment (*). Distal to the obstruction, PA lumen is open (light blue in D) and communicates with a bronchial artery (red) running between the hypertrophied PA wall (transparent black) and the bronchial (green), via a recruited IBA (white arrowhead, right). Evaluation of the serially sectioned block confirmed findings visualized in A-D and registration of the representative H&E section (A) to the 3D reconstruction confirms the above findings. (E) Schematic drawing summarizing the complex vascular remodeling in TBX4-associated PAH and proposing an adaptive blood flow pathway via IBA to bypass the obstructed PA.
Discussion
The T-box 4 gene domain, located on Chromosome 17q23, is part of a highly conserved family of T-box genes involved in regulation of embryologic developmental processes and organogenesis. TBX4 genetic variants were first identified in association with small patella syndrome (SPS) or ischiocoxopodopatellar syndrome, a rare and heterogeneous autosomal dominant condition with phenotypes ranging from small to absent patellae, irregular ischiopubic ossification to severe pelvic and femoral anomalies, and increased spacing between the first and second toes.11,14 Animal models with monoallelic and biallelic TBX4 knockout modifications support a connection between normal Tbx4 protein expression and hindlimb outgrowth.12,14,18,19 Homozygous loss-of-function mutations have been found in human fetal cases with complete pelvis and lower-limb agenesis. 14
More recent cohort studies of patients with PAH have detected TBX4 variants in both pediatric and adult populations, with concurrent SPS features in up to 80% of TBX4-associated PAH cases, as seen in this case.3,4,8-10,23 However, disease presentation in patients with TBX4 mutations is highly variable and may include lung vascular disease only, skeletal disease only, or a combination of both.7,21,28 TBX4 influences proliferation, migration, and differentiation of pulmonary mesenchymal cells.15,16,20 Detrimental variants include deletions of the coding exons, supportive introns, or the entire gene locus, as well as point mutations. While disease severity does not necessarily correlate to the level of Tbx4 protein expression, it does likely correlate to the degree of disordered embryologic development.7,13
Heritable PAH is defined as precapillary pulmonary vascular disease with either familial inheritance or, more commonly, a de novo mutation in those genes known to be associated with pulmonary arterial function: TBX4, BMPR2, ACVRL1, BMP9, SMAD9, and SOX17.3,5,6,9,10,25,26,33 Within familial TBX4-associated PAH, penetrance and disease manifestation is independent of mutation type. 9 Genetic variants are present in approximately 30 to 45% of PAH patients presenting during childhood compared to 11% presenting in adulthood.4-6 The presence of identifiable genetic variants jumps to 80% in familial cases while de novo variants were identified in approximately 25% of cases previously characterized as idiopathic. 10 While TBX4 variation is seen approximately twice as frequently in pediatric-onset PAH than adult-onset, the most frequent PAH-associated genetic variant in both groups is BMPR2.6,8,10
Persistent pulmonary hypertension of the newborn (PPHN), presents during the early postnatal period as the pulmonary vasculature fails to make the neonatal conversion from the high-resistance environment of the fetus to a low-resistance setting necessary for alveolar oxygenation.7,22 For term or near-term neonates who are poorly responsive to therapy or require prolonged cardiorespiratory support, further evaluation for genetic developmental lung disease is appropriate given that TBX4-related lung maldevelopment has been linked to lethal neonatal PAH cases. 22 In addition to progressive pulmonary hypertension, survivors of PPHN have an increased risk of developing PAH later in childhood, as well as persistent patent ductus arteriosus, atrial septal defect, and other congenital heart defects.6,7
Histopathologic variation in lung tissue affected by TBX4-associated PAH is widespread with abnormal distal lung development of not only the vascular structures, but also the airways, alveoli, and interstitium. These findings include dilated distal airspaces comprised of immature-appearing alveoli without secondary septa, interstitium thickened by fibrosis, back-to-back bronchiolar profiles, recruitment of IBAs, and mesenchymal maldevelopment including ectopic bone formation.4,7,9,18 The endothelium, which is susceptible to shear force injury, likely experiences increased blood flow through these transformed pathways as lung injury progresses, resulting in thromboses and subsequent parenchymal infarcts. Pulmonary hypertensive arterial remodeling induces adventitial thickening, medial hypertrophy, and fibrointimal proliferation resulting in concentric laminar fibrosis and obliteration of vascular lumina. Vascular plexiform lesions are a severe end-stage form of remodeling seen in PAH of different etiologies that have not previously been reported in TBX4-related lung disease but were identified in this autopsy case (Figure 2).28,34 The ensuing systemic hypoxia following lung remodeling fuels a cascade of physiologic responses including increased right heart work and eventual heart failure. In neonatal-onset PAH cases with TBX4 variants pulmonary growth arrest is demonstrated by acinar dysplasia and simplified alveolar development.13,17,22,25
In addition to disrupted respiratory tract development and hindlimb hypoplasia, neurodevelopmental disorders and dysmorphic facial features are common in patients with TBX4 variants. It is hypothesized that TBX4 has a supplemental role in the formation of external genitalia, anorectum, and body wall. 8 A possible Tbx4 physiologic pathway contributing to lung and hindlimb development has been promoted by in vitro experiments. Tbx4 protein has been shown to regulate expression of fibroblast growth factor 10 (FGF10), as well as activation of the Wnt pathway to induce fetal lung branching and lobe separation, as well as chondrogenesis and limb bud outgrowth.8,12,13,17 TBX5, a neighboring T-box family gene, has an analogous role in upper limb development but has no identified role in lung development or long-term pulmonary health.8,13
The complex pathobiology of the Tbx4-PAH pathway allows for multiple potential therapeutic targets, though there are currently no drugs available on the market that provide a cure for PAH. Current research appears focused on BMPR2 therapies; however, a broadened focus including more genetic variations and their pathways should be considered to reach more patients especially given the relatively high frequency of TBX4 mutations identified in the pediatric PAH population.35,36
Conclusion
This is a unique case of a 24-year-old male who died from complications of a heterozygous germline TBX4-variant highlighting the strikingly complex pulmonary histopathology including severe hypertensive arterial remodeling with vascular plexiform lesions, prominent IBAs, and evidence of severe right ventricular failure. This is the first-time plexiform lesions have been described in TBX4-associated PAH and suggest that these remodeling lesions may be present in other genotype-phenotype. 25 Over time and under the influence of abnormal Tbx4 protein signaling, all compartments of the lung continued to remodel, resulting in worsening PAH, impaired airway and distal airspace structure, and hypoxemia, which caused end-stage lung disease. The maldeveloped background lung, in the setting of vascular restriction from thromboses and PAH, demonstrated innumerable infarcts which additionally impaired gas exchange. The oxygen demand of the lungs likely exacerbated the patient’s known congestive heart failure and hemopericardium, which led to patient death.
Current pulmonary hypertension drug therapies are often ineffective at arresting the progression of PAH in this heterogeneous disease.35,36 The only long-term treatment option for these patients with late-stage PAH, TBX4-associated or not, is lung transplantation to prevent demise from heart failure. Therefore, greater understanding of molecular mechanisms of normal and aberrant signaling pathways associated with Tbx4 protein-related lung maturation and function are needed in order to develop targeted therapeutic interventions to prevent continuation of destructive pulmonary remodeling and attenuate progression of PAH.
Footnotes
Acknowledgements
The authors thank Paul Scherrer Institute, Villigen, Switzerland, for provision of synchrotron radiation beam time at the X02DA TOMCAT beamline of the Swiss Light Source and Goran Lovric for excellent technical support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.