
Abstract
Nonclinical safety studies are typically conducted to establish a toxicity profile of a new pharmaceutical in clinical development. Such a profile may encompass multiple differing types of animal studies, or not! Some types of animal studies may not be warranted for a specific program or may only require a limited evaluation if scientifically justified. The goal of this course was to provide a practical perspective on regulatory writing of a dossier(s) using the weight of evidence (WOE) approach for carcinogenicity, drug abuse liability and pediatric safety assessments. These assessments are typically done after some clinical data are available and are highly bespoke to the pharmaceutical being developed. This manuscript will discuss key data elements to consider and strategy options with some case studies and examples. Additionally, US FDA experience with dossier(s) including WOE arguments is discussed.
Keywords
Introduction
Weight of evidence (WOE) is an inherent part of the role of a toxicologist in risk assessment – to assess all available data and make a judgment (or at least provide an informed opinion) on the level of risk. Here we are using WOE to decide whether further data are needed to adequately assess, characterize, and communicate risk, or whether the risks are understood and can be managed or mitigated without further data or the need to conduct additional nonclinical studies. WOE can also inform on aspects of study design and some components of WOE assessment may carry different “weight”.
Guidance Title and Context.

A Guide to the Gudiances.

Weighing the Weight of Evidence: A CDER Perspective
Ron Wange
FDA regulations require that sponsors submit nonclinical data to support the clinical development and marketing of drugs and biologics. With limited exceptions (e.g., products developed under 21 CFR 314.600-650 for drugs; 21 CFR 601.90-95 for biologics – often referred to as the “Animal Rule”) the precise nature of what constitutes adequate nonclinical data is not defined by the regulations. Rather, FDA communicates recommended approaches for generating and communicating nonclinical safety data by publishing guidance documents that reflect FDA’s current thinking. A review of the guidances concerning the nonclinical safety testing for Center for Drug Evaluation and Research (CDER)-regulated products found that 15 guidances refer to the potential of using a weight of evidence (WOE) approach as a viable means of making various regulatory decisions (Table 1). (NB – Table 1 has more than 15 entries, since it also includes guidances related to clinical investigations and guidances from other regions.)
But what does CDER mean when it refers to a WOE assessment? Notably, this is not a term that is defined in U.S. statute or regulation, nor is it defined in most guidances in which the term appears. What it is, at least in the sense that FDA typically uses it, is a term used to indicate a scientific judgment call based on the totality of the available data germane to a particular regulatory decision or action. Borrowing from the definition provided in ICH S11
1
, a generalized definition might read as: “An approach that evaluates information from several sources to decide if there is sufficient evidence to support the [assessment and communication of risk] or whether additional nonclinical testing is warranted to address potential safety concerns. The weight given to the available evidence depends on factors such as the quality of the data, consistency of results, nature and severity of effects, and relevance of the information. The weight of evidence approach requires use of scientific judgment and, therefore, should consider the robustness and reliability of the different data sources.”
The way in which CDER uses nonclinical WOE assessments can generally be broken down into four, somewhat overlapping categories: 1. As a method for determining whether additional, more specialized toxicology studies are warranted (e.g., juvenile animal, abuse potential, Tier 2 immunotoxicity) 2. As a method for assessing whether all components of a “typical” full complement of nonclinical assessments are warranted to assess a particular risk (e.g., Developmental and Reproductive Toxicity [DART], carcinogenicity) 3. As a method for summarizing and communicating risk via labeling without relying on a full complement of “typical” empirical studies/endpoints (e.g., DART, carcinogenicity) 4. As a method of summarizing and communicating risk via labeling, based on integrating multiple endpoints and data elements (e.g., genotoxicity, immunotoxicity, DART, carcinogenicity).
Use of WOE in first sense, that is determining whether additional, more specialized toxicology studies need to be conducted in the setting of assessing a particular risk are discussed below by Dr. Laffan (Nonclinical Pediatric Development: One Size Doesn’t Fit All) and Drs. Kruzich and Compton (Preparation of Drug Abuse Liability Assessment Documents). The fourth category was not directly addressed during the workshop, but statements in labeling about genotoxicity, based on results from a battery of assessments or based on mechanism of action would be an example of this category. The use of WOE assessments for Categories 2 and 3 are further considered here, in this section, in the contexts of assessing embryofetal developmental (EFD) risk and the risk of carcinogenicity. The use of WOE in assessing carcinogenicity risk is further elaborated on in Dr. Manetz’s section on “Carcinogenicity WOE Assessment Overview: Principles and Methods,” below.
Use of Weight of Evidence in Assessing Embryofetal Developmental (EFD) Toxicity Risk
Most pharmaceuticals being developed in the United States need to be assessed for EFD toxicity prior to enrollment of women of childbearing potential in Phase 3 clinical trials, as well as to support the approval and labeling of the product. General guidance that provides recommendations on how this risk should be assessed is provided in ICH S5 (R3) Detection of Reproductive and Developmental Toxicity for Human Pharmaceuticals. 2 In addition to describing the more typical approach of conducting in vivo EFD studies in two species, a rodent and a nonrodent, this guidance identifies circumstances under which available evidence may be adequate to characterize and communicate risk, without the need to conduct one or more in vivo EFD studies. In Section 4.2 of the guidance, Strategies to Address Embryofetal Development, the guidance states that “The design of an appropriate testing strategy relies on a cumulative weight-of-evidence approach” and “…there can be adequate information to communicate risk without conducting EFD studies. Evidence suggesting an adverse effect of the intended pharmacological mechanism on EFD (e.g., mechanism of action, phenotypic data from genetically modified animals) can be sufficient to communicate risk.”
Similar concepts also appear in the FDA guidance on Oncology Pharmaceuticals: Reproductive Toxicity Testing and Labeling Recommendations,
3
which also provides a relatively thorough list of the types of data that can inform the WOE for EFD risk. This includes: – Reproductive findings in humans, such as when a drug is in a class of pharmaceuticals with extensive publicly available information on potential reproductive effects – Reproductive findings from genetically modified animals or models employing pharmacologic inhibition, as appropriate – Information from a surrogate molecule, when a surrogate is available and the target biology in the animal species is relevant to humans – Literature-based assessment of target biology in humans or animal species, which can describe the following: • Expression and the role of the molecular target during embryo-fetal development • Any other relevant information, such as the role of the target in placental development, placental transfer, maternal tolerance, etc. – Use of alternative assays, such as fit-for-purpose in vitro or ex vivo, or nonmammalian in vivo assays can be used
For biotechnology-derived pharmaceuticals (biologics), ICH S6 Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals also provides guidance relevant to DART risk assessment. 4 Though not specifically identified as WOE provisions, the core guidance indicates that “The need for [DART] studies is dependent upon the product, clinical indication and intended patient population.” This can be interpreted as a type of WOE assessment that focuses on whether there is a reasonable basis for concluding that there is negligible risk to reproduction and development, and therefore no need for DART studies, or perhaps not a need to study all DART endpoints. Another statement that can be considered a form of WOE assessment can be found in Note 2 of the guidance, which can be paraphrased as: If there are extensive public data available to indicate a clear DART risk for a particular class of compounds, and the NHP is the only relevant species, mechanistic studies for a new member of the class may be sufficient to assess the risk, with scientific justification. In this case, the risk can be characterized on the basis of the mechanism of action of the class.
The addendum to ICH S6 explicitly refers to WOE as being sufficient to communicate DART risk in some instances 5 : “e.g., when mechanism of action, phenotypic data from genetically modified animals and/or class effects suggest that there will be an adverse effect on fertility or pregnancy outcome.” It also indicates that it can be appropriate to forego DART assessment for monoclonal antibodies or related products directed towards foreign targets, if there is no cross-reactivity with any endogenous molecules. This can be considered as another example of a type of WOE assessment that focuses on determining whether there is a reasonable basis for concluding that there is a negligible risk to reproduction and development.
To get a sense of the degree to which weight of evidence assessments are used in communicating EFD toxicity risk, Section 8.1 (“Pregnancy”) of the US product labels for CDER-approved original biologics were analyzed for the 7-year period from 2015-2021, inclusive. 6 Looking across all indications, 62% of labels are informed by in vivo assessment in standard models (evenly split between NHPs and rodents/rabbits), 25% use mechanism of action alone to communicate risk, 5% use a rodent surrogate or genetically modified rodent as part of a WOE assessment, and 8% are silent regarding animal EFD studies or state that no animal studies were conducted. This latter category, I would argue, is also reasonably viewed as being informed by a WOE assessment, where the assessment was that the product represented negligible risk based on product- or indication-specific factors.
When examined separately, Section 8.1 of the labels for non-oncology products were informed by in vivo assessments in standard models 78% of the time, mechanism of action alone 3% of the time, WOE assessment that included studies with a rodent surrogate or genetically modified rodent 7% of the time, and 12% of labels were silent regarding animal EFD studies or stated that animal studies were not conducted. For oncology products, only 20% of labels were informed by standard in vivo models, 76% were informed by mechanism of action, and 4% relied on a WOE analysis involving a genetically modified rodent.
This begs the question of why there is such a large difference in the number of labels the rely on WOE for oncology versus non-oncology products? To a large degree this is because biologics intended to treat advanced cancer, by their very nature, are generally expected to have teratogenic activity. Examples of such products include: (1) drug-antibody conjugates, where the drug component is toxic to rapidly dividing cells, (2) monoclonal antibodies (mAbs) that disrupt immune tolerance, and (3) inhibitors of kinases known to play a role in embryofetal development. Also, for oncology products, the risk:benefit is different than most non-oncology products due the severity of the indication and there may also be less focus on the need to calculate safety margins, since clinically efficacious exposures are generally expected to result in some degree of toxicity, including EFD toxicity. This may make a qualitative assessment of risk, without the need to identify a no observed adverse effect level, more acceptable.
Most of the non-oncology biologic products that could be considered as being labeled based on a WOE approach, fall under the category of having a negligible risk of EFD toxicity based either on their mode of action or the indicated patient population. Examples include: (1) enzyme replacement therapies, (2) foreign targets, and (3) male-only treatment population for an X-linked recessive trait. However, a couple of labels were identified that used a WOE approach, based on the mechanism of action, to characterize and communicate risk. These included inhibitors of vascular endothelial growth factor and of interferon alpha.
Use of Weight of Evidence in Assessing Carcinogenicity Risk
Looking first at biologics, guidance supports conducting a WOE assessment, based on the totality of the published data related to the target (e.g., studies conducted in transgenic or knock-out animals, animal models of disease or human genetic diseases), class effects, and mechanism of action, as well as product-specific in vitro, nonclinical and clinical data. The end goal is to be able to determine whether the available data are adequate to support a conclusion that a biologic either is, or is not, likely to be tumorigenic in humans. In either of these cases, no rodent carcinogenicity studies would be expected, as the risk is already considered to have been characterized. However, in those cases where the WOE analysis provides ambiguous results or is otherwise insufficient to characterize the risk, additional studies, which could include a carcinogenicity study in a single species, can be appropriate if a pharmacologically responsive rodent is available, and the study is otherwise technically feasible. The view that WOE alone is typically sufficient for characterizing the carcinogenicity risk for biologics is based on having a thorough understanding of the potential carcinogenic impact of the intended activity of the biologic, and knowledge that there is no off-target pharmacology or toxicology – generally a valid assumption for most biologics.
When CDER receives one of these carcinogenicity assessments for biologics, it is reviewed by the review division in consultation with the Executive Carcinogenicity Assessment Committee. In most cases there is agreement between the Agency and the sponsor that a rodent carcinogenicity study would not add value to assessing the carcinogenicity risk. In those rare cases where a rodent carcinogenicity study has been recommended, this typically reflects poorly characterized target biology at either the primary or secondary targets (off target).
For small molecule pharmaceuticals, traditionally the assessment of carcinogenic risk has relied on measuring apical endpoints (tumor development) in rodent studies, typically with a 2-year study in the rat and a mouse study of either 2 years in normal mice or 6 months in transgenic mice.
7
The recent implementation of the addendum to ICH S1B,
8
however, opens up the potential for using a WOE approach for concluding that the rat 2-year study is unlikely to add value in assessing the human carcinogenicity risk and therefore need not be conducted. The guidance identifies 6 key factors that should be considered in the WOE analysis: 1. Drug target biology and the primary pharmacologic mechanism of the parent compound and active major human metabolites 2. Secondary pharmacology of the parent compound and major metabolites (rodent and human) 3. Histopathology data from repeat-dose rat toxicity studies (especially chronic) 4. Evidence for hormonal perturbation 5. Genetic toxicology study data 6. Evidence of immunomodulation
These factors are then integrated into an overall assessment of whether the 2-year rat study is likely to be informative in assessing the carcinogenic potential of the pharmaceutical (Figure 1). The end goal being to come to one of three conclusions: 1. Carcinogenic potential in humans is likely 2. Carcinogenic potential is unlikely 3. Carcinogenic potential in humans is uncertain Integration of key WOE factors to inform on the value of conducting a 2-year rat study for assessment of human carcinogenic risk. When all WOE attributes align towards the right side of the figure, a conclusion that a 2-year rat study would not add value is more likely. Note that for the genotoxicity WOE factor a 2-year rat study is less likely to be of value either in cases where there is no genotoxicity risk or in cases with unequivocal genotoxicity risk. Similarly, for the immune modulation WOE factor, a 2-year rat study is less likely to be of value in cases where there are either no effects on the immune system or in cases where there is broad immunosuppression.

In the first two categories, a rat study would be considered unnecessary for further characterizing the risk, while a rat study would be warranted if the carcinogenic potential in humans is uncertain. In general, a carcinogenicity study in mice would still be expected for compounds falling in categories 2 and 3, but not for compounds falling in category 1. There can also be scenarios where a mouse study may not be warranted for compounds falling in category 2, for example if only subtherapeutic or inactive exposures can be achieved in the mouse.
As a practical matter, sponsors should keep in mind that there are no statutory or Prescription Drug User Fee Act (PDUFA)-mandated deadlines regarding the review timeline for the adequacy of a submitted carcinogenicity WOE dossier. This is unlike the review process for a Carcinogenicity Special Protocol Assessment, which is subject to a 45-day review clock. Internal data suggests that most carcinogenicity WOE submissions are being reviewed between 3 to 5 months of receipt.
To summarize, many CDER guidances make reference to the use of WOE assessments. This is primarily in the context of (1) assessing developmental and reproductive toxicity risk, (2) assessing immunotoxicity risk, (3) assessing genotoxicity risk, (4) assessing carcinogenicity risk, (5) assessing the need for dedicated specialized toxicity studies (e.g., juvenile animal study [JAS], immunotoxicity, abuse liability). In order to help reduce unnecessary animal use, those developing pharmaceuticals should routinely consider whether there are sufficient data to inform and communicate risk in any of these areas before defaulting to conducting an animal study, especially for studies involving nonhuman primates or large number of animals.
Carcinogenicity WOE Assessment Overview: Principles and Methods
T. Scott Manetz
Carcinogenicity is the occurrence of induction of tumors, increased tumor incidence, or shortened time to tumor occurrence. For pharmaceuticals that are intended to be regularly administered for a minimum 3-6 months duration, it is important to understand the potential carcinogenicity risk of a candidate pharmaceutical. Duration of intended treatment is an important criterion outlined in available regulatory guidance. According to ICH S1A guidance, 9 for Japan, in vivo carcinogenicity studies are expected if clinical use is expected to be continuous for 6 months or longer. However, if there is a cause for concern, candidate pharmaceuticals used continuously for less than 6 months may still need carcinogenicity studies. For the United States, in vivo carcinogenicity studies are expected when used for 3 months or longer. In Europe, the expectation includes administration over a substantial period of life, i.e., continuously during a period of at least 6 months or administered frequently in an intermittent manner such that the total exposure is similar.
Although in vivo carcinogenicity studies are intended to identify the tumorigenic potential of a candidate pharmaceutical, these studies are typically conducted later in the candidate drug development program timeline. However, in vivo assessment should not be the sole data used to provide an assessment of the potential risk to humans. To address potential for malignancy risk earlier in a candidate drug development program timeline, and to augment the interpretation of risk from any later performed in vivo carcinogenicity studies, a paper exercise risk assessment should be performed. This paper exercise is typically referred to as a Weight of Evidence (WOE) risk assessment. Of note, in the case of a biotherapeutic (biologic) where often there is no pharmacology in rodents (the typical carcinogenicity study species), this WOE paper exercise may represent the primary data source for determining human malignancy risk for eventual communication of risk in drug labeling. As noted previously, the view that WOE alone is typically sufficient for characterizing the carcinogenicity risk for biologics is based on having a thorough understanding of the potential carcinogenic impact of the intended activity of the biologic, and knowledge that there is no off-target pharmacology or toxicology – generally a valid assumption for most biologics.
There are many aspects to consider when preparing a carcinogenicity weight of evidence assessment, whether for a small molecule or biologic. One key component is to build a weight of evidence assessment and review of available key data resources, including literature (pharmacological target over-expression and gene-deficient mouse characterization/phenotypes, gene deficiency in humans should it exist, etc.) and relevant guidance documents. Quite often, given the likely breadth of published information on a target worthy of pharmaceutical intervention, a weight of evidence risk assessment will include data that indicate there is not a risk, and data that indicate there is a risk. The WOE assessment could give ambiguous results or otherwise be insufficient to characterize the risk. As the WOE case study below will show, there can still be a significant ambiguity to the final outcome of a weight of evidence assessment.
A weight of evidence approach utilizes available data from many sources to form a picture of the overall malignancy risk. For a carcinogenicity weight of evidence risk assessment (Figure 2), the following components (as available) could provide useful information in terms of determining risk and include: Prior experience with other molecules in the same drug class, experience with genetic polymorphisms of the target or pathway, available clinical trial data, experience with genetically engineered rodent models or animal disease models, unintended pharmacology, evidence of immune modulation and/or hormonal perturbation, genetic toxicology results, histopathology results from repeated dose rodent toxicology studies, and metabolic profile, among others. Although the data can be mixed with some data suggesting a malignancy risk, some data suggesting the opposite, often an overall picture will form with regards to malignancy risk from these various data sources (See below for a theoretical example). It is important that the weight of evidence includes presentation and consideration of all available data, not just the data that supports a desired conclusion/position. Integration of key WOE factors to inform on the value of conducting an in vivo assessment of human carcinogenic risk for a biotherapeutic. When all WOE attributes align towards the LEFT side of the figure, a conclusion that an in vivo assessment would add value is LESS likely. Alignment to the RIGHT suggests an in vivo assessment would add value is MORE likely. Note the ‘left-less likely’ and ‘more likely’ choices by the authors are reverse of that for the ICHS1B(R1) figure from which this figure is adapted. As noted in the figure, rodent cross-reactivity of the clinical candidate, or availability of a suitable surrogate molecule is a key logistical consideration for the practicalities of in vivo assessment options.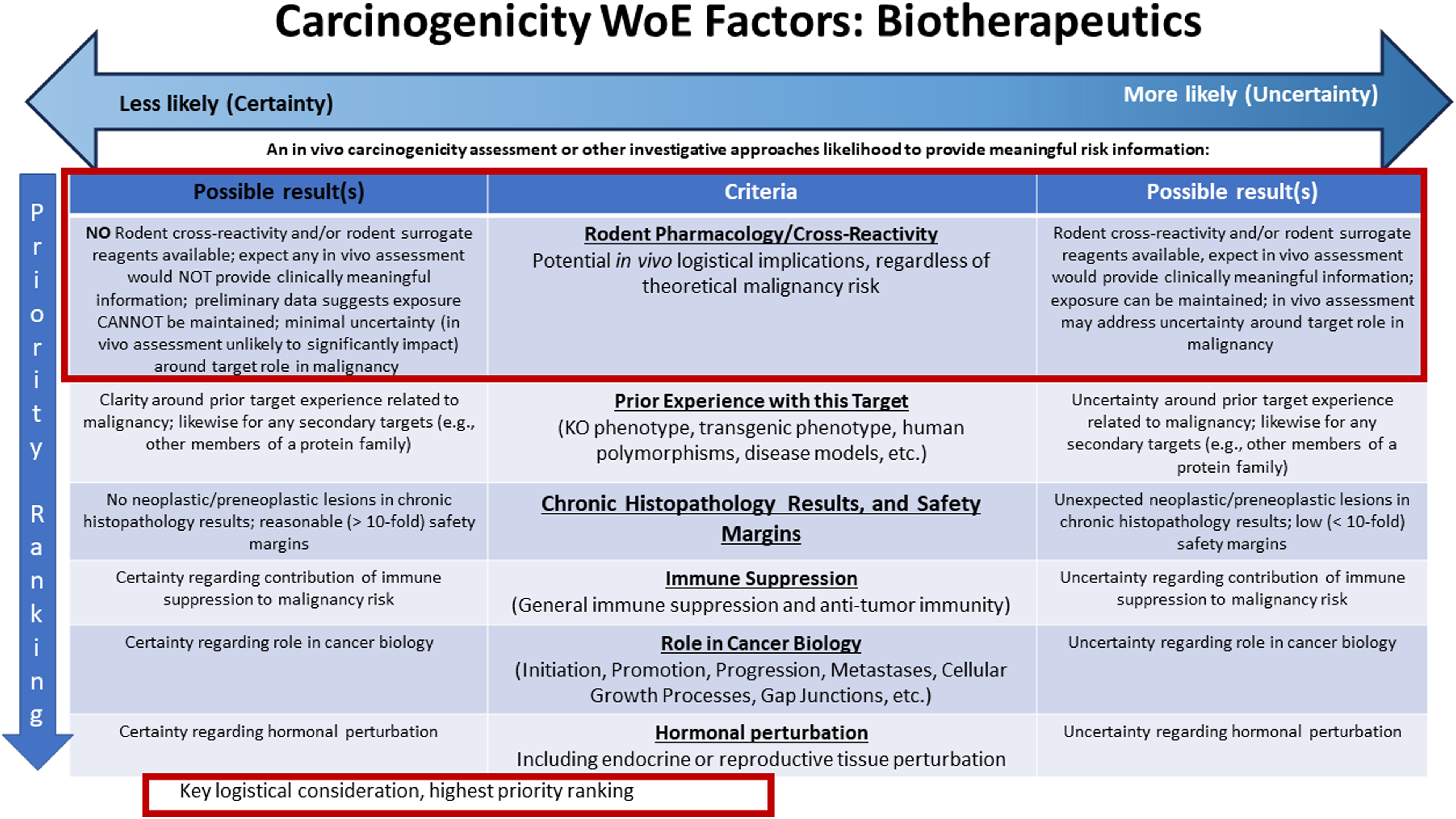
For example, in the below theoretical WOE example for an anti-cytokine monoclonal antibody, the following data were available: 1. The toxicology program conducted showed no neoplastic or pre-neoplastic lesions in a 6-month cynomolgus monkey study (there was no rodent cross-reactivity/pharmacology, so a single species toxicology program was conducted consistent with ICHS6R1 guidance expectations
4
). 2. Granted, there are limitations from concluding carcinogenicity risk from a 6 -month cynomolgus study. For example, 6 months of dosing in a cynomolgus monkey is not nearly a lifetime exposure study in this species
10
(cynomolgus monkey lifespan is < 20 years), nor are cynomolgus monkey general toxicology studies using the typically desired numbers of animals
11
adequately statistically powered for an assessment of carcinogenic potential.
12
3. While there was some literature showing an increase in malignancy risk when blocking this pathway, the majority of the literature suggested that blockade of this pathway could decrease malignancy risk. 4. Target gene deficient mice showed mixed results, which appeared to be tumor model and context dependent, and possibly also to reflect some differences in target pharmacology between rodents and man. 5. The few available human genetic polymorphisms that have been described do not address malignancy risk. 6. Though some studies have suggested that the target may have antiproliferative properties towards certain human tumor cells (therefore target blockade could increase tumor cell proliferation), many other studies suggest the target is a negative regulator of anti-tumor immunity, such that target blockade would be beneficial for anti-tumor immunity. 7. Blockade of the target in nonclinical safety studies was not associated with generalized immune suppression, nor was T cell function impaired.
In terms of logistical considerations for in vivo carcinogenicity risk assessment options, the clinical candidate does not cross-react to rodents (no pharmacology in rodents), so conducting a classical rodent carcinogenicity study was not an option. Transgenics that express the human target and use of a surrogate reagent in mice were both considered, but ruled out over concerns about immunogenicity and the resultant hypersensitivity/anaphylaxis and loss of exposure/pharmacology from repeated dosing, which has been experienced by others. 13
Finally, gene deficient mice were considered for in vivo carcinogenicity risk assessment, but ultimately not considered a viable option. One anticipated disadvantage is that lifetime inhibition of target in a gene deficient model likely represents a worst-case scenario, and therefore is not reflective of the circumstances of clinical intervention later in life. Under these circumstances the hazard will be overestimated since gene deficient models represent an all or none scenario. Gene deficient models do not offer the ability to test different doses of test article, thus preventing any potential for dose response characterization of risk. As well, a lifetime deficiency phenotype may also be impacted by yet unknown/uncharacterized compensatory mechanisms, or redundant pathways, and this may not be reflective of clinical intervention. Finally, the resultant phenotype of gene deficient models may also be influenced by background strain traits not reflected in the same gene deficient model produced on a different background strain. (For examples of the impact of strain on gene deficient mouse phenotype.14–16)
Therefore, the final outcome of this theoretical WOE was that the majority of the available information did not indicate a general malignancy risk, while acknowledging there are some available data that suggest the opposite. After careful consideration of the available models/reagents, performance of in vivo carcinogenicity studies with the available approaches was considered unlikely to provide clinically meaningful information for carcinogenicity risk assessment and no in vivo studies were proposed.
Finally, one must also consider for logistical purposes strategies for the engagement of regulatory agencies on this topic. Engaging regulatory agencies on a WOE would be the first step, ideally once chronic toxicology data are available (and prior to having an ongoing carcinogenicity study since the 3Rs are a component of this process) and can be included in the WOE. If the outcome of a WOE assessment (including any discussion with regulators) is that a carcinogenicity study would not be appropriate, the next step would be to continue monitoring the risk clinically, continue monitoring available literature, and plan for an updated carcinogenicity WOE at the time of intended submission for approval and labeling.
If instead the outcome of a WOE assessment (including any discussion with regulators) is that a carcinogenicity study would be appropriate, the next step would be a carcinogenicity special protocol assessment. This is the process for submission of draft carcinogenicity study protocols for agency review. There is an available guidance when submitting a study protocol to the Center for Drug Evaluation and Research (CDER) at the United States Food and Drug Administration. 17 As outlined in the guidance, while the primary protocol review responsibility lies within the review division, the review division may consult the CDER Carcinogenicity Assessment Committee or Executive Carcinogenicity Assessment Committee. The protocol will be generally reviewed for the appropriateness of the protocol from the agency’s perspective on approaches to in vivo testing, including the study type, doses employed, and other study design aspects. In addition to submitting a study protocol, it is recommended to submit (as available) additional information to aid this assessment: the study report for the preceding dose selection study, information on metabolic profile in humans and in the intended carcinogenicity testing species, toxicokinetic data on the drug and metabolites, pharmacokinetic information on the drug and metabolites in humans, plasma protein binding information for the drug and metabolites in humans and the intended carcinogenicity testing species, and genetic toxicology results summary. The protocol review follows a 45-day review cycle.
In conclusion, in the case of a biotherapeutic, in vivo carcinogenicity assessment may not be logistically feasible based on species cross-reactivity restrictions. Therefore, the weight of evidence assessment may serve as the primary data source used for human risk assessment. As the theoretical carcinogenicity WOE case study presented shows, there can be uncertainty or ambiguity with a WOE assessment, even one containing a wealth of information. Therefore, it is beneficial to sponsors to engage with regulators on this topic in an effort to remove as much uncertainty from the WOE conclusions (and obtain consensus on any actions the WOE outcome might trigger) as feasible.
Preparation of Drug Abuse Liability Assessment Documents
Paul J. Kruzich and David R. Compton
Drug Abuse Liability Assessment (DALA) WOE: Timing and Characteristics
An abuse liability assessment as required by regulatory guidances is similar conceptually to a WOE, as the DALA is a reiterative process with summarization of current data available pertinent to the specific toxicological issue of abuse potential. The goal, similarly, is to determine whether additional data are necessary and more specifically to determine whether specific abuse liability studies are necessary as well as, if so, the study design(s). However, guidances pertaining to abuse liability assessment do not specifically utilize the WOE terminology. These authors contend that the abundance of guidances (see Table 1) supporting the WOE concept are applicable to the DALA process, so we adopt this terminology here as an appropriate descriptor.
The initial submission of the DALA, a document summarizing all available data pertinent to abuse potential, is ideally provided at the end of phase 2 meeting (EOP2) with the Agency. This is so that nonclinical dosing and concentration data can be placed into a proper clinical exposure context (AUC/Cmax) based on doses from the Phase 2 trial. At this time the sponsor should propose for Agency agreement the abuse potential assessment plan moving forward; either (a) no further in vitro or in vivo studies are necessary, or (b) propose the abuse-specific in vivo nonclinical study(ies) to be conducted, while the potential conduct of a human abuse liability (HAL) clinical trial would only be determined in conjunction with agency agreement after abuse-specific nonclinical study results are available. The goal would be to complete all nonclinical DALA studies in time to allow for the possible HAL trial conduct and completion prior to the end of the Phase 3 trials already planned. Key findings, as summarized in a DALA document, leading to heightened concerns regarding potential for abuse and an Agency meeting for concurrence will usually include central nervous system (CNS) activity, CNS relevant clinical signs in animals, and CNS-relevant adverse events in patients in clinical trials. Specific abuse-related studies should not be conducted until after the therapeutic dose range is determined and following discussions and Agency approval of the proposed plan including nonclinical study protocols with detailed dose levels. For clarification, abuse-related studies include intravenous self-administration, drug discrimination, and physical dependence studies, usually in rat, outlined in the abuse liability guidance. 18 The expectancy is the DALA will be an integrated summary of the most current data on chemistry, receptor-ligand binding, PK/TK, metabolism with specific concern for the presence (or not) of major metabolite(s), nonclinical safety studies (safety pharmacology), nonclinical general toxicity studies (focusing on nontoxic dose levels), and characterization of abuse-related adverse events in clinical safety and efficacy studies (Phase 1 through the end of Phase 2).
It is of value to note, regarding nonclinical data, that even exploratory studies assessing possible pharmacological effects of a drug substance other than the eventual therapeutic target can be of value. Early exploration of a novel compound could lead to a variety of assessments such as an analgesic test, despite the fact the compound in question is being developed in clinical trials for a non-pain indication (e.g., asthma). An FDA IND for said compound may not initially include the “off-target” analgesic data as it is not an appropriate efficacy or pharmacodynamic data set pertinent to the target therapeutic indication. However, such data are of significant value to the DALA assessment, and such data (analgesia, locomotor activity, anxiety, etc.) should be considered within Secondary Pharmacology of an FDA IND and certainly should be included in the DALA document. Finally, sponsors should be cognizant of the fact that a ‘DALA’ or ‘DALA document’ does not necessarily have to be an arduous effort resulting in a 100-page product. It could be long, if the data that need to be summarized are extensive, however based on the experience of one author (DRC) a single-page DALA submitted with an NDA can be both sufficient and accepted by the Agency with agreement that ‘no further studies are necessary’. It is, however, best to follow an outline of data (presented below) that should be discussed/addressed by the sponsor in any submitted text/document. The last sentence introduces one more key aspect to the ‘timing’ of a DALA. It is best/optimal for the first version of the DALA to be provided by EOP2, but DALA is an iterative process. A subsequent version may be required when abuse-related nonclinical study results are available, especially if a HAL clinical trial is proposed. A final DALA will be part of the registration submission package summarizing all data to date, including the latest Phase 3 clinical trial information.
Information Used for the DALA
The key criteria affecting the outcome of a DALA document recommendation/conclusion, regarding whether or not additional studies should be performed to adequately determine risk of abuse potential, are provided both below (with detailed discussion) and summarized in Figure 3. This figure also provides indications for whether these criteria are likely to make further studies more, or less, likely. Integration of key WOE factors to inform value of conducting any additional abuse-specific studies for assessment of potential abuse liability. When all the WOE attributes align towards the LEFT of the figure, a conclusion that a study would add value is LESS likely. Alignment to the RIGHT suggests studies are MORE likely. Note the ‘left-less likely’ and ‘more likely’ choices by the authors are reverse of that for the ICHS1B(R1) figure from which this figure is adapted.
Chemistry data provided should include chemical structure, molecular formula and molecular weight (free base and salt, as appropriate), chemistry abstracts service (CAS) number if available, and chemical name. Other useful chemistry information includes description of chemical isomers of the active pharmaceutical ingredient (API), solubility, physiochemical properties, and comparison of the structure of the API with known drugs of abuse. Solubility information is important for determining liability of possible diversion to unintended use via dissolution in commonly available aqueous solvents besides water such as ethanol or other simple organic solvents which are influenced by the physiochemical properties such as melting point, boiling point, dissociation equilibrium, and stability. The degree to which the API can be extracted into ethanol suggests possible abuse pathways via drinking (such as from a ‘punch bowl’ party) or even intravenous injection after either simple aqueous dilution or solid resuspension in aqueous medium after evaporation of the ethanol to recover solid API which could then also be abused via insufflation (nasal inhalation). Similar concerns exist for an API that is water soluble or (besides ethanol) soluble in other commonly available organics. Last, an in silico structural comparison with the API against known drugs of abuse (and armed with solubility and physiochemical information) can provide insight into whether there is a potential for altering the API for illicit use or if the unaltered API shares any molecular properties in common with one or more drugs of abuse which then heightens concerns for a direct potential for abuse of the API. Proprietary software is available for determining quantitative structure-activity relationships (QSAR) between the API and known drugs of abuse. A very common chemical structural evaluation methodology results in a similarity score (to known abused drugs) referred to as the Tanimoto Index or coefficient or simply the Tanimoto similarity, 19 though other methodologies (e.g., PHASE) have been described,20,21 analyzed sets of active compounds selected from scientific articles and showed that a Tanimoto coefficient of ≥0.85 reflected a high probability of two compounds having the same activity. Should the API in question have a high degree of similarity to one or more DEA scheduled compounds, then the issue should be addressed in the DALA.
Ligand binding studies, with a focus on receptors, transporters and/or enzymes, provide information regarding the primary mechanism of action (MOA) and any secondary pharmacology activity at targets associated with drugs of abuse, amongst others. The secondary pharmacology screen generally utilizes in vitro cell culture. There are several commercially available assays for abuse liability screening and contract laboratories that can conduct these assays. To fully address possible abuse liability, these studies of the API should seek to initially describe the binding to, and then understand the activity (agonist/antagonist) at the dopamine, serotonin, GABA, opioids, cannabinoids, and NMDA receptors, ion channels, neurotransmitter transporters (especially the biogenic amines dopamine, serotonin, and norepinephrine), and P-glycoprotein (Pgp) transporter for CNS. A crucial procedural nuance to remember is to ensure that the API is evaluated at a concentration of at least 10 μM at each target. If significant activity is determined, usually considered to be ≥ 50% binding/displacement at a given target, then follow-up assays utilizing an expanded range of concentrations are conducted to determine IC50 or EC50 values at the target and to verify the veracity of the initial screening results plus to better understand mechanisms and abuse potential. Note that one of these authors (DRC) have witnessed examples in which an initial screening assay provided “greater than 50% displacement” at the 10 μM concentration, thus raising a potential concern, but the more thorough IC50 evaluation with an expanded concentration range failed to replicate the screening displacement value observed and therefore the initial concern was unfounded (as the final IC50 was much greater than the 10 μM concentration). Additionally, note that the suggested 10 μM concentration is not a limit per se, and if there is reason to believe the API will attain peripheral and/or CNS concentrations in great excess of this target, then the ligand binding screening concentrations should be similarly increased. Note that one of these authors (DRC) have evaluated an anti-infective API for which systemic concentrations were >30 μM (and CNS concentrations similarly exceeded 10 μM), so the ligand binding screening assay was conducted at much higher concentrations than simply at the 10 μM minimum. The importance of these steps cannot be overemphasized, as they frequently serve as a trigger for additional abuse-related studies, which can impact milestones and timing of drug applications (authors’ individual unpublished observations from over 13 years of experience in abuse liability assessments, including the period between when the 2010 Draft FDA Guidance was available and applied by the FDA prior to the 2017 Final Guidance). The determination of IC50 or EC50 values will help characterize potential risk from CNS penetrance as a function of systemic exposure of the API which may result in possible off-target effects/liabilities relative to therapeutic dose. One may be able to better characterize abuse liability risk based on feasibility or tolerability of the API to achieve secondary pharmacology-relevant concentrations given known side-effect profiles. If the exposure levels of a drug necessary to engage secondary (abuse) targets far exceed what is clinically relevant or even physically tolerated for CNS and peripheral exposure of the API, then this information should be integrated into the DALA and used for an integrative risk assessment.
Pharmacokinetic studies in animals and humans are essential for the DALA. Specifically, peak plasma concentration (Cmax) and time to maximum concentration (Tmax) are important kinetic parameters for drug abuse. The DALA document should present these critical animal data in context of the human equivalent Cmax/Tmax data and do so based on dose comparisons between animals and humans on the traditional AUC basis. That is, express the animal suprapharmacological (but not toxic) dose as a clinical dose multiple in the common AUC-based fashion used in regulatory documents. Then at these specified multiples, compare the Cmax/Tmax data between humans and animals, and discuss the animal Cmax results relative to the ligand binding IC50/EC50 data. Most abused drugs have rapid onset (Tmax) and the interoceptive cue or subjective effects of drugs of abuse are modulated by their plasma concentrations and ability to be absorbed into the brain rapidly. 22 Total exposure (AUC) is also a crucial pharmacokinetic parameter for understanding the relationship between pharmacodynamic effects and possible toxicities gleaned from nonclinical studies and clinical trials. Lastly, note that some indication of CNS penetration, crossing the blood-brain barrier (BBB), is critical for any analysis leading from peripheral PK/TK values to estimates of brain concentration for comparison relative to in vitro ligand binding IC50/EC50. If data show there is no penetration (none whatsoever) of the BBB to the CNS, then a Sponsor has a very short and succinct case to make to health authorities – if the API doesn’t get into the CNS, then the FDA-required DALA assessment is complete and there should be no (or very limited) further assessment required beyond the brief DALA document/text. However, the ‘no penetration’ argument that no further DALA assessment is necessary is one that has failed in the past when even 1% (or less) of the API is present in the brain, though at such small levels the DALA assessment is generally easy to make but must be made/documented by the sponsor. The sponsor cannot simply contend “the regulation isn’t applicable” and then fail to include an explanation in the appropriate section of a filing document or leave that section blank. In such circumstances, brain concentrations become vanishingly small compared to ligand binding at the micromolar level and the sponsor can successfully make the case that ‘no further studies are necessary’ for abuse liability assessment. An example of this by one author (DRC) is the need for some level of sponsor abuse-related assessment of monoclonal antibodies (for additional background, see Ref. 23). Though this is applicable to many (but not all) small molecule and biologic therapies, in those cases where very high blood concentrations are required to be therapeutic (e.g., anti-infective) then even CNS penetration of only 1% to 10% can be comparable to the low micromolar IC50/EC50 values generated in the ligand binding assessment of secondary pharmacology.
Related to the PK/TK characterization of the API is gaining a thorough understanding of any significant drug metabolites. The metabolites relevant for the DALA are drug metabolites identified only in human plasma also known as disproportionate drug metabolites, or metabolites that are ≥10% of the total parent exposure, as measured at steady-state plasma sampling. 24 Characterization of the metabolite also includes assessment of chemistry, secondary pharmacology, and measurement of plasma exposure during safety pharmacology, nonclinical toxicology, and clinical testing. This is conducted in the same manner and to the same extent as for the parent therapeutic API.
Another crucial component of the DALA is an integrated summary of nonclinical observations recorded during development of the API. Specifically, a thorough understanding of single-dose CNS safety pharmacology25,26 and other less subjective assessments (to which the Irwin/FOB [functional observational battery] has been criticized) such as locomotor activity (spontaneous, open-field activity or forced-activity, such as the rotarod). The locomotor activity assessment is equally capable of defining whether an API has any sedative or stimulatory properties. Either points a direction for which further consideration needs to be focused. Also valuable are clinical observations recorded following acute and repeated administration of the API during the exploratory dose-range finding and pivotal IND-enabling toxicology studies. Specific behaviors or observations to scrutinize may include activity, reactivity to handling, interactions with cage mates, physical signs such as Straub tail or hunched posture, stereotypical movements, catalepsy, gait abnormalities (e.g., duck walking), and body temperature changes (core/rectal). Trends in food consumption and body weight changes can be informative of physical dependence, especially in the recovery period after completion of the dosing regimen. Manifestation of physical dependence can be rapid decreases in body weight after acute administration followed by progressive increases over days. If the study includes a recovery period (observations during periods of cessation of administration), attention should be given to alterations in gastrointestinal function (rapid onset of diarrhea correlated with clearance of API), changes in activity (increases or decreases), reactivity to technicians (handling, sounds) and cage mates, or fluctuations in body weight and food consumption as the API clears from previously treated animals.
Abuse-related data from clinical studies that should be incorporated in the DALA include PK data generated from Phase 1 and 2 trials, abuse-related adverse events in safety and efficacy studies, and indications of drug tolerance and withdrawal. The Medical Dictionary for Regulatory Activities (https://www.meddra.org/) contains specific drug abuse, dependence, and withdrawal queries/Standardized MedDRA Query (SMQ) to guide the DALA authors. The proper characterization and classification of abuse-related AE’s is essential for the DALA, as incorrect characterization may result in follow-up requests from the Agency or lack of Agency agreement with considerations and interpretations of AE’s. The AE’s should be interpreted on a weight-of-evidence basis, not a single ‘positive’ event. Single, isolated AE’s can typically be explained in relation to the condition or disease state experienced, plus the therapeutic effect of the API, and abuse potential discounted. As mentioned previously, the DALA process is reiterative. The early version should contain Phase 1 and 2 AE analyses. Subsequent DALA text and/or document at the time of registration filing should provide an overview of additional data gathered during Phase 3. Note that it may not be necessary to rewrite and update the entire clinical section of the prior DALA, but at the very least confirm within the updated DALA that the same analysis as conducted previously for Phase 1and 2 was also performed for Phase 3 trial results and indicate what, if any, change was observed. Be sure to seek agreement with the Agency on this last point as there may be reasons for a complete revision of AE summary tables and a need to include the newer more complete Phase 3 result details.
The final section of the DALA to be addressed is the 8-factor analysis, at least with respect to a US FDA submission. Note that the 8-factor analysis is required by US regulations. As such, some sponsors choose to develop a single DALA document with this component at the very end so that it can be easily removed from the US-specific submission document to create a rest-of-world DALA document. The 8-factor analysis is a medical and scientific evaluation of characteristics of drugs determinative of control under the US Controlled Substance Act (21 U.S.C 811(c)). This information is provided to the US Drug Enforcement Agency (DEA) for the determination of scheduling. Ultimately, the DEA schedules the new chemical entity (NCE) if necessary; meaning placing the NCE in one of five categories restricting use, from Schedule I (heroin, LSD) to Schedule V (cough suppressants). One should be proactive and take this process seriously – delays and disagreement between Sponsor, FDA, and DEA can lead to the loss of patent exclusivity time. Most of the 8-factor analysis information will be understood prior to submission of the final application (back-and-forth with the Agency, comment from regulators, etc.) but still needs to be addressed in the DALA and ultimately scheduling is decided by the DEA.
The 8-factor analysis includes: 1. Its actual or relative potential for abuse 2. Scientific evidence of its pharmacological effect, if known 3. The state of current scientific knowledge regarding the drug or other substance 4. Its history and current pattern of abuse 5. The scope, duration, and significance of abuse 6. What, if any, risk there is to the public health 7. Its psychic or physiological dependence liability 8. Whether the substance is an immediate precursor of a substance already controlled
A full explanation of the 8-factor analysis can be found on the DEA’s website: https://www.dea.gov/drug-information/csa and Section 201 (c), [21 U.S.C. § 811 (c)] of the Controlled Substance Act https://uscode.house.gov/view.xhtml?req=granuleid:USC-prelim-title21-section811&num=0&edition=prelim.
The scheduling of a compound is a complicated process involving a multiplicity of steps not generally common knowledge to those in the pharmaceutical industry. This is an important aspect to be aware of because it represents a significant amount of time which can negatively impinge upon eventual approval and marketing, and thus upon sponsor patent exclusivity. IF the sponsor thinks scheduling is appropriate, then that should be explained clearly in the submission to the FDA. If the FDA concurs, then the process will proceed more quickly. The FDA, part of Health and Human Services (HHS) for which a Presidential Cabinet level Secretary is responsible, will confer on the sponsor/FDA proposed scheduling with experts at the National Institute on Drug Abuse (NIDA) which is part of the National Institutes of Health (NIH), and also part of HHS. However, for FDA to share proprietary sponsor information with NIDA, specific approvals must be granted within HHS to allow the review and there are time limits on this approval. Any delay, and a new approval to continue sharing must be obtained. Any disagreement on scheduling would necessitate extra time to resolve the issue. Once the FDA, with NIDA concurrence, has reached a recommendation for scheduling, then the proposal and the 8-factor analysis are sent to the DEA for review. The DEA is part of the Department of Justice (DOJ) with a Presidential Cabinet level appointee (Attorney General) separate from that of HHS. Official communications between US departments with separate and distinct Cabinet level leadership requires approval at the level of, for example, the undersecretaries of those functions. Recognize that sharing of any information, much less a sponsor’s proprietary information, is carefully controlled within the US government when sharing across differing Cabinet level functions. As stated previously, the DEA ultimately has the final decision on the scheduling of an API. But any difference in opinion with that provided by FDA would require resolution, taking yet more time. The approval by an FDA reviewing division and the marketing of that API as a novel therapeutic medicine cannot occur until after the final DEA scheduling decision. Thus, sponsors want this process to go as smoothly and quickly as possible. There has been an example (as part of public information) of a sponsor losing up to a year of patent exclusivity due to DEA-related scheduling delays prior to FDA marketing approval.
Are There Classes of Drugs for Which It Is Known a Priori that ‘No DALA’ Is Necessary?
No. The FDA Controlled Substance Staff (CSS) has repeated numerous times in multiple public scientific forums on the abuse liability topic, that ‘if the API is a CNS penetrant and crosses the BBB, then it is subject to the abuse liability guidance’ and should be evaluated to the level appropriate. Basically, a reiteration of the guidance itself. There has never been a statement in any forum or document that ‘monoclonal antibodies’ or ‘cancer chemotherapeutics’ were “exempt” from the abuse liability guidance. If an API crosses the BBB, then it is subject to guidance assessment. This is emphasized above regarding compounds for which CNS levels are only 1% (or less) of systemic levels. As described above, this makes the DALA easier and shorter, but an assessment, no matter how brief, is still necessary and one author (DRC) has conducted such assessments for monoclonal antibodies, which were very short assessments (a few pages). One author (DRC) has experience with a small molecule CNS penetrant that required a complete DALA when justification by the sponsor contented ‘no DALA was necessary’ for the NDA. Why would the sponsor think this? The compound in question was an Rx-to-OTC switch (prescription drug being converted to an over-the-counter pharmaceutical). The Rx was one with over 2 decades of clinical trial and post-marketing data with no indication of abuse liability. It was an OTC therapeutic for which multiple other OTC products of the same category (antihistamine) were already marketed without any known abuse potential. Yet, a complete assessment was required to be included in the OTC NDA. Besides these two examples of products requiring a DALA, there is one final class to address: oncology or cancer therapies. Until the 2018 publication of the ICH S9 Q&A (Nonclinical Evaluation for Anticancer Pharmaceuticals Questions and Answers), there was no specific regulatory guidance on the issue of abuse assessment of oncologic agents. The Q&A text is provided below from within section II. Studies to Support Nonclinical Evaluation (2) of that document. Question 2.5 Is there any guidance on the need for abuse liability studies for drugs developed under ICH S9? Answer: Nonclinical studies for abuse liability are generally not warranted to support clinical trials or marketing of pharmaceuticals for the treatment of patients with advanced cancer.
Note that this includes the important word “generally” in the response, as well as “advanced cancer”. Note also that this response indicates that, typically, there is no need for ‘nonclinical studies’ which has been broadly interpreted by those working in this field since 2007 (DRC) to mean there will be (“generally”) no need to conduct ‘abuse-related’ in vivo nonclinical studies (as defined earlier). This does not state that ‘no assessment’ (of currently available data) is necessary. There may be instances when such an assessment is appropriate for an oncologic agent. The authors (DRC) have direct experience with a small molecule oncologic agent, not a classical chemotherapeutic, having binding activity at multiple off-target CNS receptors/transporters of concern. A full DALA was prepared for the NDA filing given concerns for an oral medication in pill form that, though intended for cancer patients, could be diverted and misused/abused by non-patients (the larger concern) as well as potentially misused/overdosed by patients themselves.
Assembling the DALA and Regulatory Steps
When it is time to compile the DALA, combine information from chemistry, pharmacokinetics, nonclinical, clinical, and eight-factor analysis into a DALA document for the agency. Provide the initial DALA document to the Agency for EOP2, if possible. Request that the CSS from the US FDA attend the EOP2 meeting or at least comment on the submitted document. Ask the agency in the questions portion of the briefing book if they agree that no further abuse-related assessment and no additional studies are required (if this is truly the case for the API in question). Alternatively, highlight what specific abuse-related studies may be necessary, and why. Subsequently, if additional studies are warranted, the specific study design including dose selection and rationale will need to be approved by the CSS. Note that, though not known to be the case recently, in the past there has been an instance in which an FDA reviewing division had completed its review and was ready to give approval to market. However, during the process the CSS had not been notified so it was not included in the original review process. CSS became aware of this situation via typical, routine internal FDA communications/notices. Quickly contacting the reviewing division, CSS indicated approval was to be delayed pending their abuse liability assessment. Once completed, the approval process proceeded, but the lack of a single coordinated review resulted in a delay that negatively impacted the sponsor patent exclusivity. So, ensuring CSS is notified and participation requested in the division review process by a sponsor is a valuable effort.
In conclusion, the DALA is a living document that should be actively updated during development and final New Drug Application milestones. The key criteria affecting the outcome of a DALA document recommendation/conclusion, regarding whether or not additional studies should be performed to adequately determine risk of abuse potential, are provided in Figure 3, which also provides indications for whether these criteria are likely to make further studies more, or less, likely. Submit the revised DALA document when scheduled updates are submitted to the Agency and ask for feedback as needed. Anticipate that even if at the EOP2 the Agency has no request for additional DALA assessment, that the DALA document will need to be updated to include additional clinical data from Phase 3 trials. This should complete the process and confirm no further DALA assessment, no nonclinical study, and no HAL trial is necessary. Note that as generating a recompiled MedDRA analysis to include Phase 3 trials can be quite a lengthy addition to an initial DALA document, it is recommended that Agency/CSS agreement is requested to allow update of the DALA document with an appropriate textual summary (only) of the additional Phase 3 analysis conduct and associated conclusion, rather that also including the tabulated results.
Nonclinical Pediatric Development: One Size Does Not Fit All
Susan B. Laffan
Defining the Clinical Pediatric Plan
It is essential to have a pediatric clinical plan well established before any determination can be made whether further nonclinical studies are warranted. Since the clinical situation varies significantly in pediatric medicine, the first step to defining the nonclinical plan is to have a well-defined pediatric clinical plan. A key task is to identify the youngest intended clinical population, which will be specific to that medicine and indication. For instance, a pediatric medicine could be intended to treat patients 12 years and above only, compared to another medicine used only in neonates, or something in between such as a chronically used medicine to treat any patient older than 2 years; hence the approach must be handled on a case-by-case basis. It is even more ideal to have the youngest intended clinical age agreed with at least one major health authority before you get underway with any nonclinical study. An excellent way to establish a clinical pediatric plan is to go through the process of obtaining agreement with health authorities as there are official processes to do so. Pediatric drug development is somewhat unique in this way – that major health authorities have required official procedures to reach agreement on the major components and timing of the clinical and nonclinical pediatric development plans. For the United States Food and Drug Administration (FDA), it is the pediatric study plan (PSP) 27 and for European Medicines Agency (EMA) it is the pediatric investigative plan (PIP). 28 Figure 5 captures the different timing of these procedures. These plans are binding agreements that have weighty financial repercussions and rewards attached to them to incentivize sponsors to make medicines available to all ages for which there is clinical need as soon as practical. These regulatory procedures were established in response to the widespread off-label use of adult medicines in pediatric patients of decades ago.
The procedures for attainment of health authority agreement of the initial pediatric plans are similar for FDA and EMA including the content required for submission. There are important differences in the expected timing against the development of the adult indication, with EMA encouraging submission at end of Ph1, and FDA requiring submission within 60 days (or as agreed) of the End of Ph2 meeting 29 (Figure 5). Within the pediatric plans, the sponsor must address all ages of pediatric population (described in ICH E11) 30 and either include, defer or waive development for every pediatric age category. A rationale for each approach must be provided. It can be the situation that it is a mix of each option for a partial waiver and/or partial deferral; this is actually common. For instance, a typical situation would be a waiver in patients younger than 6 years because there is no clinical need, a deferral for 6 to 12 years, and inclusion of those older than 12 years in Phase 3 ‘adult’ trials so they are included in the initial file for marketing authorization. Even medicines that have no intention to develop for any pediatric patient population should submit a PIP and/or PSP so that agreement of the full waiver is officially obtained. A key component of the dialog is to agree on the youngest intended population – an age that, in the author’s experience, often trends younger after dialog with authorities. The pediatric plan should be based upon current knowledge of the medicine, disease epidemiology and clinical need. An explanation of the structure and contents of regulatory dossiers for can be found here.27,28 For planning purposes, be aware it can take several months in-house to internally agree and draft a pediatric plan, and then the regulatory procedures easily take 9 months or longer.
Deciding Whether Further Nonclinical Juvenile Animal Studies Are Warranted
ICH S11 provides a framework for conducting a weight of evidence evaluation to determine whether further nonclinical studies are warranted and if so, when and how it should be conducted
1
(Figure 4). The framework is set to evaluate different factors independently and when combined provide an integrated evaluation of the total situation taking into account the different weights of the factors. When drafting the nonclinical section of the pediatric plans, it’s helpful to refer to ICH S11 framework and to directly address each of the factors identified in the WOE figure. In the framework, there are two highly weighted factors – the youngest intended clinical population and whether there are any concerns for effects on developing organ systems. Of course, the younger the patients and more identified concerns for developing organ systems, the more likely further studies are warranted, and vice versa. The youngest intended patient population is a key factor, and generally as the age gets younger the level of uncertainty is higher which can highlight a need to conduct a further study. It is notable in the figure that a juvenile animal study is more likely needed if the patient population is a neonate or infant. For the effects on developing organ systems, this should be kept in context for the age group of the clinical trial that is being considered for the WOE (see next section about study timing). These effects could have been evaluated in the existing nonclinical toxicology studies paying close attention to the age of the animals at the beginning and the end of the study. Late developing organ systems to carefully consider are CNS, skeletal, pulmonary, reproductive, and immune. In addition to the developmental tables in the different species in ICH S11, there are several references for each of the major organ systems and species of interest. Useful reviews and reference for the eyes
31
; for the ears
32
; respiratory system33-36; for the cardiovascular system37-39; for the gastrointestinal system40-42; for the renal system42-45; for the immune system46-50; for central nervous system51-59; and for the reproductive systems.60-67 Integration of key WOE factors to inform value of conducting any juvenile animal studies for assessment of potential effects on pediatric patients. When all the WOE attributes align towards the RIGHT of the figure, a conclusion that a study would add value is LESS likely. Alignment to the LEFT suggests studies are MORE likely. From ICHS11. Copyright International Counsel on Harmonisation of Human Pharmaceuticals. The most important factors that should be highly weighted (listed first) are the youngest intended patient age and whether there are suspected adverse effects on developing organ systems of the patients. The other factors are not listed in order of importance.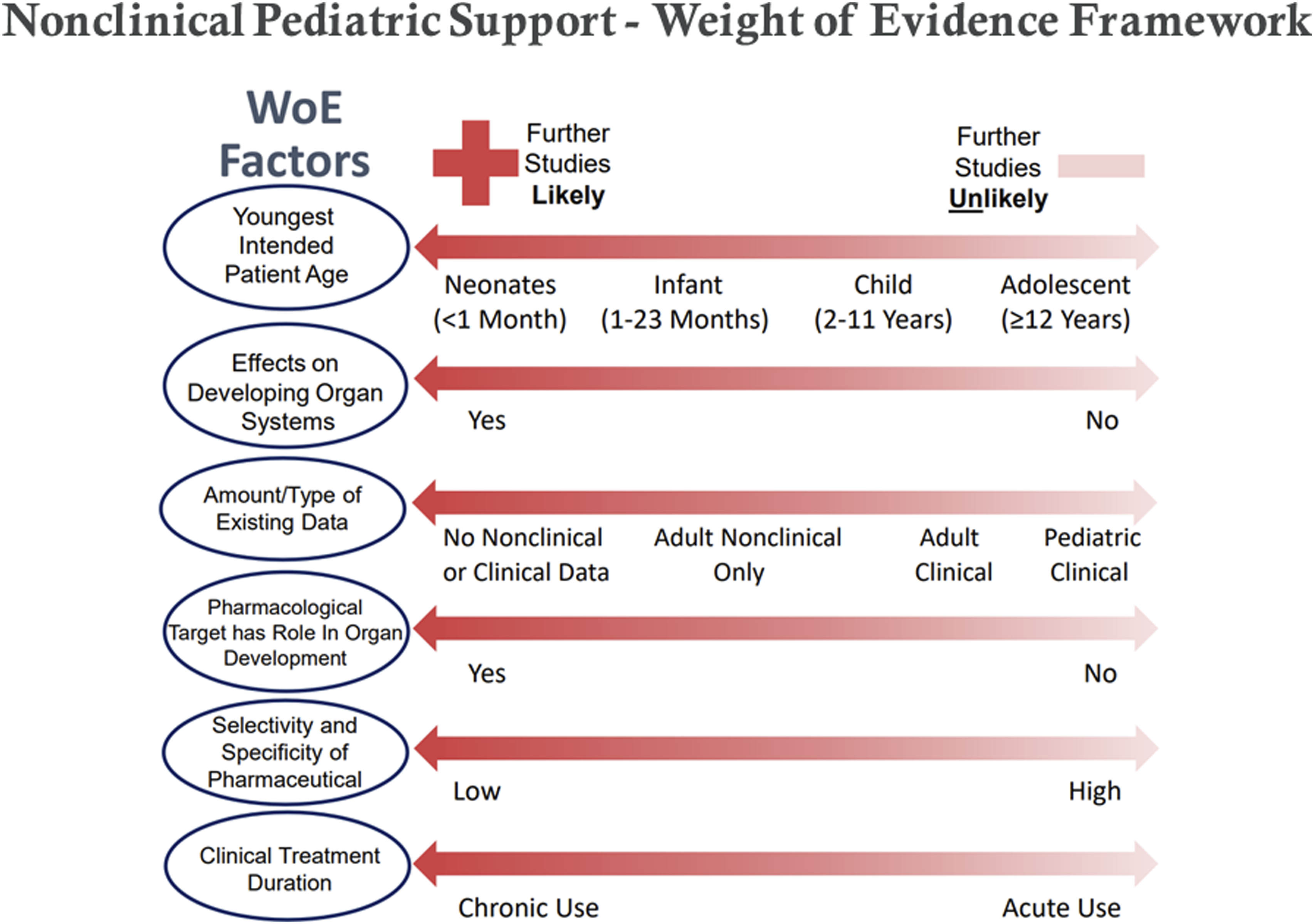
For the WOE factor of ‘Amount and Type of Data’ refer to the entire toxicology data package and whether or not there is any existing clinical data. Any clinical data is paramount to nonclinical data and in pediatric medicine it is not uncommon for off-label pediatric use to be reported in case studies. It’s recommended to consider all data for the class of medicine under development. The strength of the data should also be considered, such as randomized clinical trials vs case studies, or dose range data vs definitive GLP toxicology studies. One exercise that is very important is to consider the starting age of the animal in your ‘adult’ toxicology studies, especially in the non-rodent species. It is quite likely that those studies actually started in immature animals and thus would support the safety evaluation of some of the pediatric population. In many cases, it often provides information to include older children and adolescents in clinical trials. Next, to understand whether a ‘pharmaceutical target has a role in development’, a focused literature review is necessary. If there is a known role in development relevant to the age group included in the clinical trial, it is more likely that further nonclinical studies would be warranted. The ‘selectivity and specificity of a pharmaceutical’ is determined by the modality it uses to modulate the pharmaceutical target, a monoclonal antibody approach would be considered a highly specific modality compared to a small molecule. With so many new and different modalities in drug development today, this is becoming a more important factor. Lastly, for the duration of clinical treatment, a chronic indication taken throughout all pediatric development would increase likeliness of further studies. Note that the clinical duration does not transfer directly to the nonclinical treatment duration because the studies are designed to cover a developmental window relevant to the age range, which varies greatly because development is very contracted in most nonclinical species tested, especially rodents compared to humans.
Timing of Nonclinical Studies
The WOE evaluation can be done for the whole pediatric program to know if a study is needed at the youngest intended age; however, as explained earlier, it is common for the pediatric plan to have some age groups deferred until after filing for marketing approval. If this is the case, then the timing of the pediatric trials is critical to the timing of the nonclinical studies. A WOE evaluation should be done for each pediatric trial should be conducted to inform on which trial would need further nonclinical data. For instance, in the example provided above, studies may not be warranted for inclusion of patients 6 years old and older but may be warranted for those 2 to 6 years old. Likely the clinical trials will be conducted in a staggered fashion as well, so the nonclinical studies would be needed to support the last clinical pediatric trial in 2- to 6-year-old.
Study Design of Nonclinical Studies
The WOE outcome that supported the conduct of the study should also influence the design of the study. For instance, if a study was warranted because of the very young pediatric population without any specific organ system concern, then the study should start at a comparative age and conduct just the core endpoints as described in ICH S11. Again, to choose the starting age of the animals, the reference for comparative development should be utilized. If there was a specific identified concern with the immune system or skeletal system, 68 then additional study endpoints to properly assess those systems should be included. The core and additional endpoints are described in detail in the guidance. Juvenile animal studies are complex and rodent studies can get very large due to the litter size; thus, every additional endpoint should be scientifically justified in the pediatric plan.69-73 Since systemic exposure can be difficult to predict in young animals, dose range studies in preweaning rodents are highly encouraged. 74 It is good practice to confirm the starting age of the animals in relation to the agreed youngest intended clinical age before initiating the dose range study since dose ranging at a different (typically younger age) than the definitive study is not recommended. If the study is in the rat, there is an ICH S11 figure which directly compares the organ system developmental timing with human.
In conclusion, it simply is not possible to know whether a juvenile animal study is warranted until the pediatric clinical plan is known. Somewhat unique for pediatric medicines, there are specific regulatory procedures to agree on the clinical and nonclinical plans. A key aspect to agree is the youngest intended pediatric population which is a major factor in the weight of evidence to inform whether a study is needed. There is no default expectation that a juvenile animal study would be warranted. The decision whether to conduct a study, and if so, the timing and design of that study are decided on a case-by-case basis on the situation of that medicine. One size, indeed, does not fit all.
Overall Conclusion
Weight of evidence (WOE) is an inherent part of the role of a toxicologist in risk assessment – to assess all available data and make a judgment (or at least provide an informed opinion) on the level of risk. A WOE is typically a written document summarizing the currently known facts pertinent to a specific toxicological issue, a judgment of their robustness and relevance to the pharmaceutical in development and a final assessment based on scientific judgment of whether the concern is sufficiently characterized and can be appropriately managed going forward to the next clinical development stage. Timing of initial WOE compilation and update/revision can vary depending on the specific toxicological issue (Figure 5). One possible WOE outcome is a conclusion that additional nonclinical studies are not justified and would avoid an unnecessary use of animals consistent with the 3R’s concept (while acknowledging that the data may also indicate that additional animal studies are justified/necessary). It is also very important that the weight of evidence includes presentation and consideration of all available data, not just the data that supports a desired conclusion/position (avoids bias). The WOE concept is one well founded in regulatory guidances as summarized relative to carcinogenicity, abuse liability and pediatric safety. Suggested timing of health authorities interactions for carcinogenicity assessment, drug abuse lability assessment and pediatric development for typical medicine development programs. EoP2 is the critical actionable phase.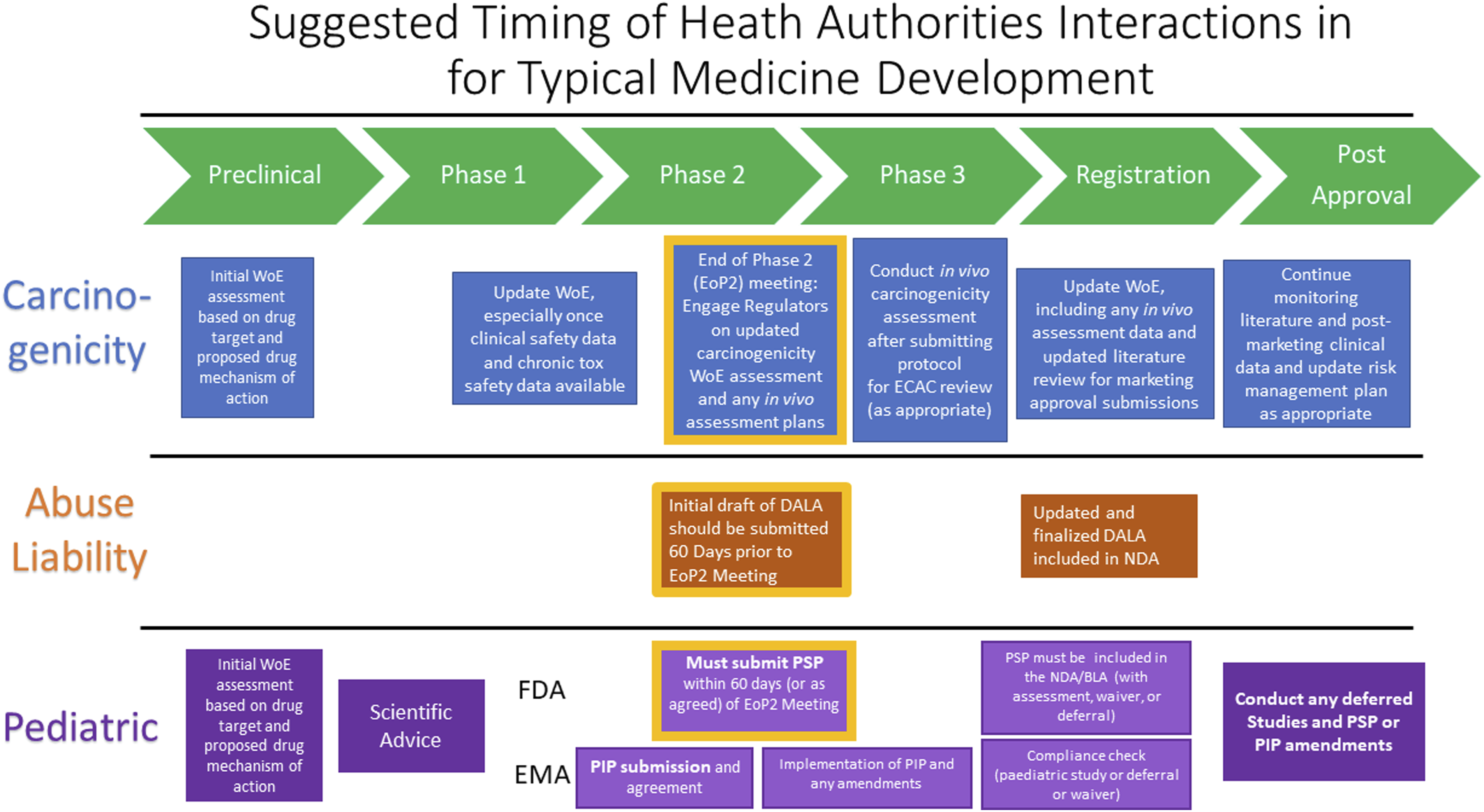
CDER Perspective
To summarize, many CDER guidances make reference to the use of WOE assessments. This is primarily in the context of (1) assessing developmental and reproductive toxicity risk, (2) assessing immunotoxicity risk, (3) assessing genotoxicity risk, (4) assessing carcinogenicity risk, (5) assessing the need for dedicated specialized toxicity studies (e.g., juvenile animal study [JAS], immunotoxicity, abuse liability). In order to help reduce unnecessary animal use, those developing pharmaceuticals should routinely consider whether there are sufficient data to inform and communicate risk in any of these areas before defaulting to conducting an animal study, especially for studies involving non-human primates or large number of animals.
Carcinogenicity
In the case of a biotherapeutic, in vivo carcinogenicity assessment may not be logistically feasible based on species cross-reactivity restrictions. Therefore, the weight of evidence assessment may serve as the primary data source used for human malignancy risk assessment. As the theoretical carcinogenicity WOE case study presented shows, there can be uncertainty or ambiguity stemming from a WOE assessment, even one containing a wealth of information. Therefore, it is beneficial to sponsors to engage with regulators on this topic in an effort to remove as much uncertainty from the WOE conclusions (and obtain consensus on any actions the WOE outcome might trigger) as feasible.
Abuse Liability
In conclusion, a strong understanding of the chemistry, ligand binding, CNS penetrance, TK/PK, nonclinical observations, clinical adverse effects, and potential class effects is necessary to determine the need for further abuse liability studies. These key criteria affecting the outcome of a DALA document recommendation/conclusion, regarding whether or not additional studies should be performed to adequately determine risk of abuse potential, are provided in Figure 3, which also provides indications for whether these criteria are likely to make further studies more, or less, likely. It is requisite to compile this information and gain agreement with the Agency regarding strategies for inclusion or exclusion of additional abuse relevant studies in a development package.
Pediatric
In conclusion, it is not an expectation that a juvenile toxicology animal study is needed a priori. Instead, each situation has to be assessed for the specific medicine in development. It simply is not possible to know whether a juvenile animal study is warranted until the pediatric clinical plan is known. Somewhat unique for pediatric medicines, there are specific regulatory procedures to agree on the main components and timing of clinical and nonclinical development plans. A key aspect to have regulatory agreement on is the youngest intended pediatric population, which is a major factor in the weight of evidence to inform whether a juvenile animal study is needed. Therefore, the sooner these regulatory processes are initiated, the smoother and more efficient the development plans are likely to be. To reiterate, there is no default expectation that a juvenile animal study would be warranted. The decision whether to conduct a study, and if so, the timing and design of that study are decided on a case-by-case basis on the situation of that medicine. The major factors in the WOE are the youngest intended clinical population and whether there are any concerns for effects on developing organ systems; other factors are based on the amount and type of available data, whether a ‘pharmaceutical target has a role in development, the selectivity and specificity of a pharmaceutical and the duration of clinical treatment. If the WOE suggests a juvenile animal study should be conducted it is important to incorporate the concerns identified in the WOE into the study design. The study design should also be customized to the medicine being development, ICH S11 offers specific considerations for all major aspects of study design. With such a large variety of new pharmacologic targets and modalities being developed it really is true that one size, indeed, does not fit all.
Overall, it’s very clear in today’s environment of so many novel therapeutic targets with an exciting variety of novel modalities that the previously established ways of assessing safety with default study designs at default times in development are no longer sufficient. In more cases, a robust WOE is needed to evaluate key components of the new pharmaceutical in the context of use related to the concern of interest. In this paper, detailed information is provided on how to conduct a WOE for carcinogenicity, abuse liability concerns and pediatric development. It is also reasonable to encourage sponsors to offer the benefits of right of reference to provide key evidence to WOE for other similar medicines, this can enable better medicines to patients quicker using fewer animals – a goal we all share.
Footnotes
Acknowledgments
Dr. Wendy Halpern provided collation of useful references and information to support comparative animal-to-human developmental biology for the pediatric section.
Author’s Note
This publication reflects the views of the authors and should not be construed to represent FDA’s views or policies. The views expressed are those of the authors and do not necessarily reflect the views or policies of their affiliated organizations.
Author Contributions
All authors contributed to conception and design, drafted the manuscript, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.