
Abstract
The iminosugar UV-4 is a broad-spectrum antiviral drug candidate with activity in vitro and in vivo against multiple, diverse viruses. The toxicological profile of UV-4, dosed as the hydrochloride salt, was evaluated in single-dose and repeat-dose oral toxicity studies in mice, rats, dogs, and non-human primates (NHP). No moribundity or deaths were associated with the drug up to the maximum tolerated dose. No treatment-related adverse effects were observed following single oral doses in dogs, rats, and mice up to 250, 400, 1000 mg/kg, respectively, and in NHP up to 180 mg/kg administered three times daily for 10 days. UV-4-related findings were generally seen at higher doses after 7- or 14-day exposure. The most common clinical pathology findings (increase in aspartate aminotransferase and decreased platelet count) were consistently found across species and each appeared dose related. The kidney, mesenteric lymph nodes, stomach including gastrointestinal tract, and thymus were identified as target organs in mice, rats, and dogs. In 14-day repeat-dose toxicology studies in mice and dogs conducted in compliance with Good Laboratory Practice regulations, the dog was considered to be the most sensitive species to UV-4 exposure based on the treatment-related adverse effects noted in the identified target organs. The results of these studies demonstrate the safety profile of UV-4 hydrochloride and supported the selection of starting and maximal doses for a single ascending dose first-in-human clinical study.
Introduction
Iminosugars are being investigated for their potential use as host-targeted broad-spectrum antiviral therapeutics mediated via a mechanism involving inhibition of endoplasmic reticulum (ER) α-glucosidases leading to misfolding of critical viral.1,2 The misfolding of viral glycoproteins reduces infectivity by producing defective virus particles or targeting the glycoproteins for degradation. Drug candidates targeting host proteins critical for viral replication often have broad-spectrum activity as multiple viruses are dependent on the same host factors for replication. By targeting the activity of a host protein such as ER α-glucosidases, iminosugars do not expose the virus to direct selective pressure, which has been shown to result in low likelihood of development of resistance.3,4,5 The recent coronavirus pandemic has highlighted the importance of broadly active, host-targeted, and orally bioavailable antiviral drugs as an important strategy to combat future viral pandemics. 6
The iminosugar UV-4 inhibits the activity of both ER α-glucosidases I and II and is a potent, host-targeted antiviral (HTAV) candidate with demonstrated in vitro activity across a diverse set of enveloped viruses. 7 UV-4, and its hydrochloride salt form UV-4B, is potent against diverse dengue virus strains in vitro and promotes complete survival in a lethal dengue virus mouse model.7,8 UV-4 also provides survival benefit in lethal and sub-lethal mouse models of influenza A and B. 9 UV-4 is highly efficacious via oral gavage against both oseltamivir-sensitive and oseltamivir-resistant influenza A (H1N1) infections in mice, even when treatment was initiated as late as 48–72 hours after infection.9,10 Lack of resistance following treatment with UV-4 has been demonstrated for dengue virus in vivo and influenza virus in vitro.3,4
The safety of antivirals targeting host cellular pathways has not been well established as most antiviral therapeutics act on essential viral components (direct-acting antivirals). It has been proposed that HTAVs may have increased risk of toxicity due to disruption of cellular pathways in both infected and healthy cells, 6 although this has generally not been confirmed in investigational new drug (IND)-enabling studies or human clinical trials. Only a few host-factor therapeutics have been approved by the FDA for viral diseases, and most are based on interferons targeting chronic infections.6,11 The α-glucosidase inhibitor celgosivir, a HTAV under development for dengue virus infection, was generally well tolerated in preclinical and Phase 1 clinical studies and however was not effective in reducing viral load or fever in patients. 12 Nonclinical studies conducted to evaluate the safety of UV-4 prior to initial clinical safety testing are described in this paper. The toxicology program for UV-4 conformed to ICH M3(R2) to support the Phase 1 clinical trial. The toxicological profile of UV-4 has been evaluated in non-GLP acute oral toxicity studies in mice, rats, dogs, and NHP; in non-GLP repeat-dose toxicity studies in mice, rats, and dogs up to 7 days; and in GLP repeat-dose toxicity studies in mice and dogs up to 14 days.
Materials and Methods
Test Article
UV-4 (MON-DNJ; N-(9-methoxynonyl)-1-deoxynojirimycin; (2R,3R,4R,5S)-2-(hydroxymethyl)-1-(9-methoxynonyl)piperidine-3,4,5-triol; CASRN 615253-61-7) has a chemical structure consisting of a 1-deoxynojiriimycin ring and a 9-carbon alkyl methoxy side chain attached to the nitrogen of the piperidine ring. UV-4 hydrochloride salt (UV-4B; CASRN 1333144-07-2) has higher aqueous solubility than the free base (solubility in water >2.4 g/mL), which is likely to impart improved dissolution characteristics for an eventual drug product, and more reliable bioavailability in humans. The concentrations and doses used in all studies were calculated and expressed as the active free base form, UV-4. This methodology is consistent with USP<1121>, which recommends that the strength of a drug product or preparation is expressed in terms of the active moiety.
Test Article Preparation
For all studies described here, except where noted, UV-4 hydrochloride salt was dissolved in reverse osmosis (RO) water to the desired concentration. The correction factor for calculation of UV-4 dose included where appropriate a correction for salt weight and assayed purity of each batch of UV-4 hydrochloride. All dose formulations were stored in a refrigerator, set to maintain 2 to 8°C (unless made on the day of dosing), until removed for dosing. Dose formulations prepared on the day of use were stored at room temperature prior to dosing. Formulations were stored protected from light.
Animals
Studies were conducted using Crl:CD1(ICR) mice (Charles River Laboratories, Portage, MI), Crl;CD (SD) rats (Charles River Laboratories, Portage, MI), beagle dogs (Advinus Therapeutics Limited, Bangalore, India, and Covance Research Product Inc., Denver PA), and cynomolgus macaques (Primate Products, Inc., Immokalee, FL). Male and females of each species were used in most studies except where noted. The Institutional Animal Care and Use Committee at each testing facility approved the use of animals prior to study initiation. All work was performed in compliance with the Animal Welfare Act and followed the principles outlined in the National Research Council’s Guide for the Care and Use of Laboratory Animals. 13 Non-GLP studies in the testing program included initial maximum tolerated dose (MTD) studies in mice, dogs, and NHP, single dose escalation and 7-day repeat dose range studies in mice, rats, and dogs, and 5- and 10-day repeat dose study in NHP. IND-enabling GLP studies in the testing program were the 14-day toxicity and toxicokinetic studies in mice and dogs.
Study Designs and Treatments for Oral Toxicity Studies in Mice, Rats, Dogs and NHP
The study designs, dose regimens and assessments for each of the seven studies conducted to evaluate UV-4 safety are shown in Table 1.
Toxicology Study Designs, Dose Regimens, and Assessments.
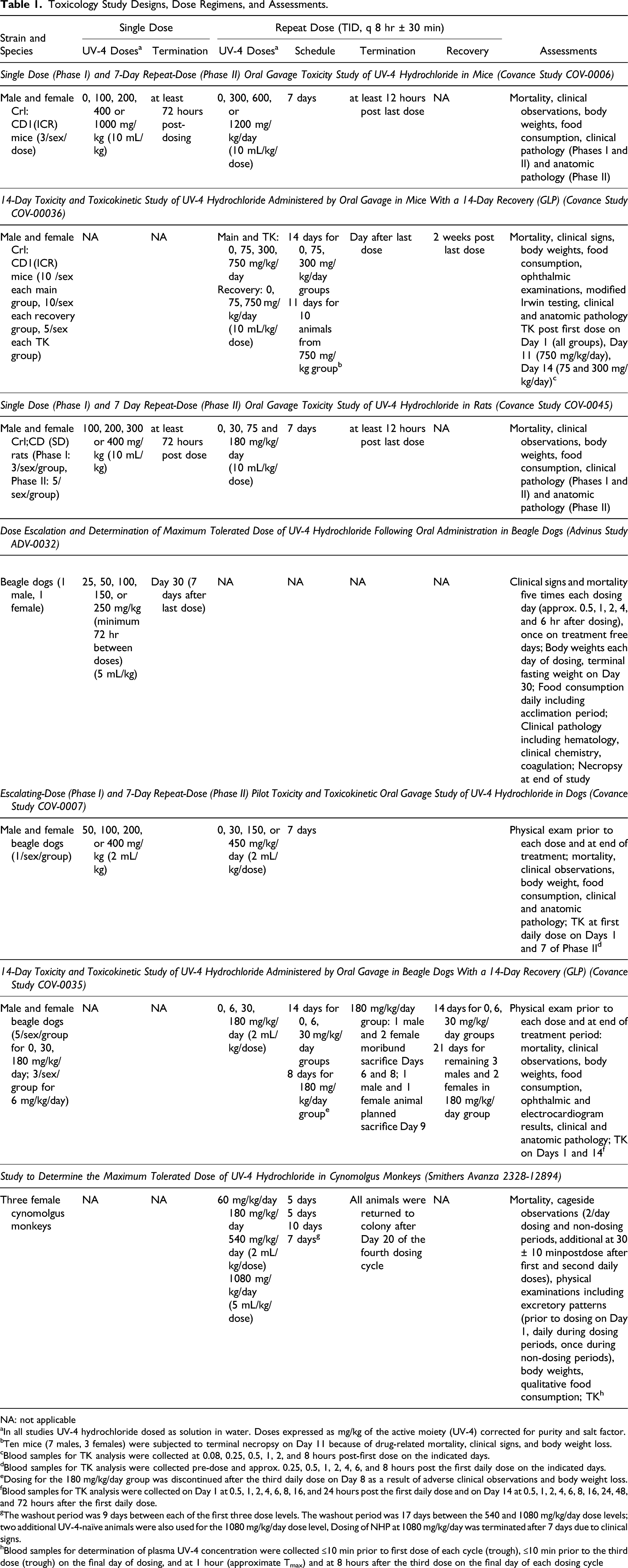
NA: not applicable
aIn all studies UV-4 hydrochloride dosed as solution in water. Doses expressed as mg/kg of the active moiety (UV-4) corrected for purity and salt factor.
bTen mice (7 males, 3 females) were subjected to terminal necropsy on Day 11 because of drug-related mortality, clinical signs, and body weight loss.
cBlood samples for TK analysis were collected at 0.08, 0.25, 0.5, 1, 2, and 8 hours post-first dose on the indicated days.
dBlood samples for TK analysis were collected pre-dose and approx. 0.25, 0.5, 1, 2, 4, 6, and 8 hours post the first daily dose on the indicated days.
eDosing for the 180 mg/kg/day group was discontinued after the third daily dose on Day 8 as a result of adverse clinical observations and body weight loss.
fBlood samples for TK analysis were collected on Day 1 at 0.5, 1, 2, 4, 6, 8, 16, and 24 hours post the first daily dose and on Day 14 at 0.5, 1, 2, 4, 6, 8, 16, 24, 48, and 72 hours after the first daily dose.
gThe washout period was 9 days between each of the first three dose levels. The washout period was 17 days between the 540 and 1080 mg/kg/day dose levels; two additional UV-4-naïve animals were also used for the 1080 mg/kg/day dose level, Dosing of NHP at 1080 mg/kg/day was terminated after 7 days due to clinical signs.
hBlood samples for determination of plasma UV-4 concentration were collected ≤10 min prior to first dose of each cycle (trough), ≤10 min prior to the third dose (trough) on the final day of dosing, and at 1 hour (approximate Tmax) and at 8 hours after the third dose on the final day of each dosing cycle
Parameters
Mortality, clinical observations, body weights, and food consumption
All animals were checked twice daily (a.m. and p.m.) for mortality, abnormalities, and signs of pain or distress. Multiple (2-5) cageside observations were made on the day of dosing for the single dose escalation studies. Otherwise, cageside observations were performed once daily during the dosing and recovery phases, except on days when detailed observations were conducted. Detailed observations were performed once during the predose phase (all animals), prior to the first daily dose on Day 1, weekly throughout the dosing and recovery phases, and on the day of scheduled sacrifice. The frequency of detailed observations was increased if adverse clinical signs were observed. Body weights were recorded prior to dosing and on the day of scheduled necropsy in the single dose escalation studies. For the 2-week toxicity studies, body weights were also recorded prior to the first dose and then weekly throughout the dosing and recovery phases. Food consumption was quantitatively assessed daily during the dosing and recovery phase.
Modified Irwin screening conducted in the in the 14 day repeat dose mouse study is reported in the companion safety pharmacology paper. 14
Ophthalmoscopic examination
In the 2-week repeat dose mouse and dog studies, ophthalmic examinations were performed during the predose, dosing and recovery phases. The eyes were dilated with a mydriatic agent prior to examination. Examinations were conducted by a board-certified ophthalmologist using an indirect ophthalmoscope.
Electrocardiogram measurements
Electrocardiogram (ECG) measurements in the dog 14-day repeat dose toxicology study were conducted once during the predose phase, 1 to 2 hours following the first daily dose during on Day 9 of the dosing phase, and once on Day 13 of the recovery phase for the 0 (control), 6, or 30 mg/kg/day dose groups, and once on Day 19 of the recovery phase for the 180 mg/kg/day dose group. Eight-lead ECGs were recorded. At least 5 consecutive waveforms were used for quantitative analysis. Routine quantitative measurements of ECGs were made on a single lead. Parameters reported included heart rate, ventricular depolarization (QRS duration), time from the beginning of the QRS complex to the end of the T wave (QT interval), the time elapsed between 2 successive R-waves of the QRS signal (RR interval), and the time from the depolarization of the sinus node to the onset of ventricular depolarization (PR interval). The heart rate-corrected QT (QTc) interval was calculated using the Fridericia method. A qualitative review for rhythm abnormalities and disturbances of the collected ECGs was performed.
Clinical Pathology
Mice were nonfasted, while rats, dogs and NHP were fasted overnight prior to blood collection. Blood was collected from mice and rats only at necropsy. Blood was collected from dogs and NHP prior to dosing, at multiple time points during the dosing and recovery phases and at necropsy. All animals were anesthetized with sodium pentobarbital for blood collection at the time of scheduled necropsy. Blood was collected by cardiac puncture (mice), from the vena cava (rats), cephalic or jugular vein (dogs) and femoral vein (NHP). The anticoagulant was potassium ethylenediamine tetraacetate (EDTA) for hematology tests and sodium citrate for coagulation tests. Samples for clinical chemistry were collected without anticoagulant. Samples for clinical chemistry were collected with lithium heparin for the initial dog dose escalation study.
A standard panel of hematology, clinical chemistry, coagulation, and urinalysis parameters were measured in all 4 species according to OECD guidelines. 15
Pathology, gross necropsy, organ weights tissue collection and histopathology
Animals euthanized at unscheduled timepoints and surviving until the time of scheduled necropsy were anesthetized with sodium pentobarbital, exsanguinated, and necropsied. Pathology, gross necropsy, organ weights, tissue collection and histopathology were conducted according to OECD guidelines 16 .
Toxicokinetics
The time points for blood sample collection in each study are shown Table 1. Blood samples for toxicokinetic (TK) analysis were collected into tubes containing EDTA as anticoagulant. UV-4 and an internal standard (hexadeuterated UV-4) were extracted from plasma samples using protein precipitation. The supernatant was diluted and analyzed using a validated liquid chromatography with tandem mass spectrometric detection. Toxicokinetic evaluations including maximum observed concentration (Cmax), time to peak concentration (Tmax), and area under the concentration-time curve (AUC0-8) were calculated using a noncompartmental analysis tool of WinNonlin Enterprise software (version 5.2). Only the AUC0-8 were calculated for the 14-day mouse and 7-day dog repeat dose studies since samples were only collected during the first 8 hours after the first dose on blood collection days.
Statistics
For mouse and rat studies, data for each sex were analyzed separately; only data collected on or after the first day of dosing were analyzed statistically. Analysis of variance (ANOVA) 17 and pairwise comparisons were used to analyze absolute body weight, body weight change, terminal body weight, quantitative food consumption, continuous clinical pathology values, absolute organ weight, organ-to-body weight percentage, and organ-to-brain weight percentage. Levene’s test18,19 evaluated equality of variances between groups. Where Levene’s test was significant (P < 0.05), a rank transformation (to stabilize the variances) was applied before the ANOVA was conducted (the Levene’s test was not applied to the rank-transformed data). Where Levene’s test was not significant (P > 0.05), ANOVA was conducted. One-way ANOVA was used (if applicable) to analyze the data. If the group effect of the ANOVA was significant (P < 0.05), Dunnett’s t-test20,21 was used for pairwise comparisons between each treated and control group. If the ANOVA was not significant (P > 0.05), no further analyses were conducted. Statistical analysis was not performed for the dog and NHP dose escalation studies due to the small sample size. Statistical analysis was limited to the calculation of means and standard deviations for the dog 14-day dose escalation study.
Results
Single Dose (Phase I) and 7-Day Repeat-Dose (Phase II) Oral Gavage Toxicity Study of UV-4 Hydrochloride in Mice
Phase I (Mouse Single Dose)
All mice in Phase I survived to their scheduled necropsy with no UV-4 related clinical signs, and no macroscopic observations were noted at necropsy. Administration of 1000 mg/kg UV-4 was associated with decreased body weight without a change in food consumption. Moderately lower absolute reticulocyte counts, moderately lower platelet counts, mildly lower globulin concentration (males only), and mildly higher triglyceride concentration (females only) were also observed in the 1000 mg/kg UV-4 dose group.
Phase II (Mouse 7-Day Repeat Dose)
Table 2 shows in-life and clinical pathology findings identified as toxicologically important in Phase II (7-day repeat dose) mouse study. No mortality or abnormal clinical observations were observed in mice administered ≤600 mg/kg/day of UV-4. Two males and one female administered 1200 mg/kg/day were euthanized in moribund condition on either Day 3 or Day 4. The cause of moribundity was undetermined, with clinical signs including hypoactivity, hunched appearance, few feces, and cold to touch-entire body. Decreased body weight was observed in surviving mice administered 600 and 1200 mg/kg/day. Decreased food consumption was observed in females administered 600 mg/kg/day and in males and females administered 1200 mg/kg/day. Clinical observations in surviving mice in the 1200 mg/kg/day dose group included a low incidence of few feces and discolored (brown) hair coat in the perineal area. UV-4 related effects included mildly to moderately lower red blood cell mass (i.e., lower red blood cell count, hemoglobin, and hematocrit), absolute reticulocyte count, platelet count and mildly to moderately higher aspartate aminotransferase (AST) activity. The findings were generally observed at doses ≥ 600 mg/kg/day and were not uniformly observed in both sexes. The mild to moderately lower red blood cell mass was also observed in males at the 300 mg/kg/day dose. UV-4 related microscopic changes in Phase II mice administered ≥ 600 mg/kg/day included changes in the kidney (necrosis of the papilla, degeneration/necrosis of the renal tubules, tubule cell vacuolation, and dilation of the tubules) mesenteric lymph node (depletion/necrosis of lymphocytes), spleen (depletion/necrosis of lymphocytes), and stomach (vacuolation of parietal cells and erosion). Minimal or slight tubule cell vacuolation in the kidney was also observed in 2 females administered 300 mg/kg/day. Based on severity and incidence, none of the changes were considered dose limiting. The 1000 mg/kg single dose and 1200 mg/kg/day dose level were considered to have exceeded the MTD for studies of longer duration.
In-Life and Clinical Pathology Summary in the 7-Day Repeat-Dose (Phase II) Oral Gavage Toxicity Study in Mice.
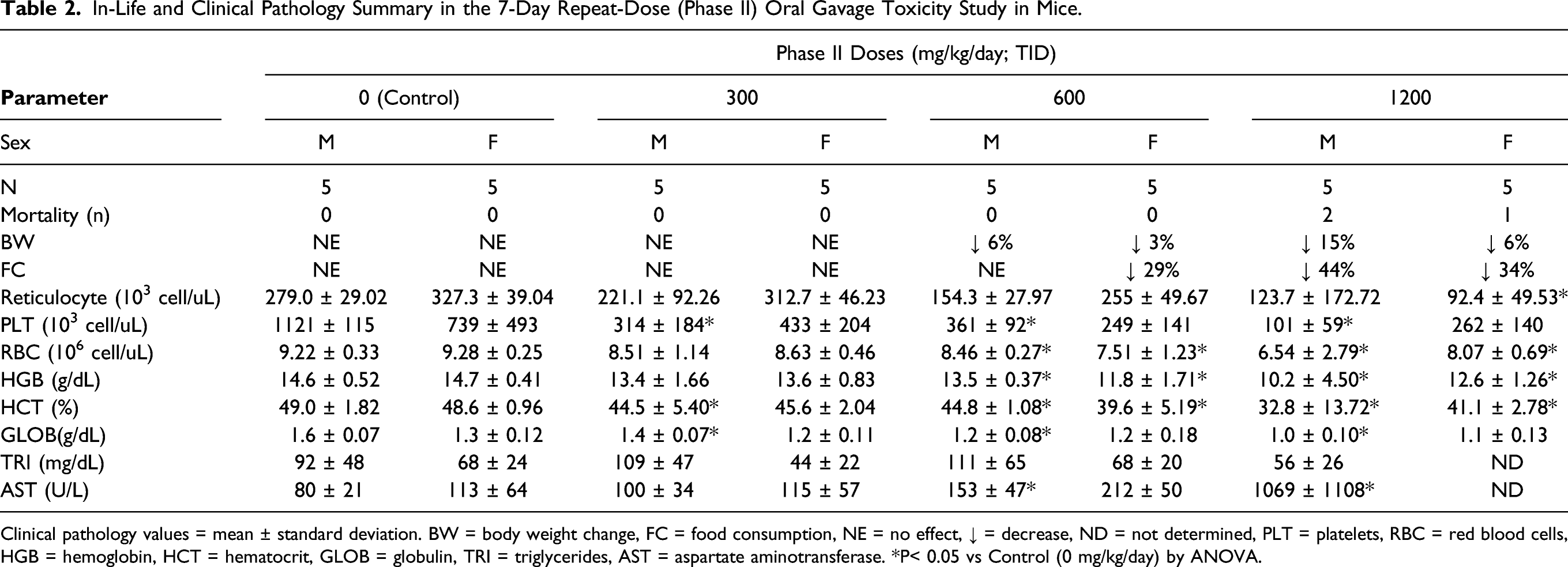
Clinical pathology values = mean ± standard deviation. BW = body weight change, FC = food consumption, NE = no effect, ↓ = decrease, ND = not determined, PLT = platelets, RBC = red blood cells, HGB = hemoglobin, HCT = hematocrit, GLOB = globulin, TRI = triglycerides, AST = aspartate aminotransferase. *P< 0.05 vs Control (0 mg/kg/day) by ANOVA.
14-Day Toxicity and Toxicokinetic Study of UV-4 Hydrochloride Administered by Oral Gavage in Mice with a 14-Day Recovery (GLP)
Table 3 shows in-life and clinical pathology findings that were identified as toxicologically important in the 14-day repeat dose mouse study. Two out of ten 750 mg/kg/day toxicity males were euthanized in moribund condition prior to the scheduled dosing phase necropsy. In addition, one 750 mg/kg/day toxicity male was euthanized in moribund condition on Day 2 of the recovery phase. These early deaths were considered UV-4 related, although the cause of death for these mice could not be determined based on microscopic examination. Four mice had unscheduled deaths attributed to gavage trauma confirmed at necropsy: two females at 0 mg/kg/day, one female at mg/kg/day, and one female at mg/kg/day. During the dosing phase, UV-4 related clinical signs, which were noted only in the 750 mg/kg/day mice, included fecal abnormalities (few, nonformed, or red discolored feces; presence of occult blood was not measured), and red discharge from the rectum), squinted eyes, pale appearance (ears and/or body), and rough haircoat (head and/or body). During the recovery phase, UV-4 related clinical signs of hunched appearance, nonformed feces, rough haircoat, swollen ventral midline, and squinted eyes were observed in the 750 mg/kg/day male euthanized on Day 2. In other males and females at 750 mg/kg/day, additional clinical signs that were present during the dosing phase and persisted during the first week of the recovery phase included hunched appearance (males only during dosing phase) and pale appearance (ears and/or body); these clinical signs had resolved by the second week of the recovery phase.
In-life and Clinical Pathology Summary in the 14-Day Toxicity and Toxicokinetic Study in Mice With a 14-Day Recovery.
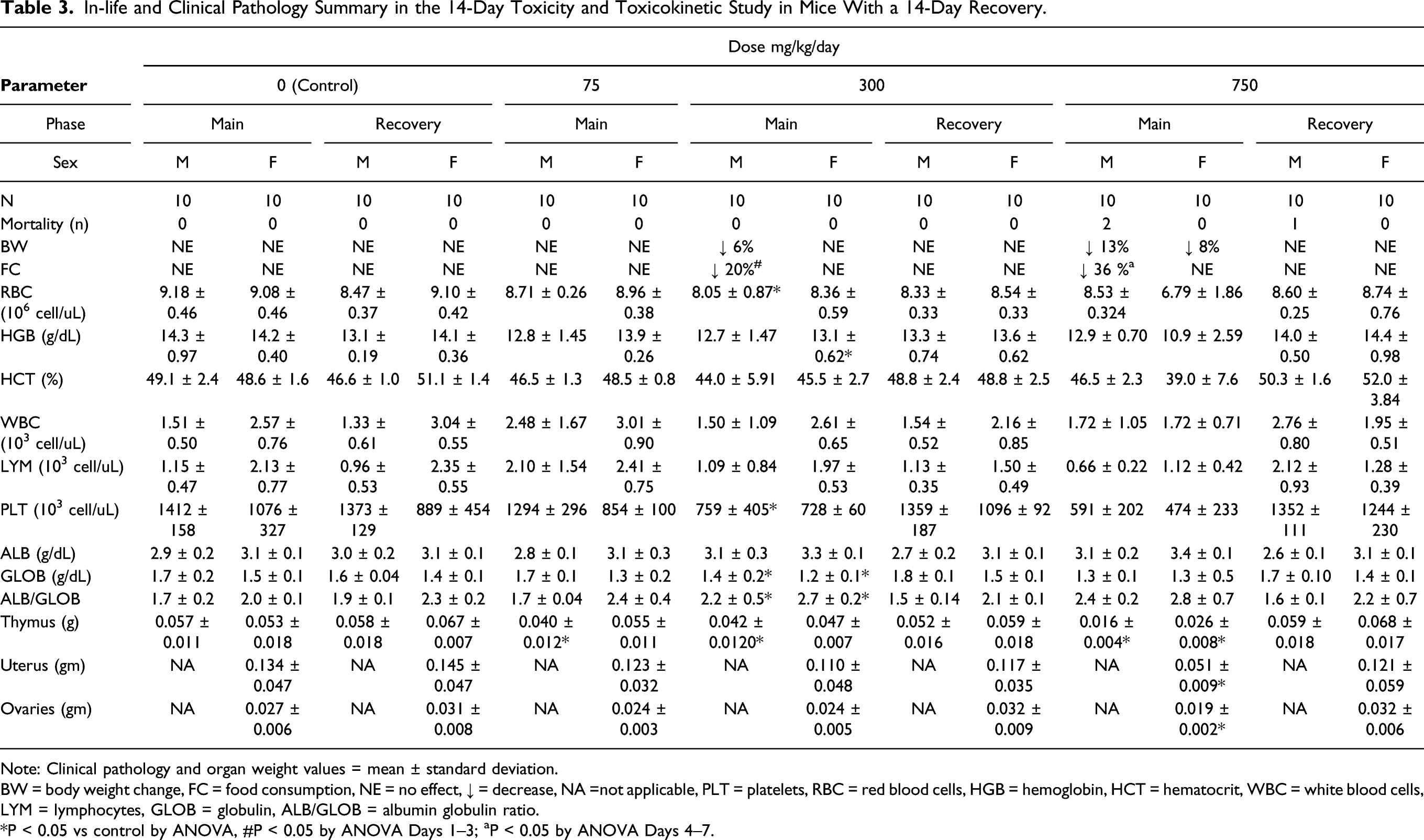
Note: Clinical pathology and organ weight values = mean ± standard deviation.
BW = body weight change, FC = food consumption, NE = no effect, ↓ = decrease, NA =not applicable, PLT = platelets, RBC = red blood cells, HGB = hemoglobin, HCT = hematocrit, WBC = white blood cells, LYM = lymphocytes, GLOB = globulin, ALB/GLOB = albumin globulin ratio.
*P < 0.05 vs control by ANOVA, #P < 0.05 by ANOVA Days 1–3; aP < 0.05 by ANOVA Days 4–7.
A UV-4 related, decrease in mean body weights was observed in male mice administered 300 and 750 mg/kg/day. The progressive decrease in body weight and magnitude of the change in male mice were considered to be adverse at 750 mg/kg/day. The decreased body weight and corresponding decrease in food consumption in the males administered 300 mg/kg/day (P ≤ 0.05 at Day 15) was considered UV-4 related but not toxicologically relevant due to the small magnitude of change. UV-4 related body weight effects demonstrated full reversibility during the recovery phase, and food consumption during the recovery phase was similar between the control mice and mice treated with UV-4.
No UV-4 related ophthalmic observations or neurologic observations (modified Irwin test) 14 were noted during the study. Only limited clinical pathology data were available from mice euthanized at an unscheduled interval. A cause of death or moribundity of these mice was not evident in clinical pathology data. One male at 750 mg/kg/day had markedly high urea nitrogen and another male at 750 mg/kg/day had a moderate decrease in albumin. Among mice that survived to their scheduled sacrifice (including mice administered 750 mg/kg/day and euthanized on Day 11 of the dosing phase), relatively minor clinical pathology findings were observed that included minimally to mildly lower red cell mass (i.e., red blood cell count, hemoglobin, and hematocrit) and mildly lower globulin resulting in higher albumin-to-globulin ratio in males and females administered ≥ 300 mg/kg/day.
Additionally, mildly lower absolute lymphocyte count was observed in males and females at 750 mg/kg/day, resulting in an overall lower white blood cell count in females at this dose. All clinical pathology findings exhibited reversibility at the end of the recovery phase. UV-4-related organ weight decreases occurred in the thymus in males administered ≥ 75 mg/kg/day and in the thymus, ovaries, and uterus of females at 750 mg/kg/day. No UV-4-related macroscopic observations were noted at the dosing or recovery phase necropsy. UV-4-related microscopic changes consisted of follicular epithelium vacuolation in the thyroid in mice administered ≥ 75 mg/kg/day, neutrophilic infiltration and erosion/ulcer in the rectum in males at 750 mg/kg/day, and females at 300 mg/kg/day; erosion/ulcer of the limiting ridge of the stomach in mice at 750 mg/kg/day, lymphoid depletion/necrosis of the thymus in males administered ≥ 75 mg/kg/day; and females administered ≥ 300 mg/kg/day; and decreased glycogen in the liver in mice administered ≥ 300 mg/kg/day. All of the organ weight and microscopic changes had reversed at the recovery phase necropsy
As shown in Table 4, exposure to UV-4 increased with the increase in dose from 75 to 750 mg/kg/dose on Day 1 and from 75 to 750 mg/kg/dose on day of termination (Day 11 or 14). The increases in Cmax and AUC(0-8) were generally dose-proportional. Sex differences were less than 2-fold in Cmax and AUC(0-8) values. No accumulation was observed in the mice after multiple dosing.
Toxicokinetic Parameters in Mice and Dogs.
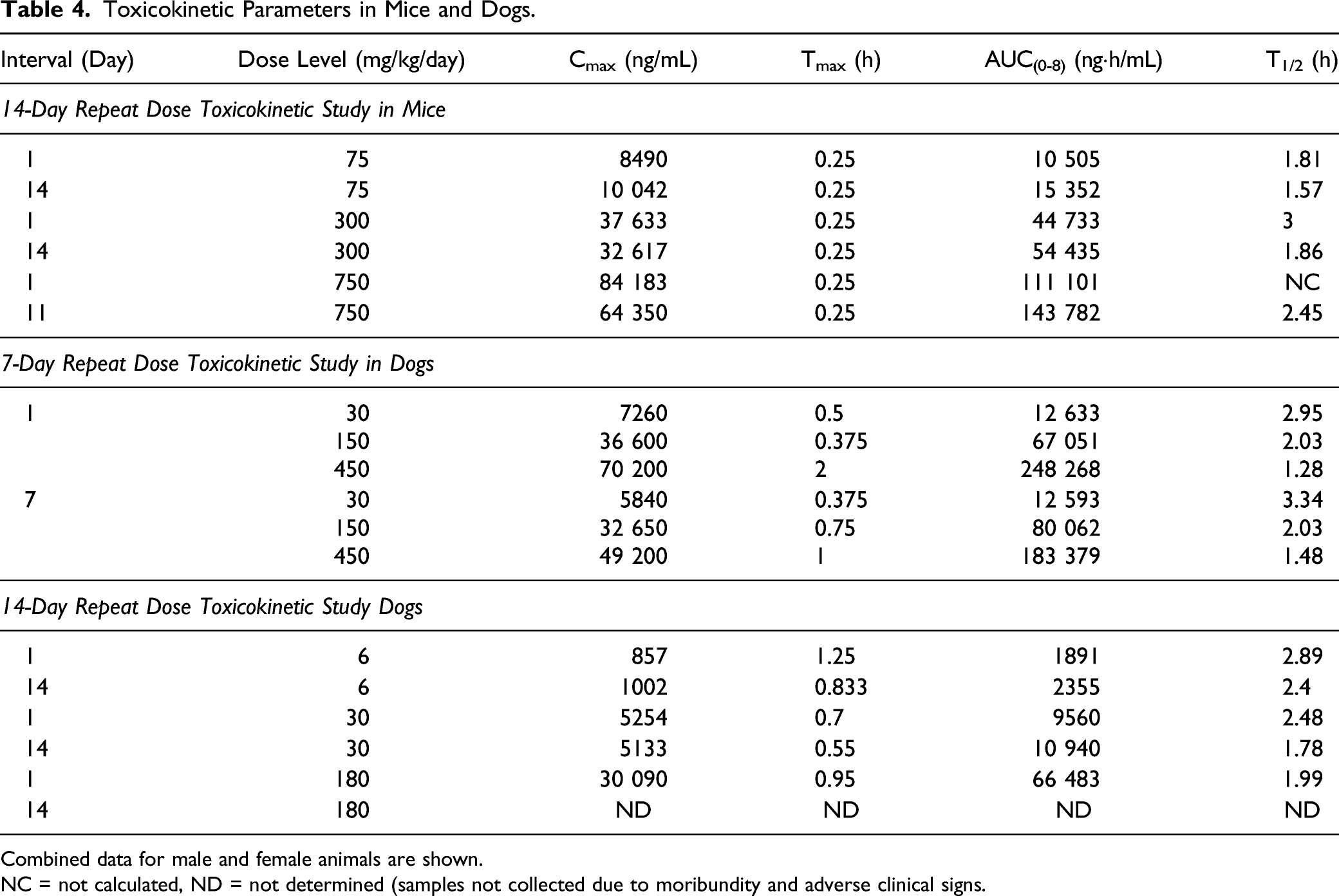
Combined data for male and female animals are shown.
NC = not calculated, ND = not determined (samples not collected due to moribundity and adverse clinical signs.
The 300 mg/kg/day dose did not result in adverse body weight changes, food consumption effects, or clinical or anatomic pathology changes; and all changes noted during the dosing phase demonstrated full reversibility by the end of the recovery phase. Therefore, based on these observations, the no observed adverse effect level (NOAEL) for UV-4 hydrochloride was 300 mg of UV-4/kg/day. This dose level corresponds to Day 14 mean UV-4 Cmax and AUC(0-8) values of 32 617 ng/mL and 54 435 ngh/mL, respectively. Daily exposure (AUC0-24) at the NOAEL (calculated as 3 times the AUC(0-8) to account for 3 doses/day) was estimated at 163 305 ngh/mL.
Single Dose (Phase I) and 7-Day Repeat-Dose (Phase II) Oral Gavage Toxicity Study of UV-4 Hydrochloride in Rats
Phase I (Rat Single Dose)
All of the Phase I rats survived until their scheduled necropsy. All rats at each dose level gained body weight during Phase I, and no UV-4 related changes in food consumption were noted. At dose levels of ≥ 200 mg/kg, observations included swollen feet and/or legs, which was transient. Swollen nose in females administered ≥ 200 mg/kg, and males and females administered ≥ 300 mg/kg along with discoloration (red) in swollen areas which was also transient. These observations were considered dose limiting. Minimal to mild UV-4 dose-related hematology and clinical chemistry changes were also observed at doses ≥ 200 mg/kg/day. The changes were not uniformly observed in both sexes. Urine volume was markedly decreased and urine specific gravity was moderately increased in males at 400 mg/kg, suggesting conservation of body water, possibly to incipient dehydration following UV-4 administration.
Phase II (Rat 7-Day Repeat Dose)
Table 5 shows in-life and clinical pathology findings that were identified as toxicologically important in the 7-day repeat dose rat study. All of the Phase II rats survived until their scheduled necropsy. No UV-4 related clinical observations were noted in rats administered 75 mg/kg/day. Decreased body weight gain and food consumption in male and female rats were considered dose limiting at 180 mg/kg/day.
In-Life and Clinical Pathology Summary in the 7-Day Repeat-Dose (Phase II) Oral Gavage Toxicity Study in Rats.
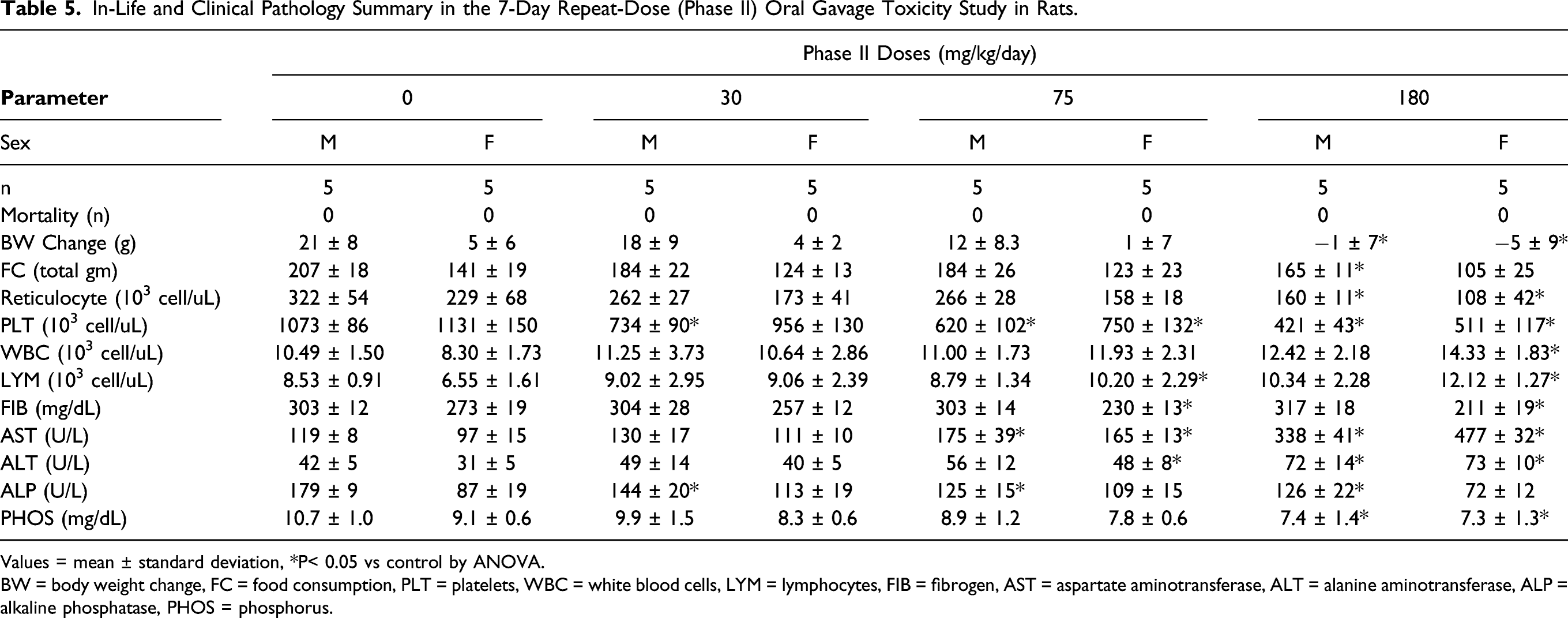
Values = mean ± standard deviation, *P< 0.05 vs control by ANOVA.
BW = body weight change, FC = food consumption, PLT = platelets, WBC = white blood cells, LYM = lymphocytes, FIB = fibrogen, AST = aspartate aminotransferase, ALT = alanine aminotransferase, ALP = alkaline phosphatase, PHOS = phosphorus.
On Day 8 of Phase II, UV-4 related hematology changes included mild to moderate, decreases in absolute reticulocyte and platelet counts in rats administered ≥ 30 mg/kg/day, and mildly increased total leukocyte and lymphocyte counts in females administered ≥ 30 mg/kg/day. A minimal UV-4 related decrease in fibrinogen concentration was also present in females administered ≥ 75 mg/kg/day. UV-4 related clinical chemistry changes included minimal to moderate, dose-dependent increases in AST and alanine aminotransferase (ALT) activities in rats administered ≥ 30 mg/kg/day, and a minimal decrease in alkaline phosphatase activity in males administered ≥ 30 mg/kg/day and females at 180 mg/kg/day. A minimal to mild decrease in phosphorus concentration was also present in rats administered ≥ 75 mg/kg/day. UV-4 had no effect on urinalysis parameters at any dose level. UV-4 related macroscopic observations were present in the stomach of 1 male at 30 mg/kg/day, and microscopic findings considered UV-4 related were present generally at ≥ 75 mg/kg/day in the liver, spleen, mesenteric lymph node, kidney, and stomach (also at 30 mg/kg/day). Extramedullary hematopoiesis was considered UV-4 related in the spleen (decreased) or liver (not present) of rats at 180 mg/kg/day. Decreased lymphocytes in the mesenteric lymph node were test article-related in females administered180 mg/kg/day. UV-4 related increased vacuoles in renal proximal tubules were noted in the kidneys of males at 180 mg/kg/day and females administered ≥ 75 mg/kg/day. A number of microscopic lesions were present in the glandular and nonglandular stomach and were generally localized to regions adjacent to the limiting ridge. Microscopic findings specific to the glandular stomach included erosion/ulcer in one male at 180 mg/kg/day and one female at 75 mg/kg/day, inflammation in one male at 75 mg/kg/day, and decreased mucous cells of the cardia region in rats administered ≥ 30 mg/kg/day. Nonglandular stomach lesions included hyperplasia/hyperkeratosis in two males at 75 mg/kg/day and erosion in one male at 75 mg/kg/day. The erosion/ulcer in the male at 180 mg/kg/day corresponded macroscopically to brown discolored mucosa. One male at 30 mg/kg/day also had brown discoloration of the mucosa that was not associated with a corresponding microscopic finding but was considered UV-4 related. Based on the findings of decreased body weight gain at 180 mg/kg/day and erosions/ulceration in the stomach in the repeat-dose phase, a daily dose of UV-4 hydrochloride at ≥ 75 mg UV-4/kg/day was considered dose limiting.
Dose Escalation and Determination of Maximum Tolerated Dose of UV-4 Hydrochloride Following Oral Administration in Beagle Dogs
Both dogs survived until the end of the study at single doses up to 250 mg/kg/day. No clinical signs were observed at 25 or 50 mg/kg. Moderate watery feces were observed in both dogs approximately 24 hours after dosing with 100, 150, or 250 mg/kg. No abnormalities were detected during physical examinations. There were no drug-related effects on body weights, food consumption, or clinical pathology parameters. There were no drug-related macroscopic observations at necropsy. The dogs tolerated UV-4 at single doses of up to 250 mg/kg.
Escalating-Dose (Phase I) and 7-Day Repeat-Dose (Phase II) Pilot Toxicity and Toxicokinetic Oral Gavage Study of UV-4 Hydrochloride in Dogs
Phase I (Dog Single Dose)
No mortality was observed in Phase I at any of the doses tested (50, 100, 200 and 400 mg/kg). No UV-4 related clinical observations were noted at doses ≤ 100 mg/kg. Vomitus was observed at ≥ 200 mg/kg and intermittent tremors in one male were observed following administration of 400 mg/kg. No body weight effects or changes in food consumption were observed. UV-4 related findings included minimal to marked increases in AST, minimal to moderate increases in ALT (except on Day 4 in the male), and minimal to mild prolongation of activated partial thromboplastin time (aPTT) at all dosing phase time points. Increases in AST appeared more pronounced than increases in ALT. These changes appeared dose-related with the most pronounced changes occurring on Day 14 at 400 mg/kg. No macroscopic observations were noted at the end of Phase I; therefore, histopathology evaluation was not conducted during this phase.
Phase II (Dog 7-Day Repeat Dose)
In Phase II, the 450 mg/kg/day (150 mg/kg/dose three times daily (TID)) male was euthanized in moribund condition prior to dosing on Day 7 and dosing was stopped following the first dose on Day 7 for the 450 mg/kg/day female. Adverse UV-4 related clinical observations in the 450 mg/kg/day male included discolored (black) and liquid feces (also noted in the female), and dehydration. Clinical pathology results for the male at the 450 mg/kg/day dose included moderate decreases in reticulocyte and platelet counts, mild to moderate increases in aPTT, ALT, and marked increase in AST. UV-4 related microscopic changes that occurred only in the male euthanized at an unscheduled interval included slight acute inflammation in the mucosa and submucosa of the rectum and minimal decreased chief cell granules in the stomach. The cause of moribundity for the 450 mg/kg/day male was gastrointestinal (GI) inflammation (acute inflammation in the rectum) and hemorrhage in the duodenum. Proximal convoluted tubular vacuolation and degeneration in the kidney sections were similar in the male and female at 450 mg/kg/day. The remaining dogs, including the 450 mg/kg/day female, survived until their scheduled necropsy.
UV-4 related clinical observations during the repeat dosing phase included excessive salivation and vomitus at ≥ 150 mg/kg/day and fecal abnormalities at ≥ 30 mg/kg/day. Fecal abnormalities (non-formed) at 30 mg/kg/day were singular occurrences in each dog. A dose-dependent decrease in body weight (net weight loss) was observed in dogs administered ≥ 150 mg/kg/day. Similarly, a progressive and dose-dependent decrease in daily food consumption was observed in males and females administered ≥ 150 mg/kg/day. Among dogs that survived until the end of Phase II, moderately prolonged aPTT, moderately to markedly increased AST, and minimally to mildly increased ALT (except male at 30 mg/kg/day) were observed at all dose levels (≥ 30 mg/kg/day). Moderately decreased reticulocyte and platelet counts were also observed in dogs administered ≥ 150 mg/kg/day. In Phase II, no UV-4 related microscopic changes were observed in dogs at 30 mg/kg/day. UV-4 related microscopic changes occurred in the kidney, GI tract (stomach, duodenum, cecum, and colon), spleen, and gut-associated lymphoid tissue (GALT)/Peyer’s patch. UV-4 related microscopic findings in the GI tract consisted of degeneration/erosion in the cecum (slight) and colon (minimal) of the female at 150 mg/kg/day and degeneration/erosion in the stomach (minimal), duodenum (minimal), and colon (slight) of the female at 450 mg/kg/day. Other UV-4 related microscopic changes in the female at 450 mg/kg/day included slight acute inflammation in the colon, presence of eosinophilic luminal content in the cecum, and minimal hemorrhage in the stomach and colon. Minimal hemorrhage in the duodenum of the female at 150 mg/kg/day was also UV-4 related. UV-4 related findings in the kidney consisted of degeneration of the proximal cortical tubules in the male and female administered 450 mg/kg/day, and minimal or slight vacuolation of the proximal cortical tubules in dogs administered ≥ 150 mg/kg/day. Degeneration of renal tubule epithelium at 450 mg/kg/day was considered dose limiting; however, vacuolation of the tubules at 150 mg/kg/day was not associated with degeneration and was therefore not considered dose limiting. Microscopic findings of degeneration of the proximal cortical tubules and degeneration/erosion in the stomach and duodenum at 450 mg/kg/day, and degeneration/erosion of the cecum and colon at ≥ 150 mg/kg/day were considered dose limiting.
Plasma concentrations of UV-4 increased with increase in dose from 30 to 450 mg/kg/day as shown in Table 4. The increases in Cmax and AUC(0-8) were variable, but were generally dose proportional. Differences in Cmax and AUC(0-8) values between male and female animals were less than 2-fold. No accumulation of UV-4 was observed after multiple dosing in dogs.
Dose-limiting toxicity was observed microscopically in the GI tract of dogs administered ≥ 150 mg/kg/day and in the kidneys at 450 mg/kg/day. The dose level of 150 mg/kg/day corresponded to Day 7 respective to the combined male and female average UV-4 Cmax and AUC(0-8) values of 32 650 ng/mL and 80 062 ngh/mL (Table 4).
14-Day Toxicity and Toxicokinetic Study of UV-4 Hydrochloride Administered by Oral Gavage in Beagle Dogs With a 14-Day Recovery (GLP)
Table 6 shows in-life and clinical pathology findings that were identified as toxicologically important in the 14-day repeat dose dog study. All of the control dogs and all of the dogs that received UV-4 hydrochloride at total doses of 6 or 30 mg/kg/day (2 or 10 mg UV-4/kg/dose TID) survived to their respective terminal or recovery sacrifice. Three dogs that received 180 mg/kg/day (60 mg/kg/dose TID; two females and one male) were euthanized in moribund condition on Day 6 or 8 of the dosing phase due to clinical observations and body weight loss (0.3 to 1.2 kg). Clinical pathology changes in the male and one of these females included mildly decreased absolute reticulocyte count, moderately decreased platelet count, moderately prolonged aPTT, markedly increased AST activity, and mildly to moderately increased ALT activity in both dogs compared with control values on Day 9 of the dosing phase. Macroscopic observations noted at necropsy were attributed to UV-4; two dogs exhibited red mucosal discoloration of multiple levels of the digestive tract and one dog exhibited dark red discoloration of the pyloric stomach mucosa and multiple mesenteric lymph nodes. Where present microscopically, the correlate for mucosal discoloration was congestion and the correlate for lymph node discoloration was sinus erythrocytes, which likely represented draining GI tract hemorrhage. Based on the clinical observations and the unscheduled necropsy of three dogs that received 180 mg/kg/day, dosing was suspended for the remaining dogs in this group following Day 8; one male and one female were euthanized as a terminal necropsy on Day 9, while the remaining three males and two females in this dose group were transferred to a 21-day recovery phase. Clinical observations in the dogs that received 180 mg/kg/day and survived until the scheduled early terminal sacrifice or recovery sacrifice were similar to those in the unscheduled sacrifice animals; and included hypoactivity, excessive salivation, vomitus (containing food, foamy, and/or yellow), and fecal abnormalities. An increased incidence of non-formed feces was also observed at 30 mg/kg/day. All clinical observations demonstrated reversibility during the recovery phase. No UV-4 related changes in body weight or food consumption were observed during the dosing phase for males or females administered ≤ 30 mg/kg/day or during the recovery phase for males or females at 30 mg/kg/day and no ophthalmic observations were noted at any dose level. UV-4 related decreases in mean body weight were observed by Day 3 of the dosing phase for males and females at 180 mg/kg/day, and this continued through Day 8. Body weight changes demonstrated reversibility during the recovery phase. Although highly variable, mean food consumption was generally lower for males and females at 180 mg/kg/day, with individual dogs having reduced food consumption during the dosing phase. Effects on food consumption demonstrated reversibility during the recovery phase.
In-Life and Clinical Pathology Summary in the 14-Day Toxicity and Toxicokinetic Study in Beagle Dogs With a 14-Day Recovery.
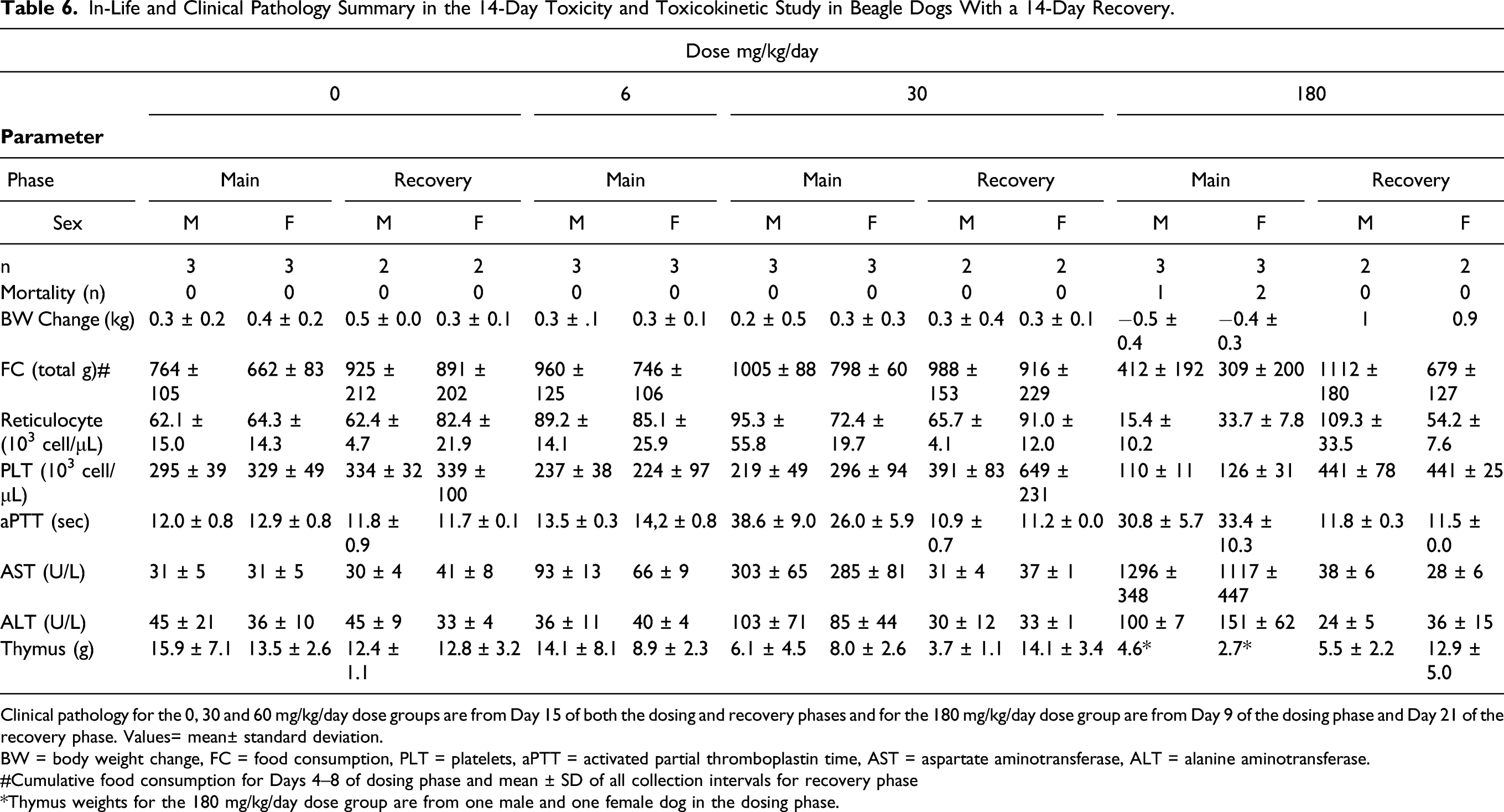
Clinical pathology for the 0, 30 and 60 mg/kg/day dose groups are from Day 15 of both the dosing and recovery phases and for the 180 mg/kg/day dose group are from Day 9 of the dosing phase and Day 21 of the recovery phase. Values= mean± standard deviation.
BW = body weight change, FC = food consumption, PLT = platelets, aPTT = activated partial thromboplastin time, AST = aspartate aminotransferase, ALT = alanine aminotransferase.
#Cumulative food consumption for Days 4–8 of dosing phase and mean ± SD of all collection intervals for recovery phase
*Thymus weights for the 180 mg/kg/day dose group are from one male and one female dog in the dosing phase.
UV-4 related hematology and coagulation changes during the dosing phase included mild to moderate decreases in absolute reticulocyte count on Days 6 and 9 in dogs at 180 mg/kg/day; minimal to mild decreases in platelet count on Days 6 and 9 in males administered ≥ 6 mg/kg/day and females at 180 mg/kg/day and on Days 11 and 15 in dogs administered 6 or 30 mg/kg/day; and mild to moderate prolongation of aPTT on Days 6 and 9 in dogs administered 30 or 180 mg/kg/day and on Days 11 and 15 of the dosing phase in dogs at 30 mg/kg/day. Hematology and coagulation changes persisted through Day 3 of the recovery phase, but reversed by Day 7 of the recovery phase.
UV-4 related clinical chemistry changes in hepatic enzyme activity during the dosing phase included a moderate to marked, dose-dependent increase in AST activity on Days 6 and 9 in dogs administered ≥ 30 mg/kg/day; mild to moderate increase on Days 11 and 15 in animals administered 6 or 30 mg/kg/day; and minimal to mild increase in ALT activity on Days 6 and 9 in dogs at 180 mg/kg/day and on Days 11 and 15 of the dosing phase in dogs at 30 mg/kg/day. Increases in AST activity reversed by Day 7 to 13 of the recovery phase, while increases in ALT activity reversed by Day 13 of the recovery phase. Macroscopic red or dark red GI discoloration associated with inflammation was noted in dogs at 180 mg/kg/day that were euthanized on Day 9 and attributed to UV-4. Decreased absolute and relative (to body and brain) thymus weights were noted for dogs at all dose levels and correlated with lymphocyte depletion (minimal to marked) for males administered ≥ 30 mg/kg/day and females administered ≥ 6 mg/kg/day. On Day 9 of the dosing phase, UV-4 related microscopic changes (degeneration/erosion, acute inflammation, and/or congestion) occurred in the stomach, duodenum, jejunum, ileum, cecum, colon, and/or rectum of dogs administered 180 mg/kg/day which were scheduled for this early sacrifice. An additional digestive system observation in dogs at 180 mg/kg/day was decreased hepatocyte vacuolation (glycogen-type), which was likely secondary to decreased food consumption. One female at 180 mg/kg/day had slight degeneration/necrosis/regeneration of renal cortical tubules, which was considered UV-4 related. UV-4 related lymphocyte depletion was noted at all dose levels. In addition to depletion in the thymus, dose-dependent depletion (minimal to moderate) was noted in the spleen, lymph nodes, and GALT/Peyer’s patch for dogs at 180 mg/kg/day. Consistent with full recovery, all UV-4 related macroscopic and microscopic findings noted at unscheduled and terminal sacrifices were absent at the recovery sacrifice or at a reduced incidence and consistent with spontaneous background changes. Recovery of microscopic thymus findings correlated with thymus weights similar to controls for females at the recovery sacrifice. Thymus weights for males did not show recovery. However, because the apparent persistent thymus weight reductions were not dose-dependent and males showed recovery of microscopic thymus findings, thymus weight differences at the recovery sacrifice for males were considered not toxicologically important.
Administration of UV-4 at 180 mg/kg/day was associated with an increase in QRS duration in males (+30%) and females (+28%) and decreased heart rate in males (−26%) on Day 9 of the dosing phase (Day 1 of the recovery phase). These changes in QRS duration in all dogs administered 180 mg/kg/day and heart rate in males at 80 mg/kg/day were reversed by Day 19 of the recovery phase. No UV-4 related rhythm abnormalities, or changes in PR interval, QT interval, RR interval, or heart rate were observed in males or females administered ≤ 30 mg/kg/day or in females at 180 mg/kg/day and no UV-4-related changes in QTc interval were observed.
Plasma concentrations of UV-4 increased with the increase in dose level from 2 to 60 mg/kg/dose on Day 1 and from 2 to 10 mg/kg/dose on Day 14 (Table 4). The increases in mean UV-4 Cmax and AUC(0-8) were generally dose proportional. Differences in mean Cmax and AUC(0-8) values between male and female animals were less than 2-fold. No accumulation of UV-4 was observed after multiple dosing in dogs.
Based on these observations, the NOAEL for this study was considered to be 30 mg/kg/day. At the end of the dosing phase, this dose level corresponded to average (male and female) UV-4 Cmax and AUC(0-8) values of 5133 ng/mL and 10 940 ng·h/mL, respectively. Daily exposure (AUC(0-24)) at the NOAEL (calculated as 3 times the AUC(0-8) to account for 3 doses/day) was estimated to be 32 820 ng·h/mL.
Study to Determine the Maximum Tolerated Dose of UV-4 Hydrochloride in Cynomolgus Monkeys
Table 7 shows in-life and clinical pathology findings that were identified as toxicologically important in the NHP study. All animals survived to the completion of dosing, although in the fourth cycle (1080 mg/kg/day, 360 mg/kg/dose) dosing was terminated on Day 7 due to adverse clinical signs. During Cycles 1, 2 and 3, treatment with UV-4 (at doses of 20, 60, or 180 mg/kg/dose, TID, for 5, 5, and 10 days, respectively) had no effect on physical examinations, post-dose, or cageside observations other than a single monkey that exhibited a rough haircoat for 3 days during Cycle 3 (from the second dose on the eighth day of the cycle until the 10th day of the cycle).
In-Life and Clinical Pathology Summary in Cynomolgus Monkey MTD Study.
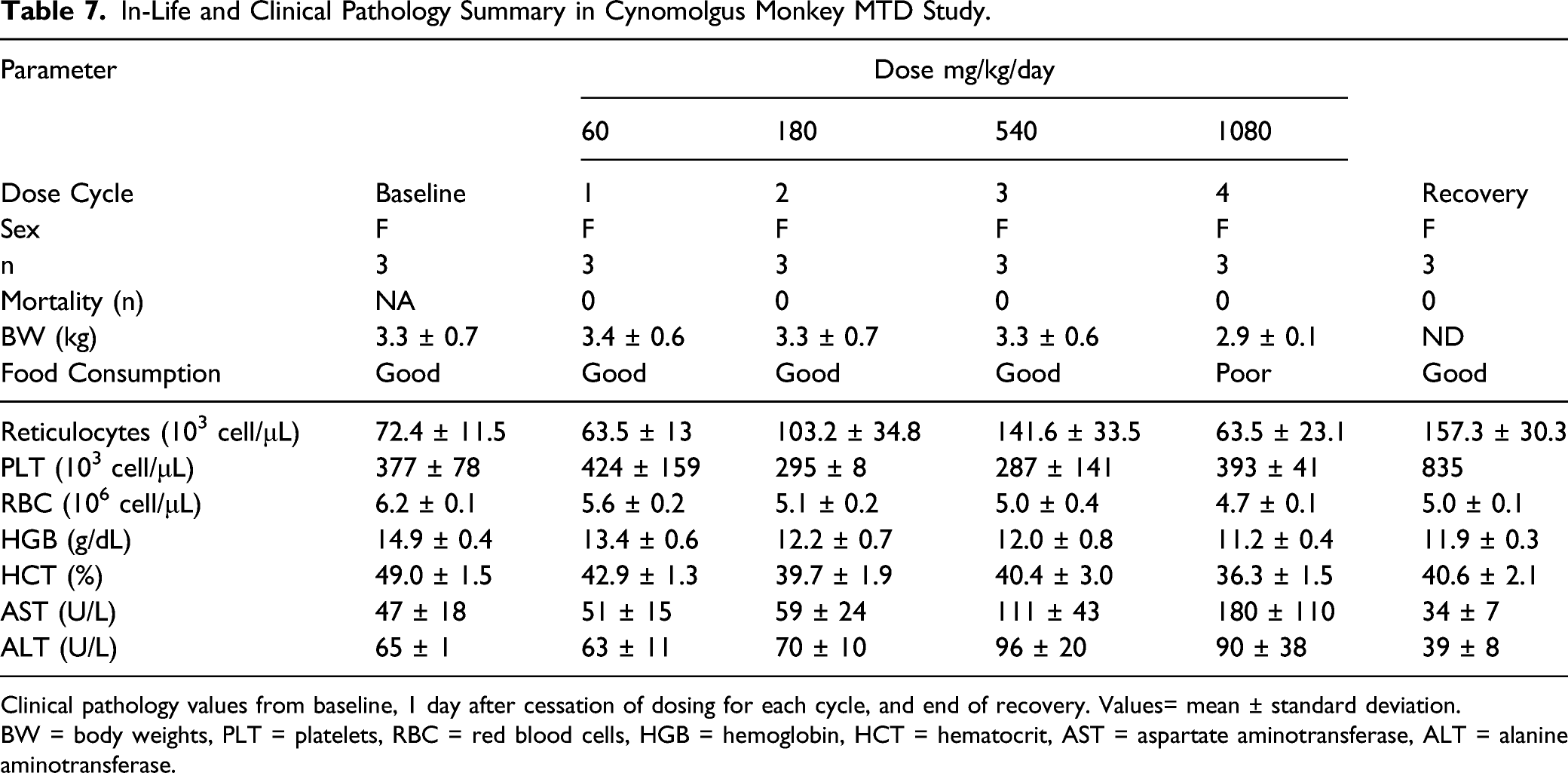
Clinical pathology values from baseline, 1 day after cessation of dosing for each cycle, and end of recovery. Values= mean ± standard deviation.
BW = body weights, PLT = platelets, RBC = red blood cells, HGB = hemoglobin, HCT = hematocrit, AST = aspartate aminotransferase, ALT = alanine aminotransferase.
Treatment with UV-4 up to and including 1080 mg/kg/day had no effect on body weights or body weight changes. Treatment with UV-4 affected food consumption during Cycles 3 and 4 (540 and 1080 mg/kg/day, respectively) with more days described as fair or poor in regard to food consumption. Treatment with UV-4 had no effect on hematology parameters at doses ≤ 540 mg/kg/day for 10 days, but resulted in treatment-related reversible changes in clinical chemistry in Cycle 4 (1080 mg/kg/day). There were slight decreases in RBC, hemoglobin, and hematocrit following all of the dose cycles; however, these were considered likely the result of the blood volumes that were collected during the study. Responses in platelet and reticulocytes were somewhat variable. In general platelets decreased following each cycle except the 60 mg/kg/dose cycle (Cycle 2) and increased during Cycle 4 and recovery period. Reticulocytes increased during Cycles 2 and 3, decreased during the Cycle 4 dosing phase and then increased following recovery. On Day 8 of Cycle 4 (1080 mg/kg/day) at 8 hours after dosing cessation on Day 7, AST increased by 217% compared to Day 1 values and ALT increased by 66% compared to Day 1 values. By Day 20 of Cycle 4 (13 treatment free days), both the AST and ALT had decreased by 56% and 20%, respectively, compared to Day 1 values.
During Cycle 4 (1080 mg/kg/day) of treatment with UV-4, affected physical examinations began on Day 1 when all three monkeys produced brown emesis. No rough hair coat or other adverse signs were observed (as had been seen in one monkey in Cycle 3). Post-dose and cage side observations were affected beginning after the fourth dose (i.e., after the first dose on Day 2) when one monkey had soft feces and the other two monkeys had few feces. Subsequent observations included few, soft, or no feces. By Day 7 of this cycle, all three monkeys displayed a hunched posture. Dosing was terminated after the third dose on Day 7 due to these clinical signs of hunched posture and abnormal feces. Clinical observations for all three monkeys returned to normal before Day 20 (13 days recovery), when the animals were returned to the stock colony.
All three animals tolerated a single UV-4 dose of 180 mg/kg and also tolerated three doses per day at this level (540 mg/kg/day) for up to 10 days. Due to clinical effects observed in Cycle 4 (1080 mg/kg/day), the MTD was considered to be 540 mg/kg/day for 10 days. A single dose of 360 mg/kg also appeared well tolerated although clinical signs became apparent after the fourth TID dose at this dose level.
To minimize blood draws from monkeys in this non-GLP study, there was minimal sampling for measurement of plasma levels of UV-4, therefore calculation of TK parameters was not carried out. Several trough (pre-dose) samples analyzed in each cycle confirmed that plasma levels of UV-4 were present throughout the cycle at all dose levels. Plasma levels greater than the upper limit of quantitation of the assay even after 100-fold dilution (that is, >50 000 ng/mL) were reported in blood samples taken at 1 hour after dosing (at the approximate Tmax). Although AUC, Tmax, and Cmax could not be calculated given the minimal sampling time points and the Cmax levels above the upper limit of quantitation, these data confirm high systemic exposure to UV-4, and rapid absorption from the gut with lower levels observed 8 hours post-dosing, as is seen in mouse and dog TK and pharmacokinetic (PK) studies.
Discussion
The goal of the toxicology program was to provide support for the dose levels to investigate the safety and tolerability of single ascending oral doses of UV-4 hydrochloride (also known as UV-4B) in healthy human subjects. The toxicology program for these studies conformed to ICH M3 (R2) “Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals” 22 to support the planned Phase I clinical trial of a Single Ascending Dose. Single and repeat dose studies in mice, rats, and dogs, and a repeat dose study in NHP to evaluate the safety of UV-4. Mice were chosen as the rodent species for IND-enabling GLP studies as in vivo antiviral activity models predominantly utilize murine strains. Dogs were selected as the non-rodent species. NOAEL doses were established in all GLP studies.
Mortality/moribundity occurred at higher doses in each species evaluated. Decreased bodyweights also decreased at higher doses in the surviving animals of each species evaluated. These decreases in body weight generally correlated with decreases in food consumption. Decreased food consumption was consistent with that noted following administration of other iminosugars in lean and obese mice. 23 Body weight and food consumption changes demonstrated reversibility during the recovery phase.
A rat specific, possible histamine-like in-life clinical sign was noted as swollen paws/legs and perioral appearance and red skin, paws, and/or feet at ≥ 200 mg/kg as a single dose or after the first or second daily dose on Day 1 or 2 of the repeat dose study at 180 mg/kg/day. This response was transient and was not observed after Day 2 following single or repeated doses. Similar findings were noted in a preliminary pharmacokinetic study in rats (unpublished data) where severe soft tissue swelling of all limbs and forefeet was transiently noted after 100 mg/kg for approximately 4 hours post-dose. These findings were not present in pharmacokinetic studies or toxicology studies in mice or dogs and appear to be a rat-specific phenomenon. Similar findings were seen in rats following dosing with miglustat, 24 an analog of UV-4 but no similar findings were seen in humans receiving doses of miglustat of up to 200 mg TID. 25 These events seen in rats administered continued doses of UV-4 resolved without any adjunct treatment and did not appear to adversely affect the animals.
UV-4-related clinical pathology changes in the 14-day studies in dogs included dose-related decreases in platelets and reticulocytes and increases in AST, ALT, and aPTT at the termination of dosing. All parameters returned to the baseline values by the conclusion of the recovery period. There was moderate to marked, dose-dependent increase in AST (up to 45-fold) activity at termination but there was no histopathological correlate, that is, hepatocellular necrosis, noted in any UV-4 dose groups. Similarly, minimal to mild increases in ALT activity (up to 4.7-fold) were noted in all UV-4 dose groups also with no histopathological correlate. Since increases in ALT may be the result of damage to skeletal muscle, 26 additional sections of skeletal muscle (biceps femoris, gastrocnemius, and quadriceps femoris) were included in the 7-day rat study and 14-day studies in mice and dogs. Except for degeneration/necrosis noted in 1 high-dose mouse at recovery, no potential UV-4-related effects were noted upon microscopic examination of these sections of muscle. The possibility that the increases in AST and ALT were associated with hemolytic effects of UV-4 was also evaluated in an in vitro hemolysis assay. UV-4 did not cause hemolysis in rabbit or rat whole blood at concentrations relevant to the in vivo studies (unpublished data). Other causes for the finding of elevated AST greater than ALT in the absence of hepatic and muscle pathology that were not investigated in this study are also possible.26,27 Based on no histopathological correlate in any tissue, the increases in AST and ALT are considered to be UV-4-related but not adverse. The underlying cause of the increase in AST greater than ALT is presently unknown.
The kidney, mesenteric lymph nodes, GI tract, and thymus were identified as the target organs in all three species in which pathology was evaluated. UV-4-related findings were generally seen at the higher doses but did extend to the lower doses in some cases after 7- or 14-day exposure. The microscopic findings in the thymus were noted at all doses and correlated with a dose related decrease in thymus weight in the mice and dogs. With the exception of the dog thymus weight, all of the microscopic findings and thymus weights showed complete reversal after the recovery period. Although the dog thymus weights did not completely reverse, the microscopic thymus findings did show some evidence of recovery. Therefore, the lack of recovery of the male thymus weight was not considered toxicologically significant.
Erosion/ulceration of the stomach was noted at 600, 750, and 1200 mg/kg/day in mice, at 75 and 180 mg/kg/day in rats, and at 150 and 180 mg/kg/day in dogs. All of these findings showed complete reversal after the recovery period. When the erosion/ulceration in the stomach of the rats at 75 mg/kg/day are scaled to the human equivalent dose (HED), the rat would be the more sensitive of the three species tested since similar findings were observed in the mouse at 750 mg/kg/day and in dogs at 180 mg/kg/day (GI tract but not stomach). However, the 7-day rat study was conducted as a non-GLP dose range-finding study; therefore, the results of the study did not satisfy regulatory requirements needed to identify the rat as the more sensitive species. The NHP study, which was conducted as a non-GLP dose range-finding study, similarly was not used to select clinical dose although the high MTD and plasma exposure may indicate higher tolerability in primates than in mice, rats, and dogs. Because erosion/ulceration observed in the preclinical studies results from prolonged irritation to the area, the risk of significant GI irritation was considered to be very low for a human SAD study. Based on these reasons, the ceiling dose and total exposure expected in the human SAD study was based on the dog GLP toxicology results.
Based on the dog NOAEL (10 mg/kg TID) and the appropriate conversion recommended in the FDA guidance “Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for therapeutics in Adult Healthy Volunteers” 28 (i.e., 60 kg body weight for humans), the HED for the NOAEL of UV-4 in dog is 333 mg TID. Considering that the lowest dose tested in dogs (2 mg/kg TID or expressed as HED 67 mg TID) was associated with decreases in platelet count (mild), increases in aPTT (very mild), increases in AST and histopathological changes, a 100-fold safety factor was applied to the NOAEL that equated to a starting dose in humans of approximately 3 mg. The ceiling dose was set as HED at the NOAEL in dogs (i.e., 1000 mg/day). This is expected to correlate to a total exposure (mean of cohort) of approximately 32 820 ng•h/mL for AUC(0-inf), based on the AUC(0-24) at the NOAEL in dogs.
If the doses used in the above-mentioned PK studies (oral doses of 100 mg/kg for rat, 50 mg/kg for dog, and 100 mg/kg for mouse) are converted to the HED, estimates for Cmax at the 3 mg starting dose range from 41 ng/mL (scaled from rat) and 54 ng/mL (scaled from dog) to 302 ng/mL (scaled from mouse). Although the NHP study was not executed under GLP, the results confirm good tolerability and high exposure in cynomolgus monkeys. Doses up to 180 mg/kg/dose TID for up to 10 days were well tolerated, with only administration at 360 mg/kg/dose TID and peak plasma levels greater than 50 000 ng/mL exceeding tolerability. The human safety and PK findings from the SAD study will be separately reported. 29
Antiviral efficacy of UV-4 has been tested in animal models using two different dosing strategies. Initial studies were conducted in mice using a dosing regimen of three times a day (TID) for 5 to 10 days with initial dosing beginning between 1 hour before and up to 120 hours post-infection.9,10 A survival benefit was seen when UV-4 hydrochloride was administered up to 120 hours post infection, a point at which the infected mice have lost ∼10% of their body weight, had adverse health changes for 2 days and 5% decrease in body temperature. Using the TID dosing regimen, the most significant benefit was observed when dosing was initiated within 48–72 hours post infection. Subsequently, studies were conducted using a high intensity dosing strategy with survival benefit observed at ≥ 250 mg/kg administered 8 hours post-infection with either dengue or influenza virus. 30
It has been proposed that host-targeted drug candidates may have increased risk of toxicity due to disruption of cellular pathways in both infected and healthy cells. 6 The nonclinical safety data reported here indicate that this hypothesis must be evaluated on a case-by-case basis, and support the potential use of UV-4 therapeutically either up to TID for multiple days or as single dose. At high doses or after multiple days of exposure, UV-4 has dose-limiting toxicities principally related to gastrointestinal effects, which was expected given the structural similarity between the active moiety UV-4 and other approved 1-deoxynojirimycin-based iminosugars such as miglustat and miglitol. Other effects are generally consistent across species and are predictable with respect to dose, and the PK and TK data indicate good absorption after dosing with the hydrochloride salt. Recent publications have highlighted the potential importance for iminosugars including UV-4 as therapeutics for SARS-COV2,31,32 and have positioned orally-bioavailable, broad-spectrum, host-targeted antiviral therapeutics as a critical component for combatting future pandemics. 6 Therefore, UV-4 is a promising candidate for further development as a therapeutic intervention against influenza, dengue, and other acute viral diseases.
Footnotes
The authors wish to thank the Study Directors and staff at Covance Laboratories Inc. (Madison WI), Advinus Therapeutics Limited (Bangalore, India), and Smithers Avanza Toxicology Services (Gaithersburg MD) for the performance of these studies.
Author Contributions
Shearer, J. contributed to interpretation, drafted manuscript, and critically revised manuscript; Wolfe, G. contributed to conception and design, contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Sampath, A. contributed to conception and design, contributed to analysis and interpretation, and critically revised manuscript; Warfield, K.L. contributed to conception and design, contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Kaufman, B. contributed to conception and design, contributed to analysis and interpretation, and critically revised manuscript; Ramstedt, U. contributed to conception and design, contributed to analysis and interpretation, and critically revised manuscript; Treston, A. contributed to conception and design, contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Kelly L Warfield is an employee of Emergent BioSolutions Inc.
Funding
This research was funded in part by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract number HHS272201100030C.