
Abstract
Research suggests that thioether analogs of vitamin K3 (VK3) can act to preserve the phosphorylation of epidermal growth factor receptors by blocking enzymes (phosphatases) responsible for their dephosphorylation. Additionally, these derivatives can induce apoptosis via mitogen-activated protein kinase and caspase-3 activation, inducing reactive oxygen species (ROS) production, and apoptosis. However, vitamin K1 exhibits only weak inhibition of phosphatase activity, while the ability of VK3 to cause oxidative DNA damage has raised concerns about carcinogenicity. Hence, in the current study, we designed, synthesized, and screened a number of VK3 analogs for their ability to enhance phosphorylation activity, without inducing off-target effects, such as DNA damage. 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) assay revealed that each analog produced a different level of cytotoxicity in the Jurkat human leukemia cell line; however, none elicited a cytotoxic effect that differed significantly from that of the control. Of the VK3 analogs, CPD5 exhibited the lowest EC50, and flow cytometry results showed that apoptosis was induced at final concentrations of ≥10 μM; hence, only 0.1, 1, and 10 μM were evaluated in subsequent assays. Furthermore, CPD5 did not cause vitamin K-attributed ROS generation and was found to be associated with a significant increase in caspase 3 expression, indicating that, of the synthesized thioether VK3 analogs, CPD5 was a more potent inducer of apoptosis than VK3. Hence, further elucidation of the apoptosis-inducing effect of CPD5 may reveal its efficacy in other neoplastic cells and its potential as a medication.
Introduction
Vitamin K can be divided into vitamin K1 and K2, which constitute the main naturally occurring forms (Figure 1A and B), and vitamin K3, which is artificially synthesized (Figure 1C). Vitamin K1 and K2, also known as phylloquinone and menaquinone, respectively, have procoagulant activity and act as cofactors for the γ-carboxylation of glutamate residues on prothrombin and other vitamin K-dependent coagulation factors.1,2 Moreover, as drugs, they are indicated for the treatment of vitamin K deficiency, osteoporosis, and warfarin toxicity. Structure of vitamin K vitamers. (A) Vitamin K1 (phylloquinone; 2-methyl-3 -phytyl-1,4–naphthoquinone), (B) vitamin K2 (menaquinone; 2-methyl-3 -multiprenyl-1,4–naphthoquinone), and (C) vitamin K3 (menadione:2-methyl-1,4–naphthoquinone).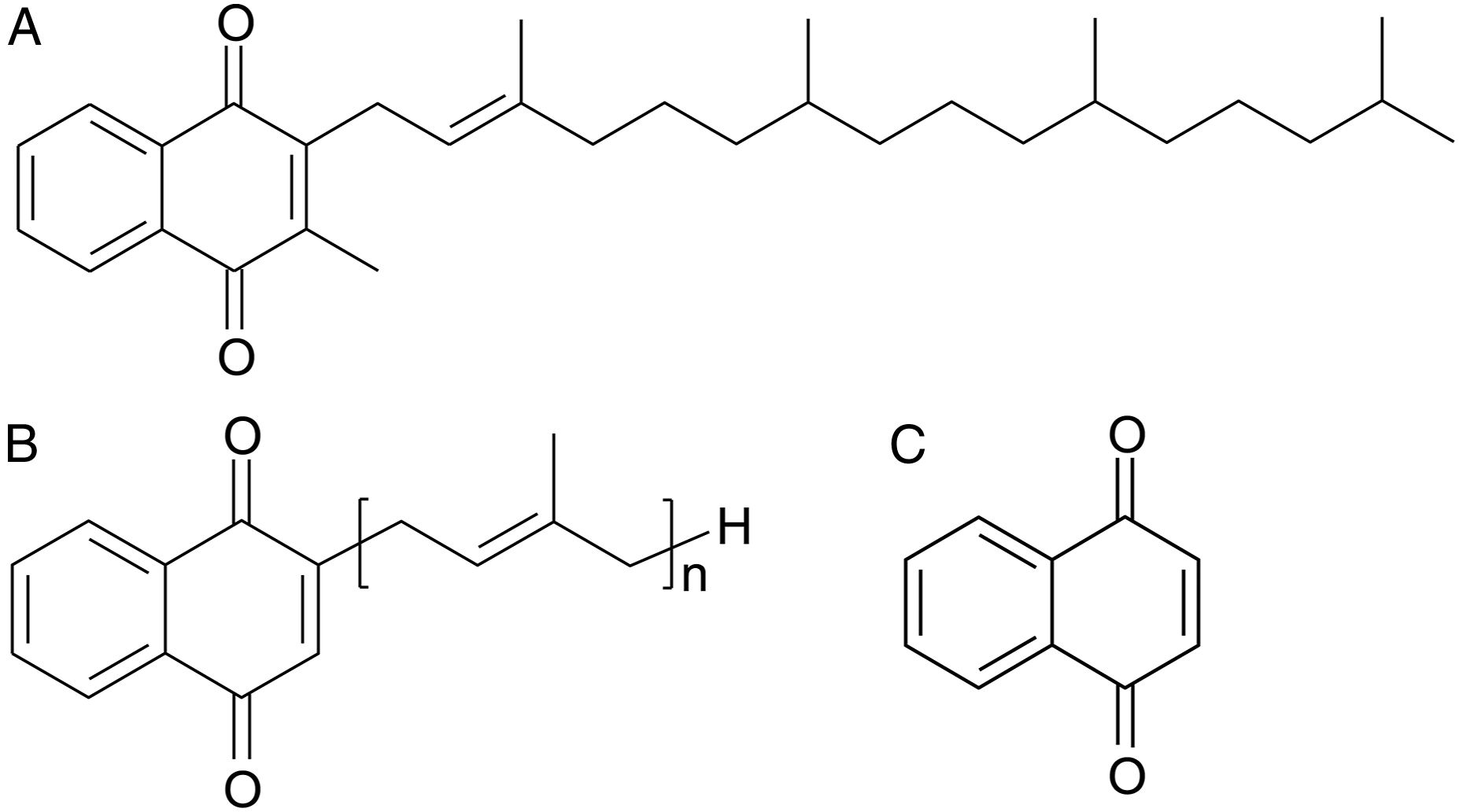
Vitamin K3 (VK3)—also known as menadione—has been shown to exhibit antiproliferative and cytotoxic effects against rodent and human tumor cells,2-5 with a particularly strong activity against neuroblastoma cells. 6 Vitamin K3 primarily acts to directly inhibit disulfide bond formation within certain proteins, thereby disrupting their three-dimensional structure and suppressing their activity.7,8 This inhibitory action has been primarily reported against cytoplasmic protein tyrosine phosphatases (PTPases), enzymes that catalyze the hydrolysis of phosphate groups from transmembrane protein tyrosine kinases (PTKs). 9 Indeed, VK3 has been tested in a phase I clinical trial for patients with advanced solid cancers (e.g., lung, kidney, and skin), in which adverse side effects were not reported. 10 However, as a fat-soluble vitamin (precursor), VK3 is metabolized more slowly than water-soluble vitamins, raising concerns about carcinogenicity risk at high doses due to residual precursors and oxidative DNA damage.11,12 Meanwhile, thioether derivatives of VK3 have been synthesized with the objective of reducing these adverse events. Such analogs directly bind to PTPases, thereby preventing them from removing phosphate groups from PTKs. Specifically, the thioether moiety binds to the sulfhydryl group of cysteine in the PTPase active site, forming a disulfide bridge, thereby maintaining PTK phosphorylation and activating downstream signaling pathways. One VK3 analog (compound 5 [CPD5: 2-(2-mercaptoethanol)-3-methyl-1,4-naphthoquinone]) with a hydroxy group at the end of its thioether side chain seems to show particular promise. In fact, this derivative is reportedly a potent promoter of sustained epidermal growth factor receptor phosphorylation 13 and exhibits antiproliferative effects through direct inhibition of the cell-cycle regulatory protein Cdc25 A, causing cell-cycle arrest. 1
Recent evidence indicates that VK3 can induce apoptosis (i.e., genetically programmed cell death) in various tumor cell lines of human, rat, and mouse origin.14-18 This physiological phenomenon is essential to organismal maintenance and can be distinguished from other types of cell death by specific morphological characteristics, including nuclear condensation/fragmentation, changes in membrane structure, and formation of apoptotic bodies. 19 Since the cell membrane is not destroyed during apoptosis, pro-apoptotic agents may represent ideal anticancer drugs capable of inducing apoptosis in tumor cells with few adverse effects. 20 Current research suggests that VK3-induced apoptosis involves reactive oxygen species (ROS) production and enhanced expression of the apoptosis-inducing Fas receptor and its ligands;14,16 however, activation of this mechanism requires mitogen-activated protein kinases (MAPKs) and caspase-3.21-23 Likewise, inhibition of PTPase by CPD5 can suppress cell proliferation through the resulting MAPK activation13,24 and induce apoptosis by prolonging the phosphorylation of extracellular signal-regulated kinases (ERKs), a MAPK subset.25,26 Hence, CPD5 may induce apoptosis by inhibiting PTPases, thereby indirectly activating MAPKs.
Cumulatively, the evidence presented above indicates that vitamin K3 analogs could serve as novel antineoplastic agents, capable of selectively inducing apoptosis in cancer cells. In the current study, novel VK3 analogs were synthesized and screened to identify those with stronger antitumor activity than VK3 (Figure 2). Moreover, existing and novel vitamin K analogs were screened for their influence on cellular DNA damage, and the intracellular signaling mechanisms associated with analog-induced apoptosis were investigated. Menadione and VK3 analogs used in experiments. (D) 2-(2-mercaptoethanol)-3-methyl-1,4-naphthoquinone (OH), (E) 2-(2-mercaptoethanol-O-methylether)-3-methyl-1,4-naphthoquinone (OCH3), (F) 2-(2-mercaptoethanol-O-methoxymethylether)-3-methyl-1,4- naphthoquinone (MOM), (G) S-(2-methyl-1,4-naphthoquinonyl-3)-β- mercaptopropionic acid (COOH), (H) 2-(2,3-dihydroxy-propylsulfanyl)-3-methyl-1,4-naphthoquinone (Diol), (I) 2-methyl-3-[2-(tetrahydro-pyran-2-yloxy)-ethylsulfanyl]-1,4-naphthoquinone (THP), (J) 2-(2,2-dimetyl-1,3dioxolan-4-ylmethylsulfanyl)-3-methyl-1,4-naphthoquinone (Acetal), (K) 2-[2-O-(β-D-glucosyl)-2-hydroxylthioethyl]-3-methyl-1,4-naphthoquinone (Glc), (L) 2-[2-O-(β-D-galactosyl)-2-hydroxylthioethyl]-3-methyl-1,4-naphtoquinone (Gal), and (M) 2-[2-O-(β-D-lactosyl)-2-hydroxylthioethyl]-3-methyl-1,4-naphtoquinone (Lac).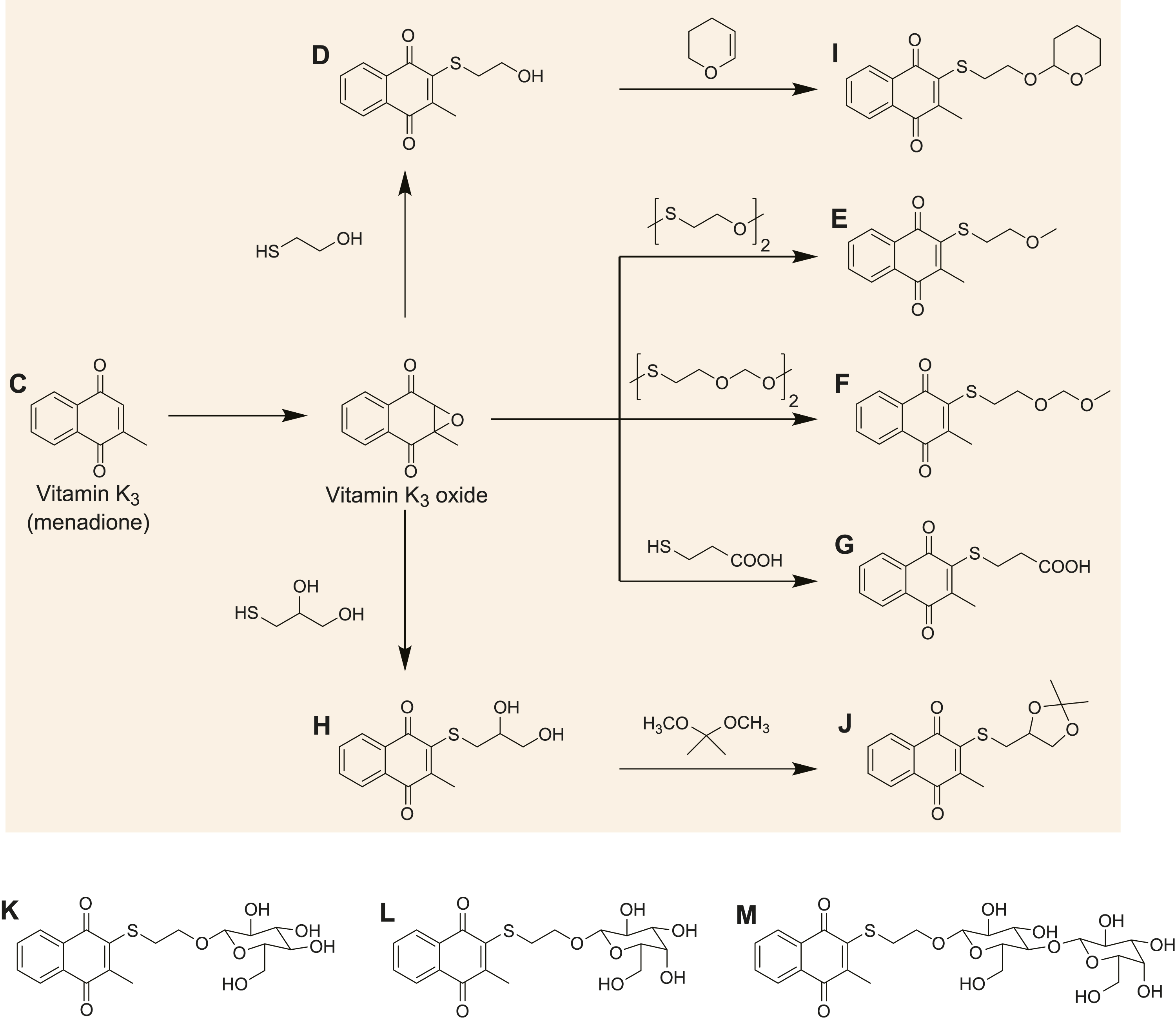
Materials and Methods
Cell Culture
Jurkat cells (RCB0806), a human non-adherent leukemia T-cell line, were obtained from RIKEN (Tsukuba, Ibaraki, Japan). Cells were grown in RPMI-1640 medium (Gibco-Invitrogen, Carlsbad, CA, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Gibco-Invitrogen), 100 μg/mL streptomycin (Gibco-Invitrogen), and 100 units/mL penicillin (Gibco-Invitrogen) at 37°C and 5% CO2.
Reagents (VK3 Analogs)
Vitamin K is a fat-soluble vitamin, the structure of which is presented in Figure 1. Vitamin K analogs (Figure 2D-H-H; verified by 1H and 13C NMR spectroscopy) were synthesized with a thiol group and a terminal oxide group so that they had the hydrophilicity of VK3. Ring structures (Figure 2I and J) were also added to analogs Figure 2D and H to alter their steric structures near the oxide group. Analog (Figure 2-K) was synthesized by attaching glucose to the OH group on analog Figure 2D. The analog Figure 2L was synthesized by attaching galactose to the OH group on analog Figure 2D, while analog Figure 2M was synthesized by attaching lactose to the OH group on analog Figure 2D. Each VK3 analog was dissolved in dimethyl sulfoxide (DMSO) to a final concentration of 50 mM and stored at −20°C in the dark. Although DMSO was used as the reagent in all experiments to ensure consistency, COOH (Figure 2G) can be dissolved in polar solvents by using saturated sodium bicarbonate. Hence, the uptake of this compound by cells might be improved using an aqueous solvent, which will be investigated in future studies.
Cytotoxicity (MTT) Assay
Characteristic features of apoptosis include cell contraction, cell membrane blebbing, chromosome compaction (pyknosis), nuclear fragmentation (karyorrhexis), DNA laddering, and cell phagocytosis. Considering that apoptotic, necrotic, and other types of cell death, arise from multiple pathways and differ morphologically and biochemically, cell death was confirmed by measuring insoluble formazan color development (blue) associated with the reduction of 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT), a tetrazolium salt.21-29 Living cells produce the formazan color product as MTT reduction is catalyzed by succinate-tetrazolium reductase, a mitochondrial reduction enzyme highly active in living cells and inactive following cell death.30,31 This experiment was performed using the MTT (M009) assay [Dojindo Molecular Technologies (Kumamoto, JAPAN)]. Briefly, Jurkat cells were seeded in a 96-well microplate (1 × 104 cells/well) and incubated with each VK3 analog for 48 h. Next, MTT dye was added to the wells, and cells were incubated for 3 h at 37°C and 5% CO2. A stop solution (0.04 M HCl in isopropanol) was then added to the wells and mixed well. Microplate absorbance was measured at 570 nm and 655 nm to eliminate background noise due to turbidity/contamination of the culture solution.
Detection of Mitochondrial Transmembrane Potential Disruption (ΔΨm) (DePsipher flow cytometry method Assay)
Mitochondrial functioning, including adenosine triphosphate (ATP) production, breaks down during apoptosis. When mitochondrial permeability transition pores open, the mitochondrial membrane potential (ΔΨm) is lost and cytochrome c is released into the cytoplasm, providing confirmation of apoptosis. 33 Hence, we sought to assess the changes in ΔΨm following treatment of cells with the most promising analog (OH (CPD5), Figure 2D), which was achieved using the DePsipher™ kit (6300-100-K; TREVIGEN, Gaithersburg, MD, USA). Briefly, Jurkat cells were seeded in a 6-well plate (1 × 106 cells/well) and incubated with OH (CPD5; Figure 2D) for 24 h. DePsipher reagent (staining solution) was added to the wells, and the plate was incubated for 20 min at 37°C and 5% CO2. Cells were then washed with reaction buffer and analyzed using a flow cytometer (Cytomics FC500; Beckman Coulter Inc, Koto-ku, Tokyo, Japan) with an Ar laser (excitation wavelength: 488 nm) and two detection filters (FL1: 525 nm, FL2: 575 nm).
Detection of Membrane Structural Changes (Annexin V–fluorescein isothiocyanate flow cytometry method Assay)
In living organisms, cells that undergo apoptosis are removed by macrophages and other phagocytes. These phagocytes have been shown to recognize apoptotic changes on the cell surface. One such change is the loss of asymmetry in cell membrane phospholipids. Specifically, phosphatidylserine (PS), normally found in the inner leaflet of the lipid bilayer, is translocated to the outer leaflet during apoptosis. Annexin V can detect apoptotic cells as it binds to PS exposed on the cell surface. Fluorescein isothiocyanate (FITC)-labeled annexin V and PI can also be used to distinguish between early- and late-stage apoptosis. To assess apoptosis in CPD5-treated cells, the annexin V–FITC Kit was used (IM3546; Beckman Coulter Inc.). Briefly, Jurkat cells were seeded in a 6-well plate (1 × 106 cells/well) and reacted with OH (CPD5; Figure 2D) for 24 h. Next, they were rinsed with phosphate buffered saline (PBS) and annexin binding buffer (ABB: 10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2; pH 7.4), after which, 5 μL of annexin V–FITC (Molecular Probes™) were added to 100 μL of cell solution in the presence of ABB. Flow cytometry was performed 20 min later using an Ar laser (excitation wavelength: 488 nm) and one detection filter (FL1: 525 nm).
Quantification of ROS Production
Reactive oxygen species generation is an unavoidable phenomenon in aerobic organisms that are well-controlled in healthy cells. However, cells have been shown to generate ROS in the presence of VK3, prompting us to measured ROS production levels in cells to investigate whether VK3 analogs induced apoptosis through ROS generation or through intracellular signaling mechanisms. To this end, the ROS Detection Cell-Based Assay kit (601520; Cayman Chemical, Ann Arbor, MI, USA) was employed. Jurkat cells were seeded in a 96-well microplate (1 × 104 cells/well) and incubated with OH (Figure 2D) for specific periods of time (30, 60, 120, 240, and 360 min). Next, DCFH-DA was added to the wells at a final concentration of 10 μM, and the cells were incubated for 20 min at 37°C and 5% CO2. Fluorescence was measured using a microplate reader (excitation filter, 485 nm; emission filter, 520 nm).
Caspase-3 Activity Assay
A characteristic feature of early apoptosis is the activation of caspases that cleave protein substrates, thereby resulting in cell degradation. Of these caspases, caspase-3 is located downstream of the intracellular signaling that is active during induced apoptosis. Hence, caspase-3 activity was assayed using the CaspSELECT™ Caspase-3 Immunoassay kit (BioVision, Inc, Milpitas, CA, USA). Briefly, Jurkat cells were seeded in a cell culture dish (60 × 15 mm; 5 × 106 cells/dish) and subsequently reacted with OH (CPD5) (Figure 2D) or VK3 for a specified length of time (1, 4, 8, and 24 h). Next, the dish was washed with PBS, and cell lysates were used as test samples. Subsequent steps were performed according to the manufacturer’s instructions.
Results
Cytotoxicity (MTT) Assay
Significant differences in Jurkat cell viability, compared to the DMSO vehicle control, were observed after treatment with VK3 and each of its derivatives (Figure 3). Cell viability was significantly higher at all concentrations >0.1 μM in the OH (CPD5, Figure 3B) and OCH3 (Figure 3D) groups (P <.001 for all), and at all concentrations >0.3 μM in the diol (Figure 3C), THP (Figure 3F), MOM (Figure 3G), and acetal (Figure 3I) groups (P <.001 for all). Meanwhile, it was significantly higher at concentrations >1 μM in the menadione (Figure 3A) group (1 μM: P < .01, >3 μM: P <.001) and the oxide group (Figure 3E μM: P <.05, >3 μM: P <.001), as well as at concentrations >3 μM in the COOH (Figure 3H) group (3 μM, P <.01; >10 μM, P <.001). Additionally, glycoside derivatives of VK3 did not impact the viability of Jurkat cells that were only significantly reduced by galactose (Figure 3K), glucose (Figure 3J), and lactose (Figure 3L) at the highest concentration (30 μM: P <.001, P <.01, P <.01, respectively). Cytotoxic activity of VK3 and its analogs. Jurkat cell viability (%) after 48 h exposure to VK3 and VK3 analogs ((A) menadione, (B) OH, (C) diol, (D) OCH3, (E) oxide, (F) THP, (G) MOM, (H) COOH, (I) Acetal, (J) Glc, (K) Gal, and (L) Lac). The exposure concentration of the respective compound is plotted on the X-axis, and the cell viability with respect to the control (DMSO-treated; vehicle control) group is plotted on the Y-axis. Results were tested by one-way ANOVA with Bonferroni correction (* P < .05, n = 3).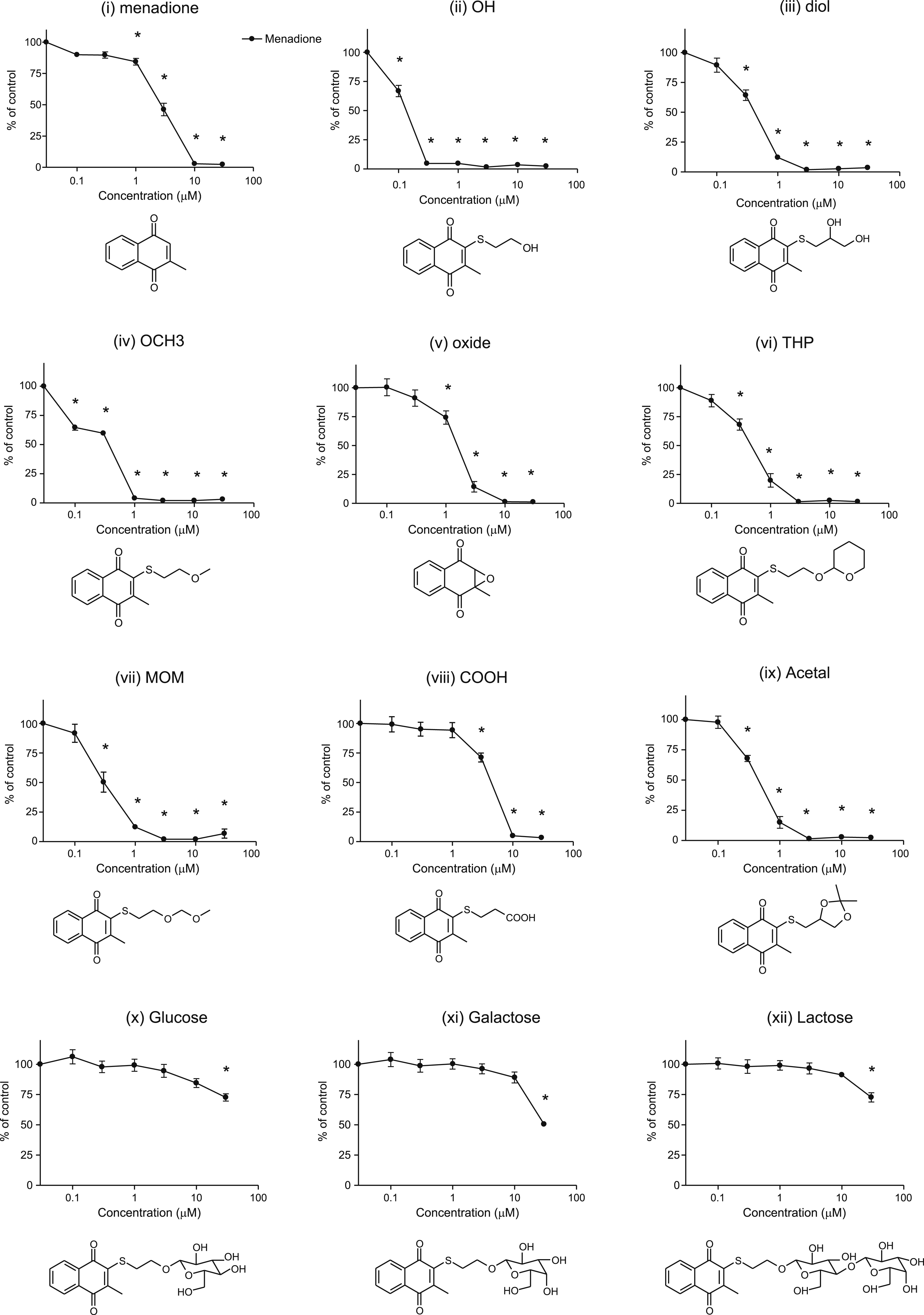
Next, sigmoidal dose-response curves were constructed based on the cytotoxicity results for VK3 and each analog, excluding the three glycoside derivatives, which are shown overlaid on the original data in Figure 4. These curves were used to estimate the half-maximal effective concentration (EC50) values for each compound, as shown in Table 1. Figure 5 presents the cytotoxicity of each VK3 analog (0.3 μM) after 48 h of exposure, compared to 48 h exposure to 0.3 μM VK3. Statistically significant differences were observed with respect to the cytotoxicity of VK3 for (ii) OH (CPD5), (iii) MOM, (iv) OCH3, and (v) Diol. Cytotoxic activity of VK3 and its analogs (sigmoidal dose-response curves). Graphs overlay the MTT assay data in Figure 3 with a sigmoidal dose-response curve. The exposure concentration of the respective compound (logM) is plotted on the X-axis; cell viability with respect to a control (DMSO-treated; vehicle control) group is plotted on the Y-axis. EC50 Values of VK3 and VK3 Analogs. EC50 values (logM, μM) of menadione and VK3 analogs. Calculated based on the data shown in Figure 4. Cytotoxic activity of VK3 analogs (0.3 μM) with respect to VK3 (0.3 μM). Jurkat cell viability after 48 h exposure to each VK3 analog (0.3 μM) calculated as a percentage of the viability measured for VK3 (0.3 μM)-treated cells. The dotted line indicates the reference viability of VK3-treated cells (100%). Significance testing was performed using one-way ANOVA with post-hoc Bonferroni correction (*P < .05, **P < .01, ***P < .001).mena, menadione; OHC3, OCH 3; Glc, glucose; Gal, galactose; Lac, lactose.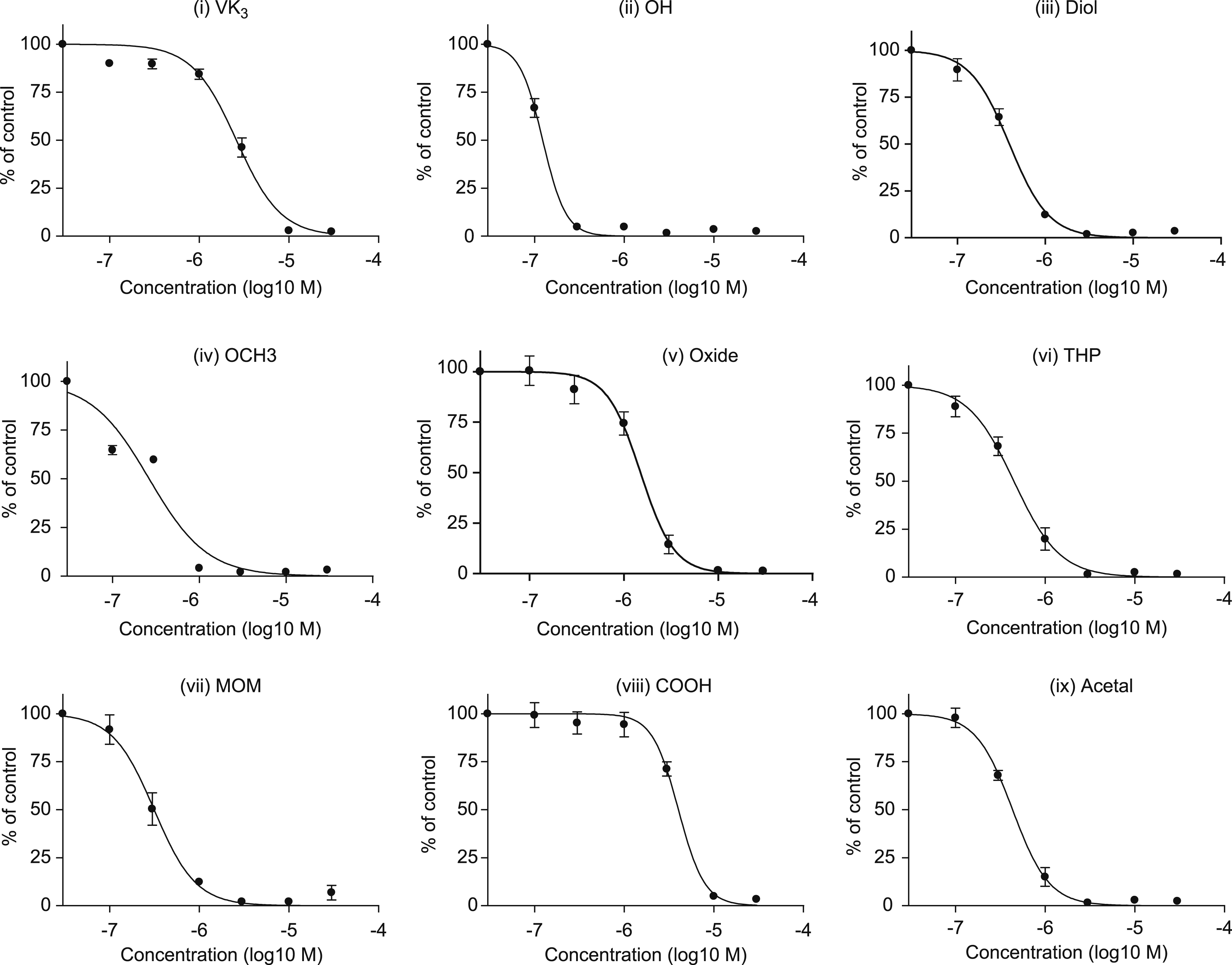
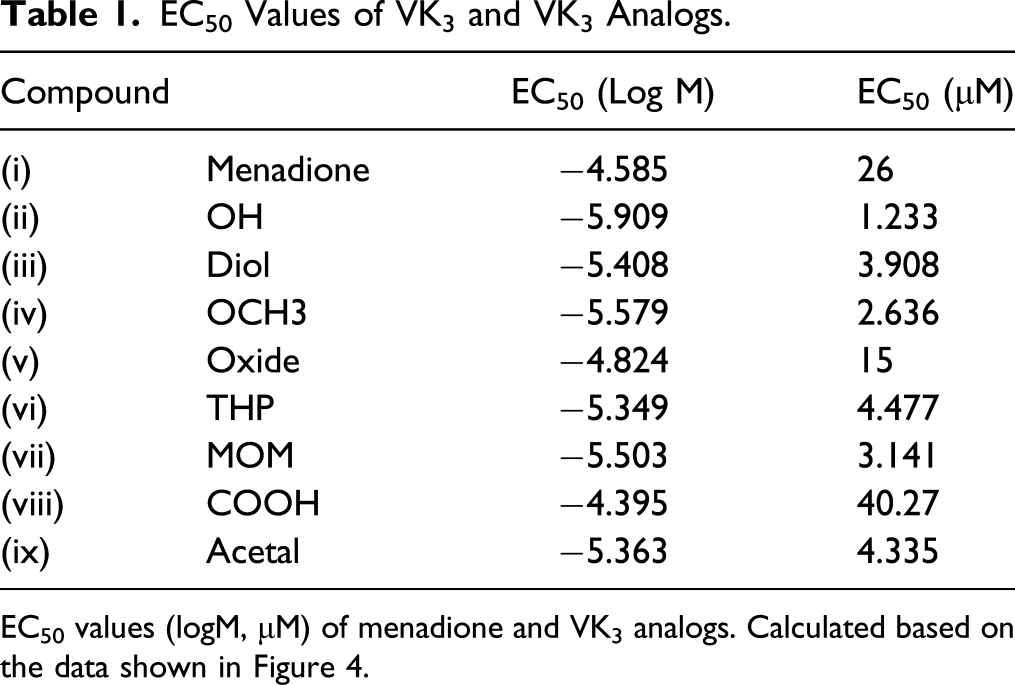
These results confirmed that VK3 and its analogs possess cytotoxic activity against Jurkat cells, and that the cytotoxicity of all analogs, except for (viii) COOH and the three glycoside derivatives (x-xii), was stronger than that of VK3. Nonetheless, as (ii) OH (CPD5) exhibited the highest cytotoxicity and the lowest EC50 of all analogs, only this derivative, designated as CPD5 from this point forward, was assessed in all subsequent experiments.
Membrane Structural Changes (Annexin V–FITC FCM)
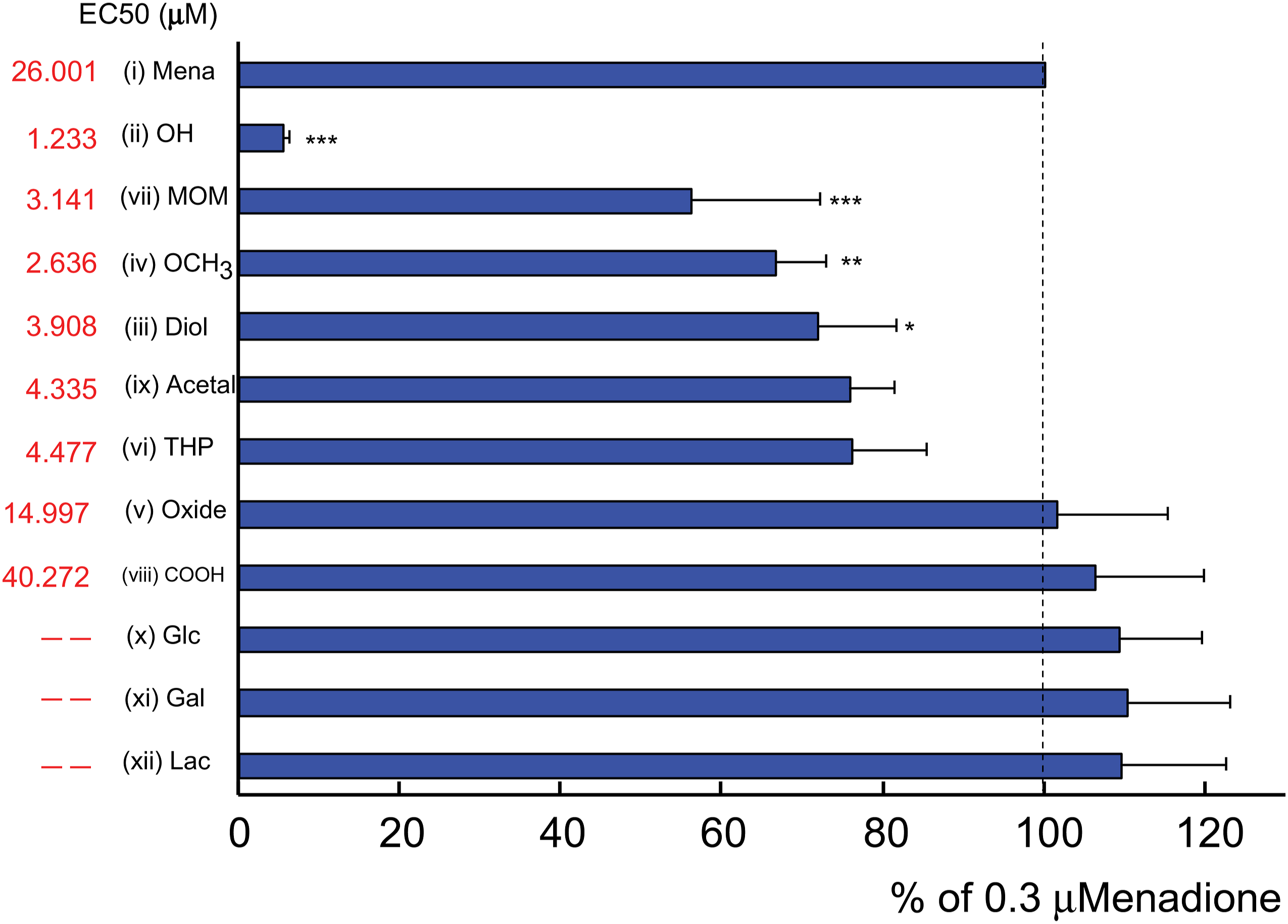
Flow cytometry was used to identify changes in cell membrane structure, a hallmark of apoptosis, to verify whether CPD5 cytotoxicity in Jurkat cells was attributable to apoptosis induction (n = 3). The forward scatter (FS)-side scatter (SS) dot plots in Figure 6A illustrate the relationship between cell size (FS Lin) and granularity (SS Lin). Cells shrink when apoptosis occurs, decreasing FS Lin while increasing SS Lin. Following the treatment of the control cells with DMSO only, a single cell population is visible in the dot plot; meanwhile, following 24 h reaction with 0.1-10 μM CPD5, two distinct cell populations are apparent (Figure 6A), one of which represents the apoptotic cell population with lower FS and higher SS, while the other represents non-apoptotic cells (“(A)” in Figure 6A). The apoptotic cell cluster grows became larger with increasing exposure concentrations, substantiating the occurrence of morphological changes characteristic of apoptosis. FS-SS dot plots and FITC histograms for CPD5-treated Jurkat cells. Jurkat cells treated with respective concentrations of CPD5 for 24 h, stained with annexin V–FITC, and analyzed by FCM. Cells treated with dimethyl sulfoxide (DMSO; vehicle control) were used as controls. (A) A distinct population of cells with hallmark morphological features of apoptosis, labeled 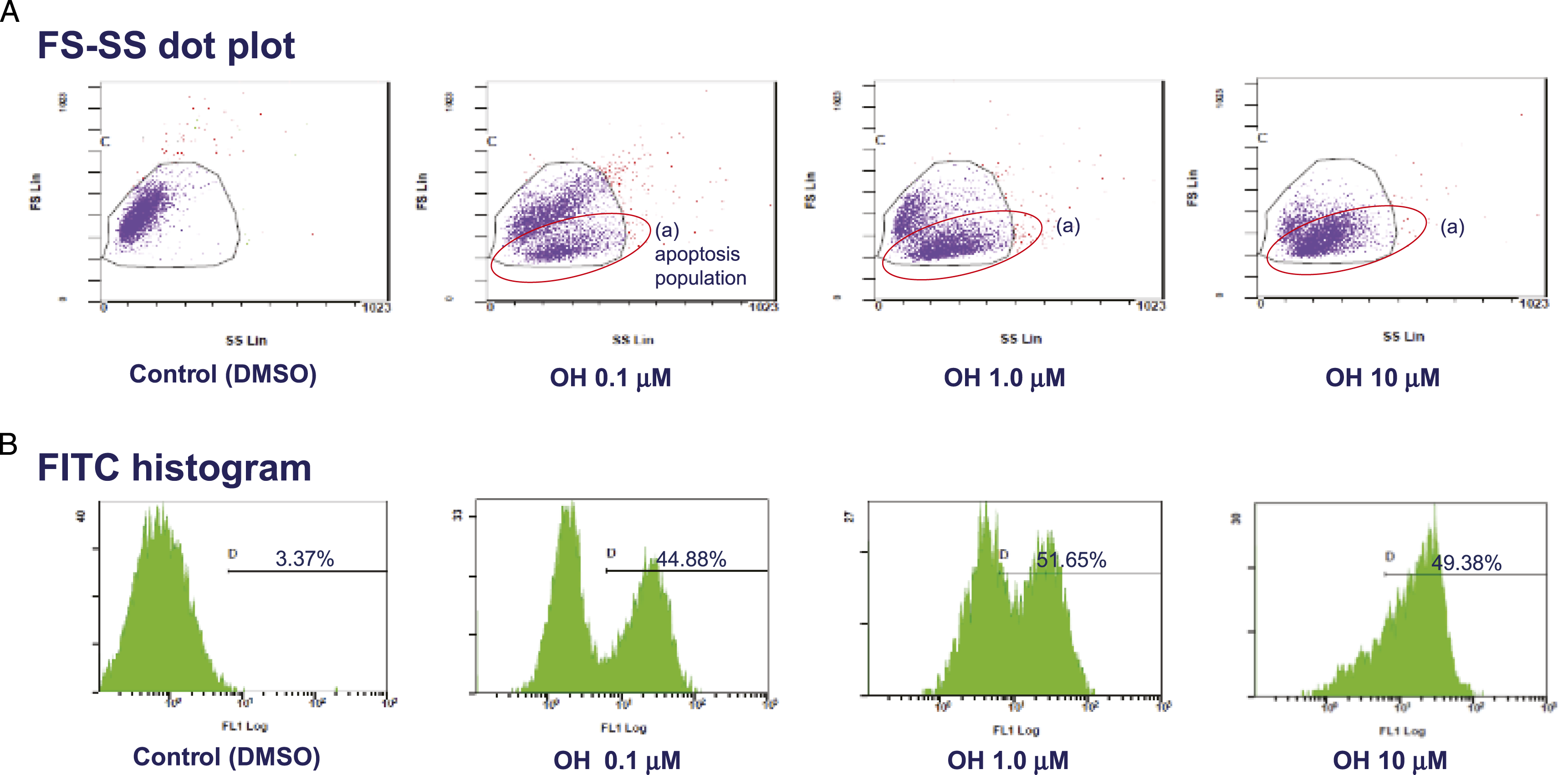
Apoptotic-related changes in the cell membrane structure include PS flipping from the inner to outer leaflet; as PS in the outer leaflet binds with annexin V–FITC, this apoptotic change increases the fluorescence intensity of FITC. This increase in intensity is shown by the FITC histograms in (Figure 6B). A gate (D) was applied to the histograms in (Figure 6B) to measure a higher range of fluorescence intensities, and FITC fluorescence intensity was determined based on the percentage of total cells detected within this gate (above D). Treatment with CPD5 resulted in a greater proportion of gated cells than with the control at any concentration (0.1 μM: 44.88%, 1.0 μM: 51.65%, 10 μM: 49.38% vs DMSO: 3.37%), thereby confirming that exposure to CPD5 caused structural changes in the cell membrane of Jurkat cells.
Mitochondrial Membrane Potential Disruption (ΔΨm)
Figure 7 shows the results of flow cytometry method (FCM) analysis of mitochondrial membrane potential in Jurkat cells treated with different concentrations of CPD5 for 24 h (0.1, 1.0, and 10 μM). The FL2 signal is enhanced when mitochondria function normally, as monomeric JC-1 (5,5,6,6-tetrachloro-1,1,3,3-tetraethylbenzimidazolylcarbocyanine iodide) is taken up by the organelles to form orange-fluorescent aggregates.32,33 However, dysfunctional mitochondria amplify the FL1 signal as JC-1 remains in the cytoplasm and retains its original green fluorescence. The FL2 signal was strongest in the control group (percentage gated in quadrant B1, 65%; B2, 27%; B4, 6.53%), whereas in the 0.1 μM CPD5 group, this signal was weaker, while the FL1 signal was significantly amplified (B1, 0.03%; B2, 0.06%; B4, 99.30%). Weak FL2 and strong FL1 signals were similarly observed in the 1.0 μM and 10 μM conditions (1.0 μM—B1: 0.02%, B2: 0.28%, B4: 99.68%; 10 μM—B1: 0.02%, B2: 0.21%, B4: 99.50%). Furthermore, the FL1 histograms illustrate that nearly all cells were detected within the corresponding gate (C), with significantly higher percentages of gated cells in all CPD5 treatment conditions than the control group (0.1 μM: 99.75%, 1.0 μM: 99.90%, 10 μM: 98.92% vs DMSO: 31.66%). Alternatively, the FL2 histograms showed markedly lower fluorescence in CPD5-treated cells than in the control group, resulting in much lower percentages of gated (D) cells (0.1 μM: 4.67%, 1.0 μM: 5.02%, 10 μM: 3.47% vs DMSO: 98.23%). In summary, the control cells exhibited high FL2 and low FL1 fluorescence as JC-1 formed aggregates within the mitochondria, as expected under physiological conditions. Meanwhile, following exposure to CPD5, FL1 intensity increased and FL2 intensity decreased as JC-1 could not enter the compromised mitochondria to form aggregates, and was instead retained in the cytoplasm. Notably, a considerable fraction of cells also fell within the FL1 gate in the control group, suggesting the presence of residual JC-1 that failed to be taken up by the mitochondria. FCM data for mitochondrial membrane potential disruption (ΔΨm). TTC coloration detected using FCM. Filters FL1 and FL2 measure green and red fluorescence, respectively. (Left) FL1-FL2 dot plot. FL1 and FL2 signal intensities plotted on the X- and Y-axes, respectively. The inset number indicates the percentage of total cells detected within the respective quadrant. (Center, Right) FL1 and FL2 histograms. Numbers signify the percentage of total (baseline) cells within the respective gates (FL1: C, FL2: D). Cells treated with dimethyl sulfoxide (DMSO; vehicle control) were used as controls. FCM: flow cytometry method; TTC: 5,5ʹ,6,6ʹ-tetrachloro-1,1ʹ,3,3ʹ-tetraethylbenzimidazolyl carbocyanine iodide.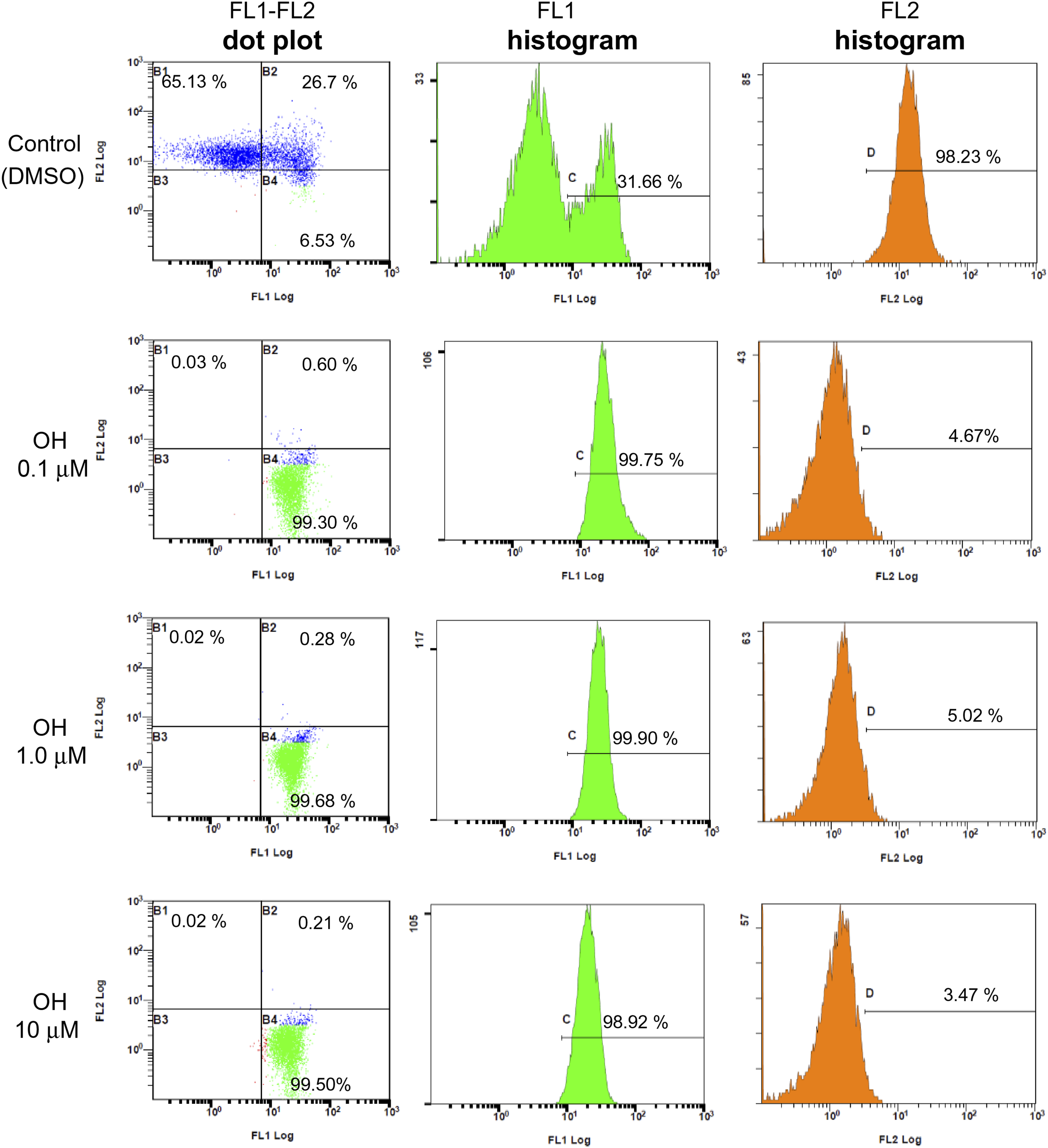
Quantification of ROS Production
Reactive oxygen species production was hypothesized to be responsible for the apoptosis induced by CPD5, as the same mechanism is known to induce apoptosis in cells treated with VK3. 14 Therefore, the ROS generated by Jurkat cells in response to CPD5 exposure was quantified (n = 3) in accordance with the experimental protocol of Jeon et al. 34
Figure 8 depicts the amount of ROS produced by Jurkat cells treated with CPD5 (0.1, 1.0, and 10.0 μM) for different lengths of time (30, 60, 120, 240, and 360 min) as a percentage of the control group. Exposure to CPD5 did not result in substantial elevation of ROS at most concentrations and time points. Reactive oxygen species production by CPD5. ROS generated by 30-, 60-, 120-, 240-, and 360- min exposure to (A) 0.1, (B) 1.0, and (C) 10.0 μM. Exposure time (min) is plotted on the X-axis; ROS production with respect to the control (DMSO; vehicle control) group (%) is plotted on the Y-axis. In addition, the ROS value measured under the normal culture conditions was used as the background. Significance testing was performed using one-way analysis of variance with post-hoc Bonferroni correction. Abbreviations: DMSO: dissolved in dimethyl sulfoxide; ROS: reactive oxygen species.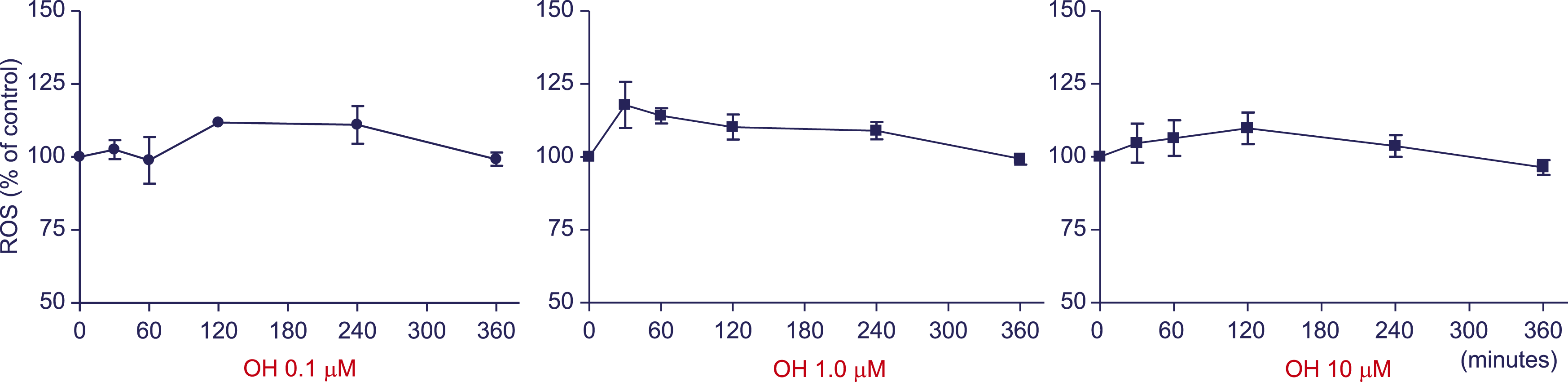
Caspase-3 Activity
Considering that caspase-3 is a major effector protein involved in apoptosis, its activation levels in response to VK3 and CPD5 were measured by immunological assay and subsequently compared to determine whether the latter was a more potent inducer of apoptosis. Caspase-3 was selectively captured from the cell lysates using caspase-3 antibodies. When captured caspase-3 is in its active form, it cleaves DEVD-7-Amino-4-trifluoromethylcoumarin (AFC) to release fluorescent AFC. Hence, caspase-3 activity was quantified in terms of AFC fluorescence intensity using a spectrofluorometer.
AFC fluorescence was more intense in the CPD5 and VK3 groups than in the control (DMSO) group; however, it was substantially stronger in the CPD5 group than in the VK3 group (Figure 9). Caspase-3 activation levels. Caspase-3 activation levels measured using DEVD-AFC, a substrate that fluoresces in the presence of activated caspase-3. The exposure time (h) is plotted on the X-axis, and AFC fluorescence intensity is plotted on the Y-axis. •: CPD5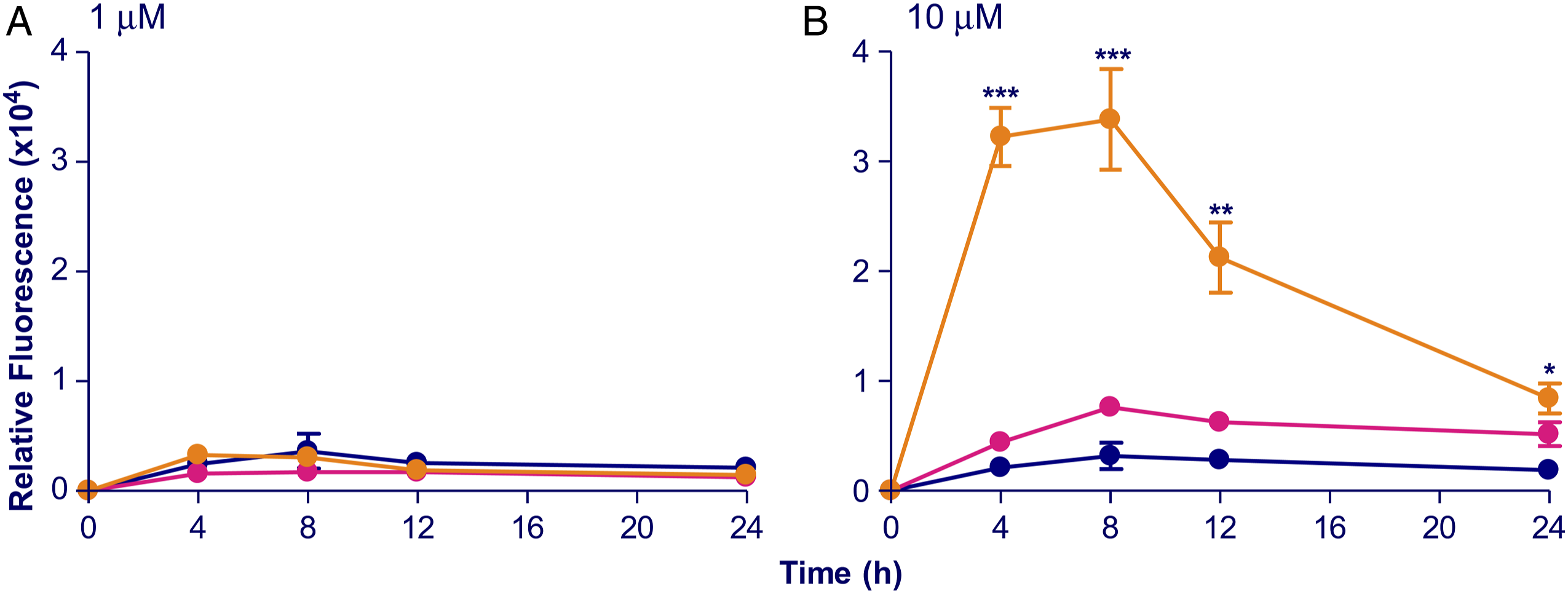
Discussion
Using an MTT protocol to screen each analog for cytotoxicity revealed that each produced a different level of cytotoxic effect; however, none elicited a cytotoxic effect that differed significantly from that of the control (DMSO) group. Of the VK3 analogs, CPD5 exhibited the lowest cytotoxicity EC50, and flow cytometry showed that apoptosis was induced even at final concentrations ≥10 μM. Furthermore, given that CPD5 exposure did not cause ROS production, we can postulate that CPD5 does not cause vitamin K-attributed ROS generation, as described in other reports. Moreover, CPD5 was the only analog associated with a significant increase in caspase 3 expression and, thus, represents the single analog with more potent pro-apoptotic activity than VK3.
We initially screened ten VK3 analogs (Figure 2D-2M), alongside VK3 to identify the compound that possesses the strongest cytotoxic activity. MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) is a yellow dye that is taken up and metabolized to purple formazan by cells with normal mitochondrial function. In this way, MTT absorbance at the corresponding wavelengths reflects mitochondrial activity. 35 Compounds’ cytotoxicity was estimated based on the findings of the MTT assay, as mitochondrial activity is lost during cell death. CPD5—a hydroxy derivative of menadione (Figure 2D: OH (CPD5)), was identified as having the strongest cytotoxic effects.
Plasma membrane inversion, another hallmark of apoptosis, was detected using annexin V–FITC FCM. Phosphatidylserine is a phospholipid species that is normally present in the inner leaflet, but when apoptosis occurs, it becomes exposed on the outer leaflet as a result of this inversion. 36 Annexin V–FITC consists of annexin V, a protein that binds specifically to PS, labeled with the green fluorescent dye FITC. Since this conjugate cannot pass through the cell membrane, FITC only fluoresces when it binds to exposed PS. FCM can then be employed to measure the intensity of this fluorescent signal and to approximate cell size and granularity by extension. Light is scattered when cells are hit by the laser beam: photons that scatter forward and perpendicular with respect to the axis of incidence are measured as FS and SS, respectively. Forward scatter is proportional to cell surface area, and therefore, reflects cell size, while SS is a product of diffraction/refraction of light within cells, reflecting granule density and the complexity of subcellular architecture. Plotting these measures yielded FS-SS dot plots in Figure 6A. When apoptosis occurs, cells shrink and form apoptotic bodies, which tend to reduce FS while increasing SS. In the present work, an apoptotic population of Jurkat cells with low FS and high SS appeared following treatment with CPD5 at all concentrations ≥10 μM, evincing these hallmarks of apoptosis. Likewise, the results shown in Figure 6B confirm the occurrence of membrane reversal. Mitochondrial damage can also trigger apoptosis if the mitochondria membranes lose their potential (ΔΨm). 37 When this membrane polarization is normal, the green dye 5,5ʹ,6,6ʹ-tetrachloro-1,1ʹ,3,3ʹ-tetraethylbenzimidazolyl carbocyanine iodide (TTC) TTC (5,5ʹ,6,6ʹ-tetrachloro-1,1ʹ3,3ʹ-tetraethylbenzimidazolyl carbocyanine iodide) is taken up by the mitochondria, where it forms red/orange-fluorescent aggregates. The results FCM analysis of the current study, confirmed that CPD5 induced apoptosis in Jurkat cells.
Reactive oxygen species production is another mechanism of VK3-induced apoptosis.14,16 To ascertain the ROS-generating potential of its derivative CPD5, this process was evaluated by the DCFH-DA assay. The dye DCFH-DA (2ʹ,7ʹ-dichlorofluorescein diacetate) is taken up by cells as dichlorofluorescein, which undergoes structural changes in the presence of ROS to fluoresce green. 38 Reactive oxygen species generation can then be evaluated based on the intensity of the fluorescent signal. However, negligible levels of ROS were showed little change in the assay results (Figure 8), while stronger cytotoxic activity was observed following exposure to 1.0 μM or 10 μM than 0.1 μM CPD5 (Figure 3B). Therefore, if the apoptosis triggered by this compound in Jurkat cells was dependent on ROS levels, their production should be higher at 1.0 and 10 μM than at 0.1 μM; however, no such tendency was observed. Taken together, these findings indicate that while CPD5 may act to generate ROS, this is not the primary mechanism by which it induces apoptosis in Jurkat cells; therefore, other factors must be responsible.
CPD5 was recently reported to inhibit the activity of PTPases and other phosphatases via direct binding.13,24 Such inhibition can effectively preserve active phosphorylation of CDK4, a cell-cycle regulator and the target of Cdc25 A, another phosphatase inhibited by CPD5 as well as the MAPK intermediary MEK, leading to cell-cycle arrest.1,23,24 Hence, these pathways may also be involved in Jurkat cell apoptosis triggered by CPD5. Previously, we discovered that a different VK3 analog (Figure 2G: COOH) induced cell differentiation by blocking the PTPase-mediated dephosphorylation of Trk, a transmembrane tyrosine kinase involved in this process, thereby prolonging Trk phosphorylation to activate downstream Ras and MAPKs. 5 Elsewhere, reported associations between MAPKs and the apoptosis effector caspase-3 39 suggest that activating MAPK may activate caspase-3 to trigger apoptosis. Thus, it seems plausible that CPD5 induces apoptosis in a similar manner to COOH, that is, through PTPase inhibition and consequent MAPK activation.
In the present study, Jurkat cells treated with CPD5 had damaged mitochondria with compromised membrane potential. Figure 7 indicates that the bulk of mitochondrial activity was extinguished after 24 h exposure to 10 μM CPD5. However, FCM data confirmed that 50% of cells survived in the same conditions (Figure 6), suggesting that the mitochondrial damage observed was directly inflicted by CPD5, and not a secondary consequence of cell death. Damage to the mitochondria frequently acts as an apoptotic trigger. When these organelles sustain damage their membrane potential (ΔΨm) becomes compromised, releasing mitochondrial cytochrome c into the cytosol, where it activates caspase-9. Once activated, caspase-9 activates caspase-3 to induce apoptosis. 40 The activation levels of this apoptosis effector protein were measured in cells treated with VK3 or CPD5 using DEVD-AFC (acetyl-Asp-Glu-Val-aspaminotrifluoromethylcoumarin), a substrate that fluoresces when cleaved by activated caspase-3. Caspase-3 activation was confirmed (Figure 9), supporting the notion that mitochondrial damage due to CPD5 drove apoptosis through the release of cytochrome c into the cytosol. In addition, CPD5 may have promoted indirect cytochrome c release similar to that reported for another compound belonging to the naphthoquinone family that was found to inhibit cdc25, thereby inhibiting the phosphorylation of Akt, a protein that suppresses cytochrome c release from mitochondria. 41 These findings underscore the plausibility of several mechanisms for CPD5-induced apoptosis in Jurkat cells, including phosphatase (e.g., PTPase) inhibition, mitochondrial inhibition, or a combination of both. Furthermore, CPD5 appears to induce apoptosis more effectively than VK3, as it resulted in greater caspase-3 activation (Figure 9). Going forward, the authors intend to further decipher the apoptotic mechanism induced by CPD5.
Nearly all currently employed antineoplastic drugs used in oncotherapy kill cancer cells as a result of their cytotoxic properties; however, due to their poor target selectivity, they can also attack normal cells, causing necrosis and inflammation. Hence, patients taking these drugs often suffer from adverse side effect, which can be quite severe and even (rarely) fatal. Targeted molecular therapies (TMTs) are being developed to address these issues by targeting antigen proteins specifically expressed by cancer cells, affording these agents high selectivity. However, their clinical application is complicated by their narrow range of indications; for example, they cannot be used in patients who do not express the target antigen. Hence, there has been a shift toward developing antineoplastic drugs that can trigger apoptosis without an accompanying inflammatory response. Killing cancer cells by inducing apoptosis allows tumors to be treated without producing inflammatory reactions, such as necrosis. In fact, apoptotic effects have been reported for several antineoplastic agents currently used in oncotherapy, such as camptothecin, etoposide, and cisplatin. 17 Unfortunately, the poor selectivity of these drugs against cancer cells means they harm normal cells as well, thereby raising expectations for antineoplastic agents that kill cancer cells by inducing apoptosis while retaining the specificity of TMTs.
Although the pro-apoptotic capacity of CPD5 was confirmed in the present study, its specificity against cancer cells remains to be assessed. For specificity examination, it is indispensable to examine the usefulness of CPD5 by increasing the number of tumor types. Similarly, there is an urgent need to elucidate the mechanism of action of CPD5. In the future, we aim to close these knowledge gaps and demonstrate the effectiveness of CPD5. Nonetheless, we presume that exploiting PTPase inhibition, or similar effects, to develop therapeutics with high specificity against cancer cells is by no means impossible.
Finally, retinoids, as well as other differentiation-induction therapies, have been shown to possess antitumor activity against undifferentiated tumors, such as leukemia and neuroblastoma. 42 Hence, retinoic acid (all-trans retinoic acid, 9-cis retinoic acid, 13-cis retinoic acid, and so on), which has both differentiation- and apoptosis-inducing properties, has been applied as an antineoplastic agent in acute myeloid leukemia. 43 In conclusion, VK3 analogs, especially CPD5, are promising therapeutic agents with antitumor properties, based on their ability to induce cell differentiation in addition to inhibiting cell proliferation via induction of apoptosis.
Footnotes
Author Contributions
Asami, S. contributed to conception and design and contributed to acquisition, analysis, and interpretation; Suzuki, M. contributed to design and contributed to acquisition and analysis; Nakayama, T. contributed to design and contributed to acquisition and analysis; Shimoda, Y. contributed to design and contributed to acquisition and analysis; Miura, M. contributed to design and contributed to acquisition and analysis; Kato, K. contributed to conception and contributed to acquisition and analysis; Tokuda, E. contributed to design and contributed to acquisition and analysis; Ono, S. contributed to conception and contributed to interpretation; Kawakubo, T. contributed to conception and contributed to interpretation; Nishizawa, K. contributed to conception and contributed to interpretation; Yamanaka, K. contributed to conception and contributed to interpretation; and Suzuki, T. contributed to conception and contributed to interpretation. All authors drafted manuscript, critically revised manuscript, gave final approval, and agree to be accountable for all aspects of work ensuring integrity and accuracy."
Declaration of Conflicting Interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.