
Abstract
Myostatin is a negative regulator of skeletal muscle and has become a therapeutic target for muscle atrophying disorders. Although previous inhibitors of myostatin offered promising preclinical data, these therapies demonstrated a lack of specificity toward myostatin signaling and have shown limited success in the clinic. Apitegromab is a fully human, monoclonal antibody that binds to human promyostatin and latent myostatin with a high degree of specificity, without binding mature myostatin and other closely related growth factors. To support the clinical development of apitegromab, we present data from a comprehensive preclinical assessment of its pharmacology, pharmacokinetics, and safety across multiple species. In vitro studies confirmed the ability of apitegromab to inhibit the activation of promyostatin. Toxicology studies in monkeys for 4 weeks and in adult rats for up to 26 weeks showed that weekly intravenous administration of apitegromab achieved sustained serum exposure and target engagement and was well-tolerated, with no treatment-related adverse findings at the highest doses tested of up to 100 mg/kg and 300 mg/kg in monkeys and rats, respectively. Additionally, results from an 8-week juvenile rat study showed no adverse effects on any endpoint, including neurodevelopmental, motor, and reproductive outcomes at 300 mg/kg administered weekly IV. In summary, the nonclinical pharmacology, pharmacokinetic, and toxicology data demonstrate that apitegromab is a selective inhibitor of proforms of myostatin that does not exhibit toxicities observed with other myostatin pathway inhibitors. These data support the conduct of ongoing clinical studies of apitegromab in adult and pediatric patients with spinal muscular atrophy (SMA).
Introduction
Myostatin is a member of the transforming growth factor-β (TGFβ) family that signals through activin receptors and functions as a negative regulator of skeletal muscle mass.
1
The protein is secreted as an inactive precursor, termed promyostatin, wherein the mature myostatin growth factor is held inactive by its prodomain, thereby preventing the access of mature myostatin to its receptors.
2
Activation of promyostatin involves two steps (Figure 1). Initial cleavage by a proprotein convertase (eg, furin) results in the formation of a latent complex, termed latent myostatin, in which the myostatin growth factor remains inactive due to noncovalent association with the prodomain. Next, the latent myostatin complex undergoes an additional cleavage step by a bone morphogenetic protein (BMP)/tolloid family protease, resulting in the release and activation of mature myostatin growth factor.
3
The mature myostatin binds to the activin receptor type-2B (ActRIIB) and leads to phosphorylation and signaling through the SMAD2/3 pathway. Activation of this signaling pathway causes a reduction in protein synthesis and an enhancement of protein degradation, ultimately leading to accelerated muscle catabolism and muscle loss.
4
Schematic of myostatin activation by proteases. Myostatin is secreted as an inactive precursor, termed promyostatin, wherein the mature myostatin growth factor is held inactive by its prodomain, thereby preventing the access of mature myostatin to its receptors. Activation of promyostatin involves two steps. Initial cleavage by a proprotein convertase (eg, furin) results in the formation of a latent complex, termed latent myostatin, in which the myostatin growth factor remains inactive due to noncovalent association with the prodomain. Next, the latent myostatin complex undergoes an additional cleavage step by a BMP/tolloid family protease, resulting in the release and activation of mature myostatin growth factor.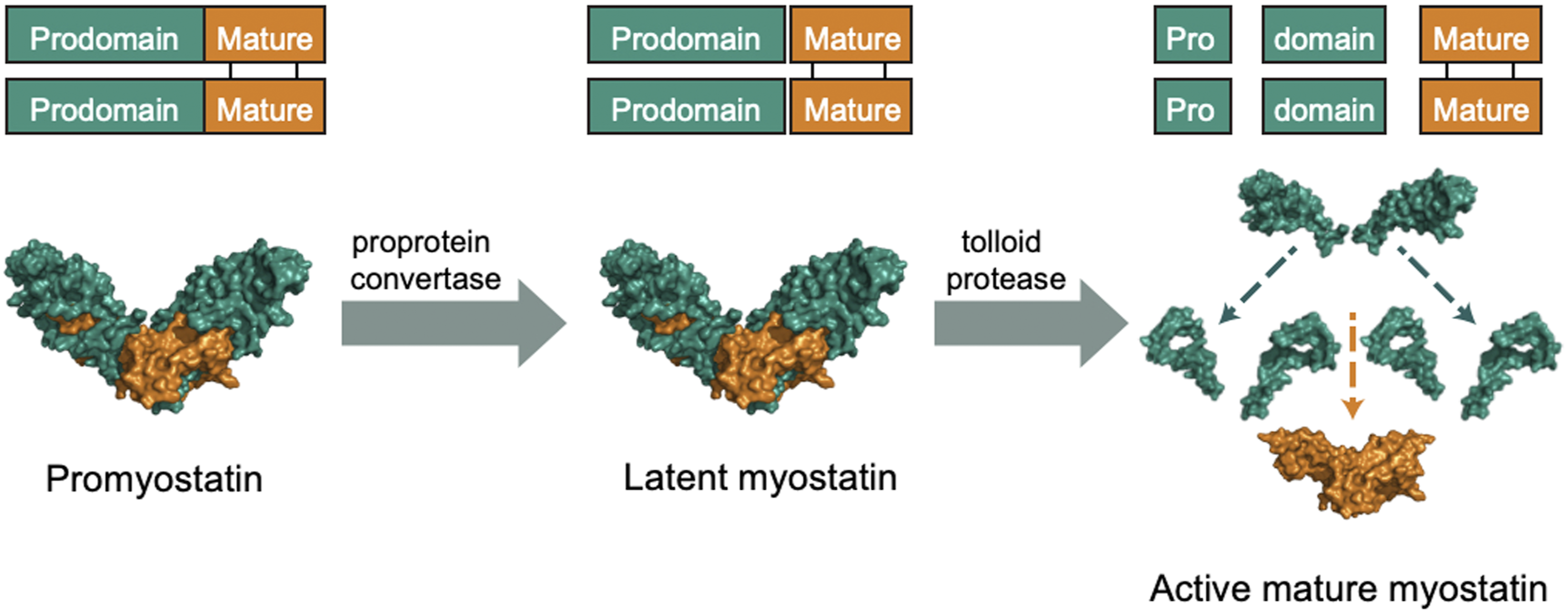
Myostatin has become an attractive therapeutic target for muscle atrophying disorders such as Becker muscular dystrophy, Duchenne muscular dystrophy, and spinal muscular atrophy (SMA) since loss-of-function mutations in the myostatin gene have been correlated with muscle hypertrophy in both animals and humans.5,6 Several therapeutic approaches have been employed to inhibit myostatin and its signaling pathway. This includes drugs targeting the myostatin growth factor or myostatin receptors, ligand traps to sequester the growth factors, and overexpression of antagonists, such as follistatin. 7 Although these therapeutic modalities have shown promising preclinical data in animal models, and have established a role for myostatin in the pathophysiology of muscle-atrophying disease, the clinical success of these therapies has been limited. This lack of meaningful clinical impact has been attributed to several possibilities: (1) differences in pharmacokinetics and drug exposure in animal models compared to humans, 8 (2) reduced levels of circulating target in pathological conditions, 9 (3) compensatory signaling through other related growth factors regulating muscle mass, 10 (4) incomplete understanding of the underlying biological rationale for the clinical indication or inappropriate clinical endpoints,11,12 and (5) undesirable on-target and off-target side effects that include nose and gum bleeding, skin telangiectasias, and modulation of follicle-stimulating hormone (FSH).13,14
In order to address the lack of specificity and potential side effects seen with the majority of previously tested antimyostatin therapies, we developed apitegromab (SRK-015), an investigational, fully human, monoclonal antibody that specifically binds to proforms of myostatin, which include promyostatin and latent myostatin, inhibiting myostatin activation. Since the prodomains of myostatin share a low sequence homology with other TGFβ-related growth factors, we were able to generate an antimyostatin antibody with a high degree of specificity, ensuring no binding to other closely related growth factors, such as growth differentiation factor 11 (GDF11), activin A, BMP 9/10, or TGFβ1. 15 In preclinical studies, apitegromab increased muscle mass and force in healthy mice while preventing muscle atrophy in a dexamethasone-induced muscle atrophy model. 15 We have also evaluated the activity of apitegromab in murine models of SMA, a neuromuscular disease caused by mutations or deletions in the survival of motor neuron 1 (SMN1) gene that leads to the reduction of the SMN protein and loss of spinal motor neurons and muscle atrophy.16,17 In this context, apitegromab in combination with an SMN upregulator, also known as SMN corrector therapy, led to an increase in muscle mass and function in these animals. 11 These studies provided the pharmacological rationale for the evaluation of apitegromab to treat patients with SMA in the Phase 2 TOPAZ trial (NCT03921528).
To complement our previous work, we present herein a comprehensive set of preclinical studies for apitegromab. These comprised pharmacology and pharmacodynamic assessments and extensive in vivo toxicology and toxicokinetic (TK) studies in adult cynomolgus monkeys as well as adult and juvenile rats. The nonclinical safety studies were designed to enable clinical trials with apitegromab across both adult and pediatric (>2 years) patients with SMA.
Materials and Methods
Test Article and Vehicle
Apitegromab is a fully human monoclonal antibody of the IgG4/lambda isotype that binds to promyostatin and latent myostatin. Apitegromab for the in vitro studies was generated by transiently transfecting Expi293TM cells with heavy and light chain expression vectors. 15 For the rat and cynomolgus monkey studies, apitegromab was expressed in a Chinese hamster ovary cell line that stably expresses the heavy and light chains. For all studies, apitegromab was provided as a stock solution at a nominal concentration of 50 mg/mL in histidine-buffered saline and stored at ≤ −60°C.
In Vitro Pharmacology and Tissue Cross-Reactivity
Sequence homology for the myostatin prodomain and mature growth factor
The sequence identity of human, cynomolgus monkey, and rat myostatin prodomains and mature growth factor peptide were obtained from the UniProt database (References O14793, Q95J86, O35312; https://www.uniprot.org/). A pairwise alignment was performed with the sequence alignment program Clustal Omega, 18 using the protein sequence without its signal peptide, followed by analysis of the percent sequence identity.
Apitegromab binding affinities
The binding affinity of apitegromab for human, cynomolgus monkey, and rat promyostatin isoforms was measured using biolayer interferometry (FortéBio Octet QKe) following the methods outlined in Pirruccello-Straub et al, 2018. Briefly, in the binding experiments, apitegromab was immobilized on human Fc capture tips and the association and dissociation of each species’ promyostatin was evaluated. All baseline steps, loading of biotinylated antigens, and antibody association and dissociation steps were performed in 1x kinetics buffer, containing phosphate buffered saline (PBS) pH 7.4 with .002% tween 20 and .01% bovine serum albumin (BSA). Association proceeded for 300 seconds, followed by dissociations in 1x kinetics buffer for at least 600 seconds. Data analysis was performed on FortéBio Data Analysis software version 8.2. A 1:1 global fitting model that considered both association and dissociation was utilized for KD determination for each antigen antibody pair.
Myostatin activation assay
Ability of apitegromab to inhibit myostatin activation was measured using a previously described cell-based activation assay. 15 A sample of human, cynomolgus monkey, or rat promyostatin at 400, 500, and 200 nM, respectively, was incubated with varying amounts of apitegromab for 30 minutes at 37°C. This mixture was then incubated at 37°C for 16 h with cell supernatants containing recombinantly overexpressed proprotein convertase (eg, furin) and a tolloid protease (eg, mammalian tolloid like-2 (mTLL2)). The extent of proteolytic release of the mature myostatin growth factor was assessed by adding samples to a culture of 293T cells, which contain a stably integrated pGL4 plasmid (Promega, Madison, WI) responsive to SMAD activation. Cells were incubated with the myostatin reaction for 6 h, followed by detection of luciferase expression using BRIGHT-GLO™ reagent (Promega, Madison, WI) according to manufacturer’s instructions.
Tissue cross-reactivity (TCR) study
A good laboratory practice (GLP) TCR study in human tissues that evaluated the potential on- and off-target tissue binding of apitegromab was conducted per International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) S6(R1) at Charles River Laboratories in Reno, NV. 19 Apitegromab was applied at two concentrations (optimal = 5.0 and high = 25.0 μg/mL) to cryosections of a panel of normal human tissues, at least 3 donors per tissue and 38 tissues (sourced from the Special Pathology Services (SPS) Human Tissue Bank). The tissue panels used included all those on the suggested list from the European Medicines Agency (EMA) guideline document for development and characterization of monoclonal antibodies and all of the tissues recommended by the US Food and Drug Administration (FDA)/Center for Biologics Evaluation and Research (CBER) and specifically included striated muscle, the organ where target expression is expected.20,21 A human IgG4 lambda antibody was used as the control article, to evaluate the specificity of apitegromab. Additionally, the Flp-In T-Rex 293 cell line (Invitrogen, cat# R78007) was transfected with a DNA construct to express promyostatin and used as a positive control, while untransfected cells were used as a negative control. Localized binding of apitegromab in the cellular tissue samples was performed using a biotinylated antihuman IgG4 secondary antibody.
In Vivo Toxicology and Toxicokinetic Studies in Cynomolgus Monkeys and Rats
Apitegromab GLP Multidose Toxicology and TK Studies in Cynomolgus Monkeys and Sprague Dawley Rats.
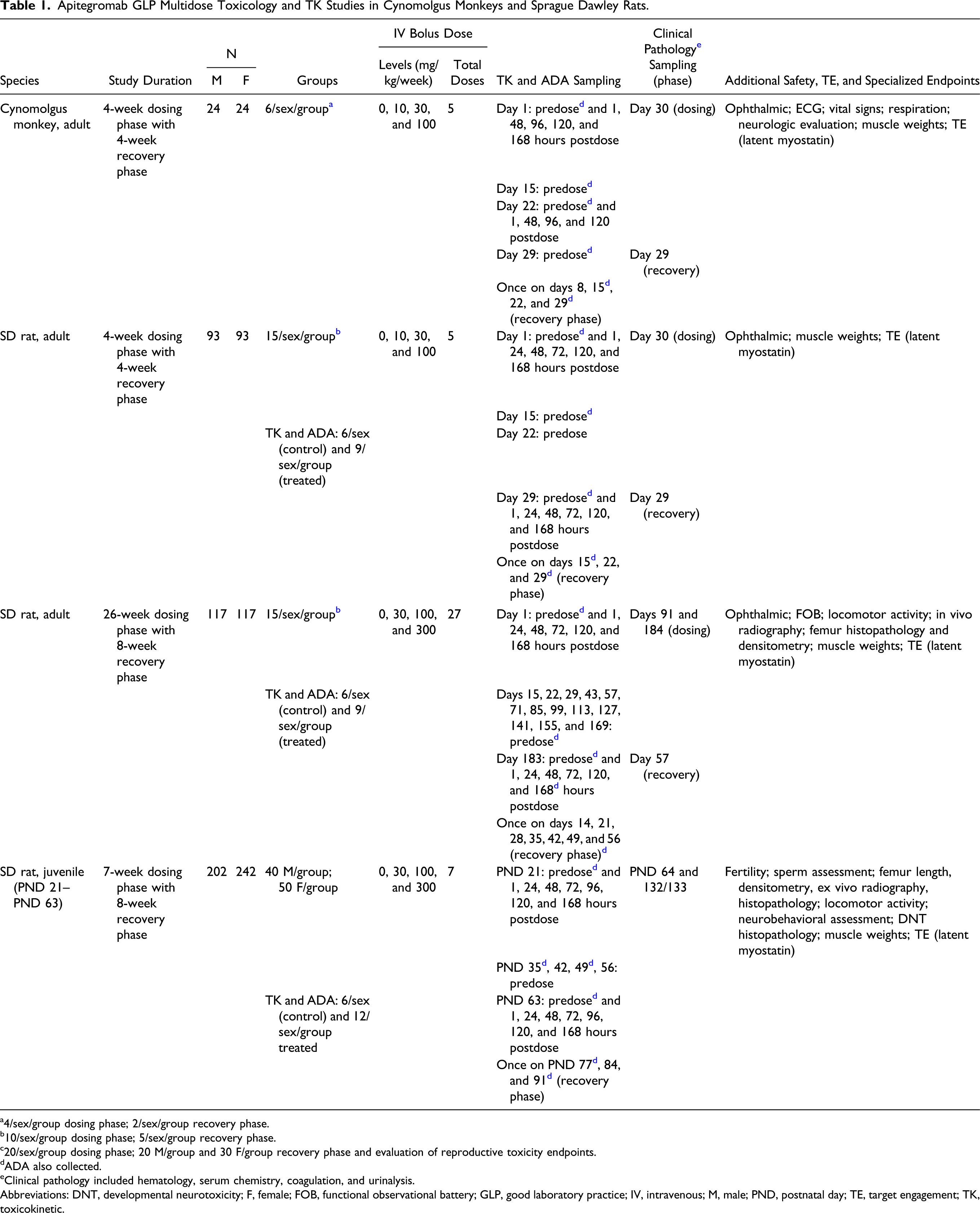
a4/sex/group dosing phase; 2/sex/group recovery phase.
b10/sex/group dosing phase; 5/sex/group recovery phase.
c20/sex/group dosing phase; 20 M/group and 30 F/group recovery phase and evaluation of reproductive toxicity endpoints.
dADA also collected.
eClinical pathology included hematology, serum chemistry, coagulation, and urinalysis.
Abbreviations: DNT, developmental neurotoxicity; F, female; FOB, functional observational battery; GLP, good laboratory practice; IV, intravenous; M, male; PND, postnatal day; TE, target engagement; TK, toxicokinetic.
Cynomolgus monkey toxicology study
Male and female cynomolgus monkeys (32 to 48 months of age) were randomly assigned to treatment groups and group housed in stainless steel cages in a controlled environment (20 to 26°C; 30 to 70% relative humidity; 12-hour light/dark cycles). Animals were offered cage enrichment devices and food, Certified Primate Diet #5048 (PMI Nutrition International Certified LabDiet®) one to two times daily, and tap water ad libitum.
Adult and juvenile rat toxicology studies
Male and female Sprague Dawley (SD) rats (adult studies, 6 to 8 weeks of age; juvenile study, postnatal day (PND) 21 for F1 developmental arm and 15–16 weeks of age for the mating arm) were randomly assigned to treatment groups. In adult studies, all animals were group housed; in the juvenile study, F1 animals were individually housed starting on PND 21, naive males for mating were individually housed (except when pairing), and naive females were group housed (except when pairing and during gestation). Housing was in polycarbonate cages with Diamond Soft® bedding in a controlled environment (20 to 26°C; 30 to 70% relative humidity; 12-hour light and dark cycles) except during mating, where animals were placed in stainless steel, wire bottom cages. Food, Certified Rodent Diet #2014C or #2016C (Envigo RMS, Inc.), and tap water were offered ad libitum, and animals were provided cage enrichment devices.
Safety and other specialized evaluations
All toxicology studies evaluated the following general endpoints: mortality, clinical observations, body weights, and food consumption. General clinical observations of animals were performed twice daily and cageside observations were conducted 2 to 3 hours postdose to assess acute toxicity in monkeys and once daily in rats. Detailed observations were performed weekly on dosing days. Food consumption was measured once daily in monkeys, once weekly in adult rats, and every 3–4 days (corresponding to body weight intervals) for juvenile animals. Measurement of body weight were taken once weekly for monkeys and adult rats and every 3–4 days in the juvenile study. All studies also included clinical pathology (hematology, serum chemistry, coagulation, and urinalysis) and anatomic pathology (gross and microscopic) evaluations with additional collection of skeletal muscle weights for the biceps brachii and gastrocnemius. Blood samples were collected for toxicokinetic, antidrug antibody (ADA), and target engagement (serum latent myostatin) analysis. Other safety evaluations performed for the monkey and adult rat studies included ophthalmic examinations (both species); and for the monkey only, vital-sign measurements; electrocardiograms; neurologic exams; and measurements of respiration rate and heart rate.
The following specialized endpoints were evaluated in the 26-week rat study: functional observational battery (FOB), locomotor activity assessments, in vivo bone analyses of the femur (ie, radiographic image), and ex vivo bone analyses of the femur (ie, mineral content and densitometry by peripheral quantitative computed tomography (pQCT) and histopathology).
The following specialized endpoints were evaluated in the juvenile rat study: development landmarks, anatomic and developmental neurotoxicity (DNT) and neuropathology, male and female fertility endpoints, neurobehavioral findings, and ex vivo bone analyses of the femur (ie, mineral content and densitometry by pQCT, radiography, histopathology, and length). DNT pathology examination involved an assessment of numerous sections from the brain, spinal cord, dorsal root ganglia, and nerves, including key neural structures as listed in the “best practice” recommendations for nervous system sampling during nonclinical toxicity studies.22,23
Quantification of apitegromab in cynomolgus monkey and SD rat serum
Serum samples were analyzed for the presence of apitegromab using an ELISA validated in both cynomolgus monkey and SD rat serum. Assay plates were coated with recombinant promyostatin and blocked with a solution of BSA. Serum samples were diluted to the minimum required dilution (MRD) (1:100) or further to ensure the analyte was within the quantitative range of the assay. The diluted samples were then incubated on the assay plate. Bound apitegromab was detected using a goat-antihuman horseradish peroxidase reagent, and the assay signal was generated using tetramethylbenzidine. The standard curve was fitted to a 5-parameter logistic function with 1/Y2 weighting. The quantitative range is 200 to 10 000 ng/mL apitegromab in serum. For all studies, sample analysis was conducted (GLP) at Biologics Development Services in Tampa, FL.
Detection of anti-apitegromab antibodies in cynomolgus monkey and SD rat serum
Serum samples were assessed for the presence of ADA against apitegromab using an electrochemiluminescent (ECL) bridging-format plate-based assay. Samples were acidified with 100 mM acetic acid to dissociate any ADA complexes, followed by neutralization with a Tris buffer and incubation with biotinylated and ruthenium-conjugated apitegromab. The neutralized samples were then incubated on streptavidin-coated assay plates and the assay signal was measured. Samples that resulted in assay signals above an established cut-point value were analyzed in a confirmatory assay, in which excess apitegromab is added to the neutralized sample to show immunodepletion of the assay signal. This assay was validated for use in both cynomolgus monkey and SD rat serum, using an MRD of 1:20. The sensitivity was measured at 23.5 ng/mL in the cynomolgus monkey assay and 4.23 ng/mL in the SD rat assay. For all studies, sample analysis was conducted (GLP) at Biologics Development Services in Tampa, FL.
Quantification of latent myostatin in cynomolgus monkey and SD rat serum
Latent myostatin concentrations were quantified using a qualified ECL assay that selectively detects total latent myostatin (ie, bound and unbound). We recently described the qualification of this assay in cynomolgus monkey serum and the protocol was used here, without modification. 24 A version of this assay was also qualified in SD rat serum. The assay sensitivity was 10 ng/mL of latent myostatin in serum and achieved a drug tolerance of up to 1 mg/mL of apitegromab. Animals determined to be ADA-positive were removed from latent myostatin analyses; however, this did not impact data interpretation. For all studies, latent myostatin was measured (non-GLP) at Scholar Rock in Cambridge, MA.
Toxicokinetics
Toxicokinetic parameters were calculated for each study using Phoenix WinNonLin (Certara, Princeton, NJ). Animals determined to be ADA-positive were removed from TK analyses; however, this did not impact data interpretation. Noncompartmental analyses were performed using an intravenous (IV) bolus dose administration route, nominal dose levels, and nominal sampling times. Concentrations below the limit of quantitation (<200 ng/mL) were treated as 0 for descriptive statistics. Area under the curve (AUC) assessments were calculated using the linear trapezoidal rule. Terminal half-life (t½) was estimated by log-linear regression of the terminal phase of the mean concentration vs time profiles. At least 3 points clearly visible in the terminal phase, an adjusted r2 value >.7, and time interval of at least 3 half-lives were required for half-life characterization. The accumulation ratio (AR) was calculated as [Cmax or AUC0-168 on the last day of dosing]/[Cmax or AUC0-168 on the first day of dosing] for all studies except the 4-week cynomolgus monkey study, where AR was calculated using the day 22 values rather than the last dosing day.
Results
Species Selection for Toxicology Studies
Prior to initiating the in vivo toxicology studies, we sought to confirm the relevance of the rat and cynomolgus monkey as the appropriate rodent and nonrodent species for assessing apitegromab toxicity, per ICH S6(R1). 19 The ability of apitegromab to increase and strengthen muscle mass has previously been demonstrated in preclinical pharmacology studies in healthy animals and in multiple rodent models of muscle atrophy, including 2 models of SMA.11,15 For the studies described here, we compared the sequence identity of myostatin prodomain and confirmed in vitro binding and activity of apitegromab across human, rat, and cynomolgus monkey.
Myostatin prodomain has similar sequence identity and binding to apitegromab across species
Sequence Identity of the Myostatin Prodomain and Cross-Species Promyostatin Binding Affinity.
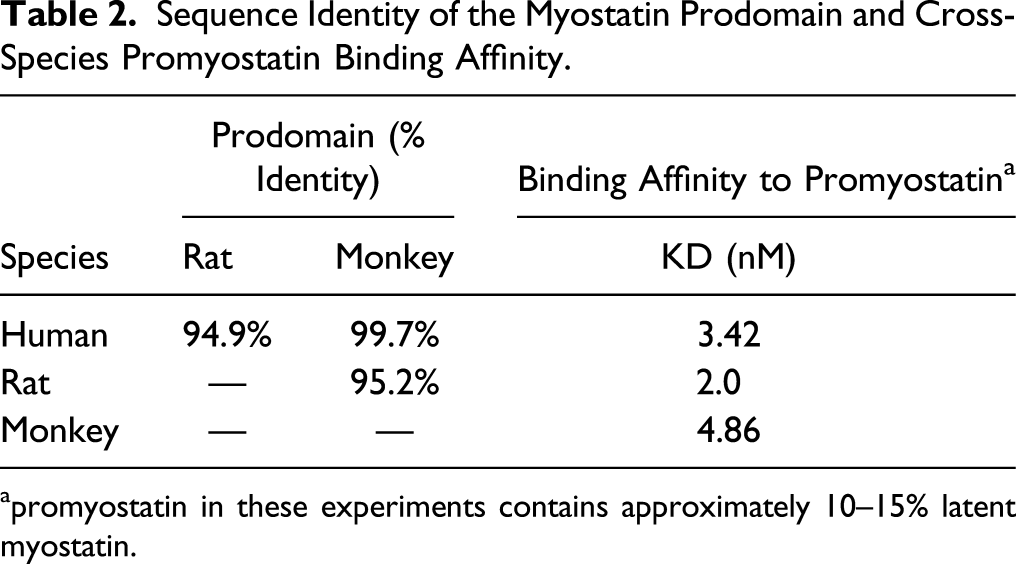
apromyostatin in these experiments contains approximately 10–15% latent myostatin.
Apitegromab inhibits myostatin activation
In vitro activity of apitegromab was assessed using a previously described cell-based activity assay.
15
To confirm similar inhibitory activity of apitegromab across species, the assay measured the ability of apitegromab to block the proteolytic cleavage of human, cynomolgus monkey, and rat promyostatin and the release of the mature myostatin growth factor. Apitegromab inhibits proteolytic activation of human myostatin (IC50 = 286 nM) and also prevents activation of cynomolgus myostatin (IC50 = 626 nM) and of rat myostatin (IC50 = 178 nM) (Figure 2). Apitegromab inhibits activation of human, cynomolgus monkey, and rat myostatin in a SMAD-responsive luciferase reporter assay. (A) Recombinant human (circles), cyno (squares), or rat (triangles) promyostatin was incubated with apitegromab, followed by incubation with mTLL2 and furin proteases, followed by incubation on CAGA cells transfected with a SMAD-responsive luciferase reporter vector to measure myostatin signaling. The ability of apitegromab to inhibit the proteolytic processing and thus release of the active growth factor was measured and plotted as % activation. One hundred percent activity was determined in the presence of vehicle control. Data shown are mean ± standard deviation. Curves are best fits to a dose–response inhibition model. (B) Apitegromab IC50 values for inhibition of myostatin activation, by species.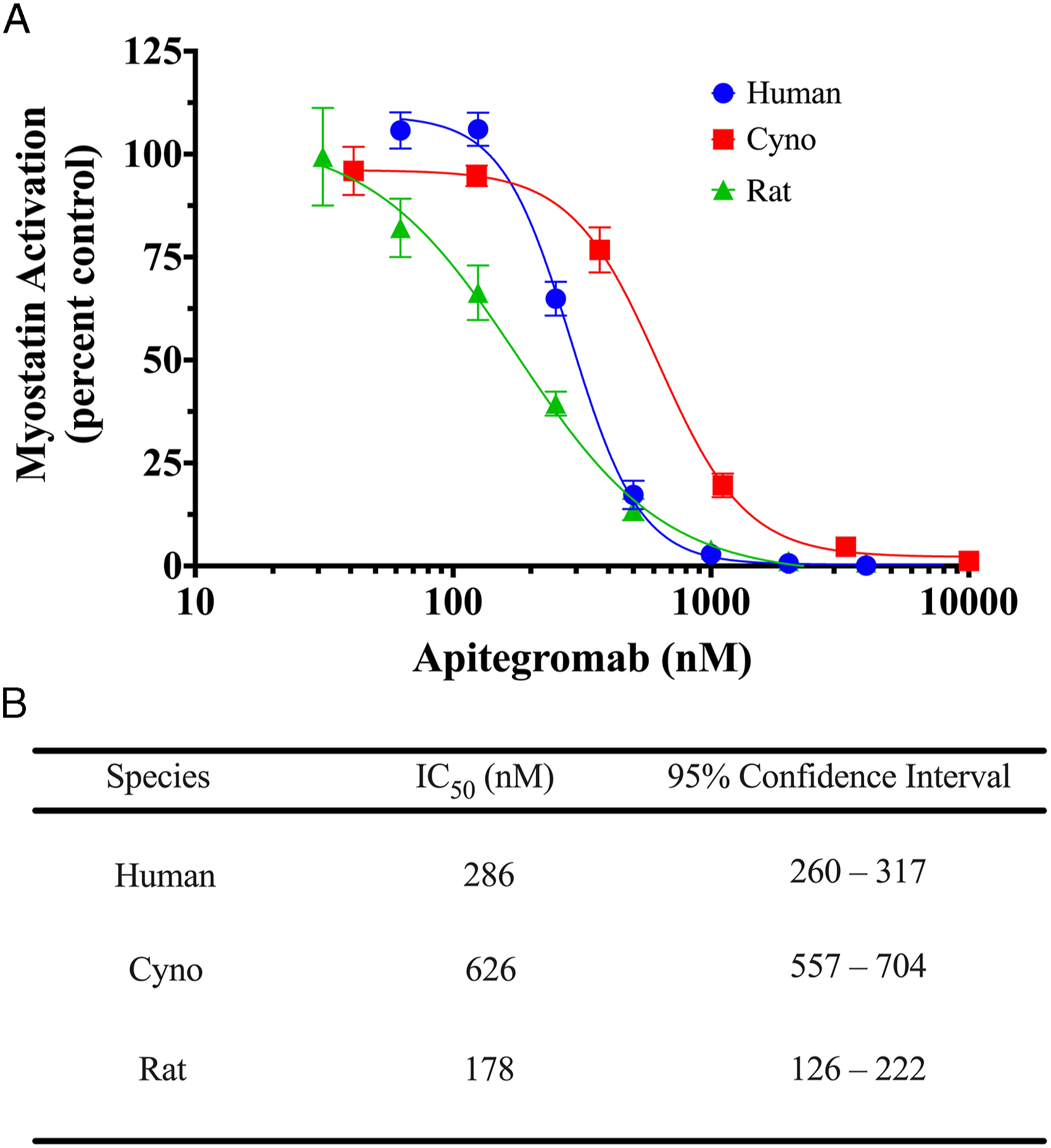
Human Tissue Cross-Reactivity of Apitegromab
Apitegromab does not exhibit off-target binding in human tissues
To evaluate the potential on- and off-target tissue binding of apitegromab, we conducted a GLP tissue cross-reactivity study in a full panel of human tissues from 3 different human donors. 19 Apitegromab bound, as anticipated, to positive and negative control cell lines; however, it did not show specific positive staining to any human tissue.
In Vivo Safety Assessment of Apitegromab in Cynomolgus Monkeys and Rats
Four-week GLP toxicology studies were conducted in cynomolgus monkeys and rats to support the initiation of a Phase 1 clinical study. 25 Additional repeat-dose GLP studies were conducted in rats, which included a juvenile rat toxicity study to support dosing in pediatric populations and a long term, 26-week toxicity study in adult animals.
Four-week toxicology study in cynomolgus monkeys
Male and female cynomolgus monkeys were administered apitegromab once weekly by IV bolus at doses of 0 (vehicle), 10, 30, and 100 mg/kg, on days 1, 8, 15, 22, and 29 (5 doses total) for 4 weeks of the dosing phase, followed by a 4-week (treatment-free) recovery phase. No mortality was observed in the study and all animals survived until the scheduled necropsy. There were no apitegromab-related adverse effects on clinical observations, food consumption, ophthalmic examinations, clinical chemistry, hematology, coagulation, or urinalysis. Muscle mass (gastrocnemius and biceps brachii) was slightly higher in apitegromab-treated animals than control animals, ranging from approximately 5 to 25% over controls, although the effects lacked a dose response. Safety pharmacology endpoints (ie, neurological examinations, respiration rates, and electrocardiography) demonstrated no treatment-related effects. No apitegromab-related macroscopic or microscopic findings, including the injection site, were observed in the dosing or recovery phase animals. Overall, there were no treatment-related adverse findings observed on any endpoint evaluated and the no observed adverse effect level (NOAEL) was 100 mg/kg, which was the highest dose tested.
Four-week toxicology study in adult SD rats
Male and female rats were administered apitegromab once weekly by IV bolus at doses of 0 (vehicle), 10, 30, and 100 mg/kg on days 1, 8, 15, 22, and 29 (5 doses total) for 4 weeks of the dosing phase, followed by a 4-week (treatment-free) recovery phase. No apitegromab-related mortality was observed. There were no apitegromab-related adverse effects on clinical observations, food consumption, ophthalmic examinations, hematology, coagulation, or urinalysis. No apitegromab-related findings were observed at the injection site. Apitegromab-related body weight increases were observed in animals administered ≥10 mg/kg, which did not correlate with higher food consumption but did correlate with increased skeletal muscle weights, at termination. Specifically, anatomic pathology findings were limited to weight increases for skeletal muscle tissues (left biceps brachii, right biceps brachii, left gastrocnemius, or right gastrocnemius) of all groups administered apitegromab, that occasionally reached significance, with a mean effect that ranged from approximately 3 to 32% over controls; however, there were no microscopic correlates. Increased muscle mass was an anticipated pharmacological effect of apitegromab, an antibody that targets promyostatin and latent myostatin, as previously reported in healthy and disease animal models.11,15 Several minor apitegromab-related clinical chemistry findings (eg, mildly higher globulin concentration, mildly higher total protein concentrations, and lower albumin:globulin (A:G) ratios) were observed at the end of the dosing phase, predominately in animals administered 100 mg/kg, and most exhibited evidence of reversibility. Increases in globulin concentrations were likely related to the high levels of apitegromab (an immunoglobulin 4) in circulation. In conclusion, there were no treatment-related adverse findings observed on any endpoint evaluated and the NOAEL was 100 mg/kg, which was the highest dose tested.
Twenty six-week toxicology study in adult SD rats
Rats were selected as the single toxicity species for further toxicology assessment of apitegromab because of similar results (ie, no toxicity observed) in the 4-week rat and cynomolgus monkey toxicology studies, per ICH S6(R1).19,26 Male and female rats were administered apitegromab once weekly by IV bolus at doses of 0 (vehicle), 30, 100, and 300 mg/kg, for 26 weeks (27 doses total) of dosing phase, followed by an 8-week (treatment-free) recovery phase. No apitegromab-related mortality was observed. There were no adverse apitegromab-related effects on clinical observations, food consumption, clinical chemistry, hematology, ophthalmic examinations, FOB testing, bone mineral density (femur), in vivo radiography (femur), macroscopic pathology, and microscopic pathology evaluations, including the injection site. Similar to the 4-week rat study, apitegromab-related clinical chemistry effects were limited to minimally higher total protein and globulin concentrations, and anatomic pathology findings were limited to slight increases in skeletal muscle weights (left and right biceps brachii). The increase of skeletal muscle mass occurred in all apitegromab-treated groups, ranged from approximately 15 to 33% over controls, and correlated with microscopic findings of minimal to slight hypertrophy of muscle fibers. These muscle changes persisted during the recovery phase but were not dose-responsive, as was expected, based on previously conducted pharmacology studies with apitegromab.11,15 In conclusion, administration of apitegromab once weekly by IV bolus administration for up to 26 weeks was well-tolerated in adult SD rats and the NOAEL was the highest dose tested of 300 mg/kg.
Juvenile toxicology study in SD rats
To support the dosing of pediatric patients (≥2 years of age) with SMA, a juvenile rat toxicology study was conducted. Juvenile rats were dosed for 7 weeks from PND 21, the equivalent of a 2-year old human, until PND 63, when animals are sexually mature and the development of major organs and the immune system are generally complete. 27 The study design also incorporated neurotoxicology, fertility, and reproductive and developmental toxicology endpoints to investigate the effects of myostatin pathway inhibition on young animal development and reproductive biology (Table 1).
In this study, apitegromab was administered by IV bolus administration at weekly doses of 0 (vehicle), 30, 100, and 300 mg/kg. There were no apitegromab-related adverse effects observed on mortality, clinical observations, food consumption, clinical pathology, ophthalmic examinations, and anatomic pathology evaluations including the injection site. No apitegromab-related effect was observed on mean body weight or weight gain in males; however, an apitegromab-related, nonadverse, significant increase in mean body weight gain was noted for females over the entire dosing period (PND 21 through 63) that persisted into the recovery phase for some animals. Several clinical pathology findings were observed in apitegromab-treated juvenile rats, though due to their overall small magnitude, none were considered adverse. These included minimally to mildly higher total protein and/or globulin concentrations, which resulted in minimally lower albumin:globulin ratio in both sexes administered ≥100 mg/kg/dose, mildly lower triglyceride concentration in males administered 300 mg/kg/dose, and minimally lower sodium concentration in males administered ≥100 mg/kg/dose and females administered 300 mg/kg/dose. The changes in globulin were likely due to apitegromab (an immunoglobulin 4) in circulation. Similar to the adult animal studies, there was an increase in absolute muscle weights (biceps brachii and/or gastrocnemius) in apitegromab-treated males and females, with a mean effect that ranged from approximately 13 to 28% over controls, and with no apparent microscopic correlates or dose response. Additionally, no adverse toxicity was observed for any of the specialized study endpoints, including anatomic and developmental neurotoxicity (DNT) and neurohistopathology, neurobehavioral evaluations (acoustic startle, locomotor activity, and Morris water maze), developmental sexual landmarks, and bone analyses (densitometry, radiography, femur length, and femur histopathology). Lastly, following the breeding of treated males and treated females with naive animals, apitegromab had no adverse effects on reproductive parameters (estrous cycles, reproductive performance, fecundity, fertility, and sperm assessment); however, an apitegromab-related increase in mean body weight was observed during gestation for some females. In conclusion, administration of apitegromab once weekly by IV bolus administration, from PND 21 to PND 63 (7 doses total), was well-tolerated in juvenile SD rats and the NOAEL was the highest dose tested of 300 mg/kg.
Multidose Toxicokinetics of Apitegromab in Cynomolgus Monkeys and Rats
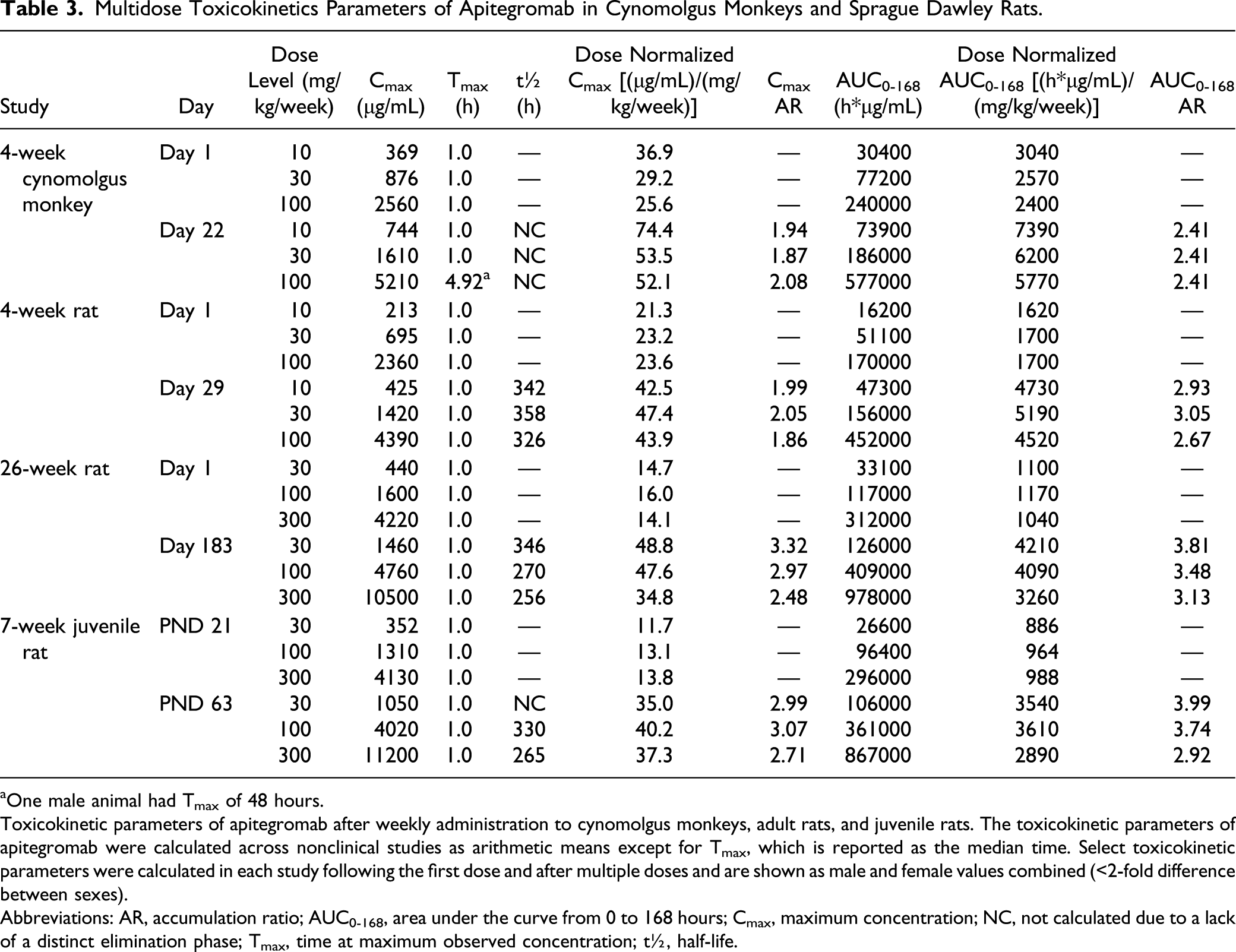
The apitegromab multidose TK profile, TK parameters, and ADA were evaluated in the 4-week studies in adult rats and cynomolgus monkeys, the 26-week study in adult rats, and the juvenile rat study. In all studies, systemic exposure to apitegromab was confirmed in treated animals (Figure 3). The Cmax and AUC0-168 parameters increased dose proportionally with increasing dose levels and showed accumulation after multiple doses (Table 3). Additionally, there were no differences in apitegromab exposure between male and female animals. Serum concentration time profile of apitegromab following multiple doses. Apitegromab was measured in the serum of male and female animals following once weekly repeat IV doses across (A) four weeks of dosing in cynomolgus monkeys at 10, 30, and 100 mg/kg, followed by a 4-week recovery phase; (B) four weeks of dosing in adult SD rats at 10, 30, and 100 mg/kg, followed by a 4-week recovery phase; (C) 26 weeks of dosing in adult SD rats at 30, 100, 300 mg/kg, followed by an 8-week recovery phase; and (D) seven weeks of dosing in juvenile SD rats at 30, 100, and 300 mg/kg, followed by a 4-week recovery phase. Across all studies and dose groups, apitegromab showed minimal-to-no differences in TK profile between male and female animals (ie, less than 2-fold), achieved dose-proportional exposure, and demonstrated accumulation following multiple doses. Data shown are mean ± standard deviation from male and female animals combined. (Note that in panel C, the large standard deviation observed in the 30 mg/kg group at the day 196 postdose time point is due to a single animal without evidence of ADA.) Dose symbols: 10 (circles), 30 (squares), 100 (triangles), and 300 (diamonds) mg/kg. Multidose Toxicokinetics Parameters of Apitegromab in Cynomolgus Monkeys and Sprague Dawley Rats. aOne male animal had Tmax of 48 hours. Toxicokinetic parameters of apitegromab after weekly administration to cynomolgus monkeys, adult rats, and juvenile rats. The toxicokinetic parameters of apitegromab were calculated across nonclinical studies as arithmetic means except for Tmax, which is reported as the median time. Select toxicokinetic parameters were calculated in each study following the first dose and after multiple doses and are shown as male and female values combined (<2-fold difference between sexes). Abbreviations: AR, accumulation ratio; AUC0-168, area under the curve from 0 to 168 hours; Cmax, maximum concentration; NC, not calculated due to a lack of a distinct elimination phase; Tmax, time at maximum observed concentration; t½, half-life.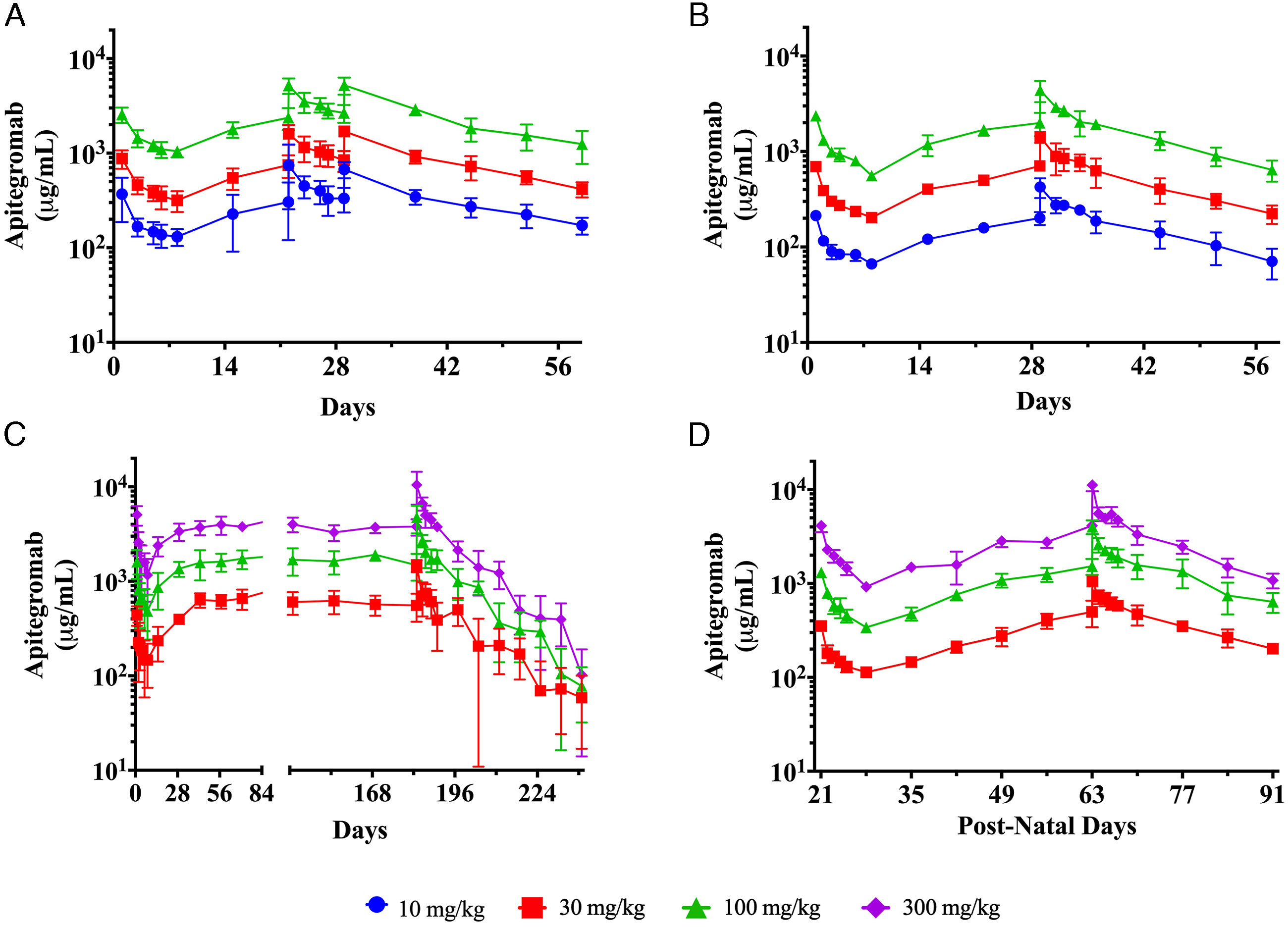
Multidose TK in cynomolgus monkeys (4 weeks)
Male and female cynomolgus monkeys were administered IV bolus doses of apitegromab at dose levels of 10, 30, and 100 mg/kg, once weekly on days 1, 8, 15, 22, and 29 (5 total doses). The TK parameters were determined after day 1 and day 22. Cmax was observed at the first sampling point of 1 hour following dosing in all treated animals, with the exception of day 22 where 1 animal in 100 mg/kg group had Cmax observed at 48 hours after dosing (Table 3). The t½ for apitegromab was not measured in this study due to a lack of a distinct elimination phase. Exposure, as assessed by apitegromab Cmax and AUC0-168, demonstrated a dose-dependent increase on days 1 and 22 and was approximately dose proportional from 10 to 100 mg/kg, suggesting that the target was fully saturated. 28 Animals maintained exposure throughout dosing and recovery phases (Figure 3A) and accumulation was observed at all doses from day 1 on day 22 (Table 3). No animals developed ADA following apitegromab administration at any dose.
Multidose TK in adult SD rats (4 weeks)
Male and female rats were administered IV bolus doses of apitegromab at dose levels of 10, 30, and 100 mg/kg, once weekly on days 1, 8, 15, 22, and 29 (5 total doses). The TK parameters were determined after day 1 and day 29. Cmax was observed at the first sampling point of 1 hour in all groups on both days and apitegromab attained a t½ ranging from 326 to 358 hours on day 29 (Table 3). Exposure, as assessed by apitegromab Cmax and AUC0-168, demonstrated a dose-dependent increase on days 1 and 29 and was generally dose proportional from 10 to 100 mg/kg, suggesting that the target was fully saturated. Animals maintained exposure throughout dosing and recovery phases and accumulation was observed at all doses, following weekly administration (Figure 3B). Two females, one in the 10 mg/kg group and one in the 30 mg/kg group, were ADA-negative for all intervals in the dosing phase but were ADA-positive on days 15 and 29 of the recovery phase, which correlated with lower concentrations of apitegromab measured following the day 29 dose (compared with other animals in the group). All other animals were ADA-negative throughout the study.
Multidose TK in adult SD rats (26 weeks)
Male and female rats were administered IV bolus doses of apitegromab at dose levels of 30, 100, and 300 mg/kg once weekly for 26 weeks (27 total doses). The TK parameters were determined after day 1 and day 183. Cmax was observed at the first sampling point of 1 hour in all groups on both days and apitegromab attained a t½ ranging from 256 to 346 hours on day 183 (Table 3). Exposure, as assessed by apitegromab Cmax and AUC0-168, demonstrated a dose-dependent increase and was generally dose proportional. Animals maintained exposure throughout dosing and recovery phases and accumulation was observed at all doses, following weekly administration (Figure 3C). In the 30 mg/kg group, ADA was detected in one male (on day 43) and two females (one each on day 42 and day 99). Serum concentrations of apitegromab were not affected for the male, but both females showed decreased exposure. ADA was not detected at any interval in any animal in the 100 and 300 mg/kg dose groups.
Multidose TK in juvenile SD rats
Juvenile male and female rats were administered IV bolus doses of apitegromab at dose levels of 30, 100, and 300 mg/kg once weekly from PND 21 to PND 63 (7 total doses). The TK parameters were determined after PND 21 and PND 63. Cmax was observed at the first sampling point of 1 hour in all groups on both days and apitegromab attained a t½ ranging from 265 to 330 hours on PND 63 (Table 3). Exposure, as assessed by apitegromab Cmax and AUC0-168, demonstrated a dose-dependent increase and was generally dose proportional. Animals maintained exposure throughout dosing and recovery phases and accumulation was observed at all doses, following weekly administration (Figure 3D). All animals were determined to be ADA-negative.
Target engagement with apitegromab in cynomolgus monkeys and rats
Target engagement with apitegromab has previously been confirmed in pharmacology studies in rodents
11
and cynomolgus monkeys,
24
wherein apitegromab exposure led to increased circulating levels of latent myostatin. In our current studies, we confirmed the target engagement with apitegromab by measuring serum latent myostatin levels across the dosing and recovery phases (Figure 4). Among all studies, the group averaged baseline levels of latent myostatin ranged from approximately 20 to 100 ng/mL. Serum latent myostatin levels were increased over baseline following the initial dose of apitegromab and remained elevated throughout the dosing phase for treated animals. The group averaged maximal levels of latent myostatin during the dosing phase ranged from 2780 ng/mL to 5218 ng/mL (between 10 to 100 mg/kg) in cynomolgus monkeys. In rats, the group averaged maximal levels of latent myostatin during the dosing phase ranged from 5594 ng/mL to 12821 ng/mL (between 10 to 300 mg/kg) in adults, and from 4517 ng/mL to 9116 ng/mL (between 30 to 300 mg/kg) in juveniles. Absolute levels of latent myostatin achieved after apitegromab dosing increased with increasing dose but were not dose proportional. During the recovery phase, latent myostatin levels in apitegromab-treated groups remained elevated compared to baseline levels. Serum latent myostatin concentration time profile of apitegromab following multiple doses. Latent myostatin was measured in the serum of male and female animals following once weekly repeat IV doses of apitegromab across (A) four weeks of dosing in cynomolgus monkeys at 10, 30, and 100 mg/kg, followed by a 4-week recovery phase; (B) four weeks of dosing in adult SD rats at 10, 30, and 100 mg/kg, followed by a 4-week recovery phase; (C) twenty six weeks of dosing in adult SD rats, followed by an 8-week recovery period; and (D) seven weeks of dosing in juvenile SD rats at 30, 100, and 300 mg/kg, followed by a 4-week recovery phase. In all studies, serum latent myostatin increased over baseline after the first dose and remained elevated throughout the dosing and recovery phases. There were minimal-to-no differences in latent myostatin levels between male and female animals (ie, less than 2-fold). Data shown are mean ± standard deviation from male and female animals combined. (Note that in panel C, the large standard deviation observed in the 30 mg/kg group at the day 196 postdose time point is due to a single animal without evidence of ADA). Dose symbols: vehicle (open circles), 10 (filled circles), 30 (squares), 100 (triangles), and 300 (diamonds) mg/kg.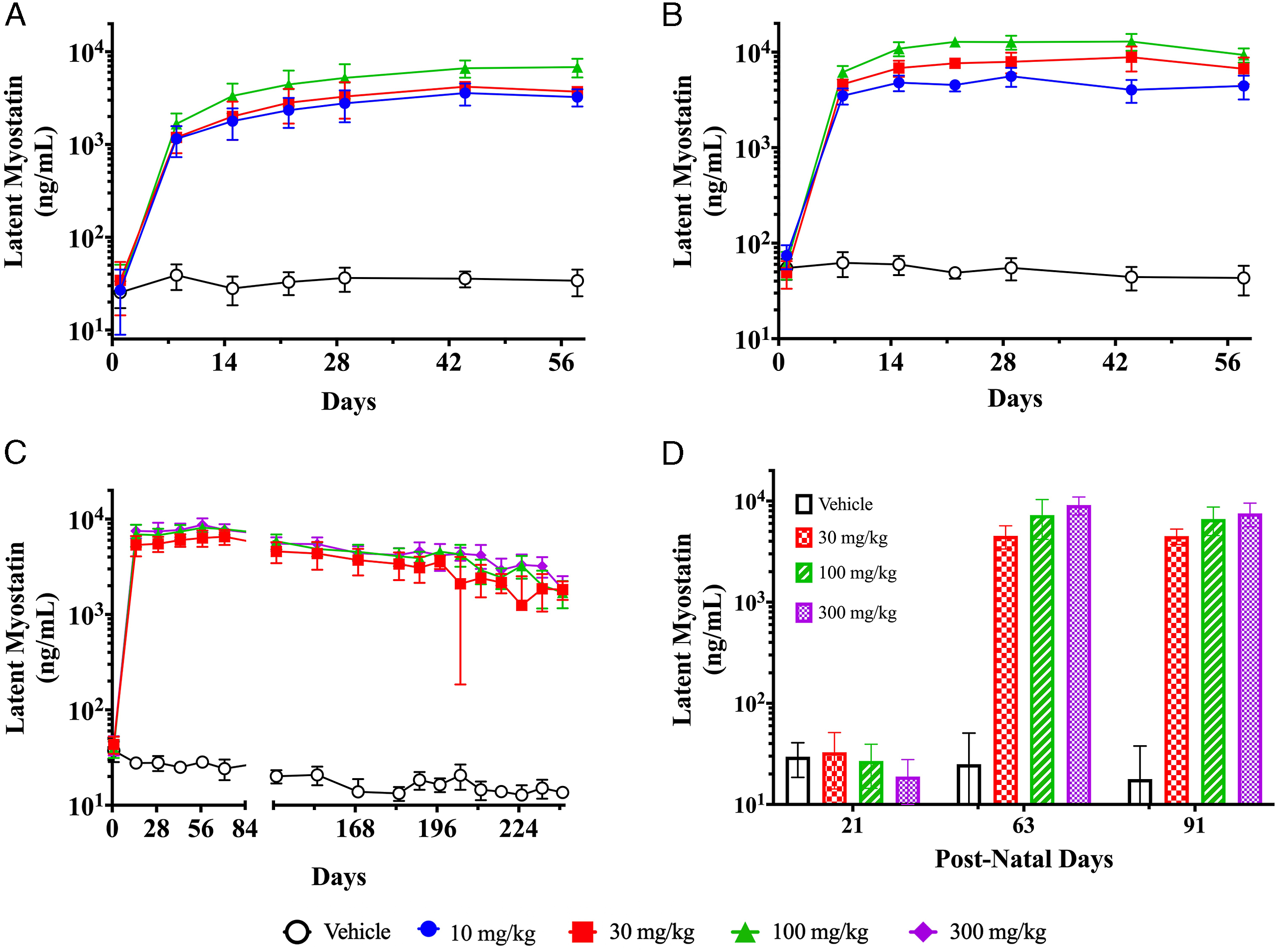
Discussion
As a potential therapeutic target for muscle wasting disease, inhibition of the myostatin pathway has been of great interest and led to the development of numerous antimyostatin treatments. 29 However, one of the major challenges in developing antimyostatin drugs has been their lack of specificity, namely, the ability to inhibit myostatin without binding other closely related growth factors that may lead to undesirable on-target and off-target side effects. For instance, ramatercept (ACE‐031) is a ligand trap for myostatin, GDF11, and activins that was discontinued in the clinic due to safety concerns, including nose and gum bleeding and dilation of blood vessels within the skin. 13 Similarly, undesirable effects of modulating the neurohormonal axes, particularly changes in FSH expression, have been reported with bimagrumab, a monoclonal antibody that blocks binding of myostatin and other ligands to ActRII receptors. 14 Given that closely related TGFβ family growth factors play an important role in normal physiological functions, there remains a critical need to identify a highly specific and potent myostatin inhibitor that avoids off-target side effects.
We have previously described the discovery and development of apitegromab, a highly specific and selective, fully human antimyostatin monoclonal antibody that specifically binds proforms of myostatin (ie, promyostatin and latent myostatin) with no binding to mature myostatin nor any related growth factors. 15 Our current work summarizes a set of preclinical toxicology and TK studies (Table 1) to thoroughly characterize the safety and exposure profile of apitegromab. The nonclinical safety data from the 4-week toxicology studies in both rats and cynomolgus monkeys showed that apitegromab was well-tolerated when administered by IV bolus injection once weekly for 4 weeks at doses up to 100 mg/kg. Based on similar results and a lack of toxicity among the two species in these studies, subsequent repeat-dose studies were conducted in rats to support clinical dosing in adult and pediatric patients, per ICH S6(R1).19,26 Apitegromab was again well-tolerated in a 26-week study in adult rats and in a juvenile rat study, each of which concluded a NOAEL of 300 mg/kg, the highest dose tested.
The preclinical safety data with apitegromab are in stark contrast with the off-target effects noted in rodents with nonspecific myostatin inhibitors. For example, activin inhibition resulting from manipulation of the myostatin/activin signaling axis by a soluble ligand trap (sActRIIB) has been shown to attenuate testis development and lead to adverse effects on sperm motility, count, and ultrastructure in rodents.30,31 Furthermore, reduced FSH levels, smaller testis, and teratogenic effects have been observed in rats treated with bimagrumab, a monoclonal antibody that blocks binding of myostatin and other ligands to ActRII receptors. 14 Notably, our studies with apitegromab did not identify alterations in the weight or histopathology of the testes in adult and juvenile rats and adult monkeys, and there were no apitegromab-related effects on caudal epididymal weight, sperm motility, concentration, or morphology in the juvenile rat study. Given the role of activins in reproductive and developmental biology, 32 we also investigated fertility endpoints in the juvenile rat study as a functional outcome, to determine any potential reproductive effects caused by promyostatin inhibition. Again apitegromab had no effect on fertility parameters such as estrous cyclicity, reproductive performance, and fecundity in juvenile rats. As per ICH S5(R3) guidance, a full battery of development and reproductive studies including embryofetal development (EFD), fertility and early embryonic development (FEED), and prenatal/postnatal development (PPND) studies are currently underway that will provide a full assessment of the potential impact of apitegromab on developmental and reproductive biology. Nonetheless, our current data indicate that apitegromab can circumvent the potential off-target reproductive organ effects observed with nonspecific myostatin inhibitors in animal studies.
Transforming growth factor β family growth factors such as activins and inhibin are also expressed in the central nervous system (CNS),33,34 where they have shown involvement in neuronal development. Modulation of activin and follistatin levels has led to anxiety-related behavior with altered locomotor activity in transgenic mice. 35 Since myostatin is expressed in the CNS, 36 we evaluated the impact of apitegromab treatment on the FOB, locomotor activity, and developmental neurohistopathology in juvenile rats. There were no apitegromab-related adverse observations on any of these endpoints, confirming that in juvenile rodents, the specific inhibition of latent myostatin by apitegromab does not result in any undesirable impact on the CNS.
Although myostatin is primarily expressed in skeletal muscle, there is minimal expression in normal cardiac tissue. 37 Animal studies have shown that the modulation of myostatin and other TGFβ family growth factor expression levels may play a role in the pathophysiology of heart disease, such as heart failure and myocardial infarction.37,38 In particular, GDF11 has been suggested to be a critical growth factor in reversing age-related cardiac hypertrophy in mice. 39 We evaluated the impact of apitegromab on cardiovascular histopathology and physiology in rats and monkeys. There were no apitegromab-related changes in the weight and histopathology of the heart. In addition, apitegromab did not have any adverse impact on heart function, as determined by quantitative and qualitative assessment of electrocardiograms. Our data suggest that specific targeting of promyostatin and latent myostatin is not associated with any unwanted cardiac toxicities in rats and monkeys. These findings are consistent with published data, wherein myostatin knockout (KO) mice did not exhibit any adverse cardiac phenotype, including cardiac hypertrophy or fibrosis.40,41
In addition to the well-understood function of myostatin for skeletal muscle growth, there is evidence to support myostatin’s role in maintaining bone homeostasis and repair. 42 Pediatric patients with SMA experience a loss of bone mineral density, may be at increased risk of osteopenia and osteoporosis, and have an increased prevalence of fractures in both the femur and vertebrae.43,44 Studies in myostatin KO mice administered a myostatin ligand trap (ActIIRB-Fc) showed that treatment led to an improvement in bone mass, regeneration, and modeling. 45 Therefore, these positive effects of myostatin inhibition on bone mass and strength may provide additional benefit to patients with SMA. In the toxicology studies presented here, the effects of apitegromab on bone were extensively evaluated in adult and juvenile animals. Apitegromab did not alter bone mineral density, bone length, and bone histopathology and showed no effect on radiographic observations. These results were not surprising as it is likely that the positive effects of myostatin inhibition on bone are only evident in pathophysiological conditions. As we have previously shown, apitegromab increases bone volume in the mouse model of SMA, suggesting that specific inhibition of myostatin might improve bone health and may reduce fracture incidence in patients with SMA. 11 Most importantly, in our safety assessments, we did not observe any apitegromab-related adverse effects on bone parameters.
Recent publications suggest additional possibilities for the lack of positive clinical outcomes noted with antimyostatin inhibitors, including limited drug exposure achieved in humans that leads to incomplete target saturation or downregulation of the target myostatin in neuromuscular disease.8,9 The multidose TK profile of apitegromab in rats and monkeys confirmed systemic exposure in all animals with approximate dose proportionality and drug accumulation after multiple doses. Apitegromab exposures achieved in these toxicology studies were similar between adult and juvenile animals (in rat studies) and sufficient to produce pharmacodynamic effects, as evidenced by the increase in skeletal muscle mass in treated animals. Furthermore, increases in circulatory levels of latent myostatin confirmed the ability of apitegromab to engage and saturate the target.
In conclusion, the in vivo toxicology assessment of apitegromab demonstrates a favorable safety profile with no toxicity identified at up to 300 mg/kg in both a juvenile rat study and in a chronic study in adult rats dosed up to 26 weeks. Our work supports the rationale that highly specific targeting of proforms of myostatin with apitegromab circumvents the on-target and off-target toxicity observed with nonspecific myostatin inhibitors. We acknowledge that these conclusions are based on preclinical studies while some of the off-target effects related to other myostatin inhibitors were observed in humans.13,14 Encouragingly, however, apitegromab has been shown to be safe and well-tolerated in a Phase 1 trial in healthy adult subjects, 25 while emerging data from a Phase 2 clinical trial (NCT03921528) suggest that apitegromab is safe and well-tolerated in patients with SMA. 46 Overall, the nonclinical toxicology data complement the preclinical pharmacology studies in mouse models of SMA and support the ongoing clinical exploration of apitegromab in adult and pediatric patients with SMA.
Footnotes
Acknowledgments
The authors thank the following Study Directors at Covance and Charles River Laboratories for the successful conduct of these studies: Shana R. Dalton (Covance, Madison, WI), Curtis Grace and Sara E. Hershberger (Covance Greenfield, IN), and Solomon Haile and Martin Guillot (Charles River Laboratories Montreal). Also, thanks to Adam Dunn (Covance, Madison, WI) for his diligent program management and to Kimberly Long for her critical review of this manuscript. In memory of Chris TenHoor.
Author Contributions
Welsh, B. contributed to conception and design, contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Cote, S. contributed to design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Meshulam, D. contributed to interpretation and critically revised manuscript; Jackson, J. contributed to conception, contributed to analysis and interpretation, and critically revised manuscript; Pal, A. contributed to interpretation and critically revised manuscript; Lansita, J. contributed to conception and design, contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Kalra, A. contributed to conception and design, contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interest
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All contributors are either employees or contractors of Scholar Rock. In addition, Mr. Jackson has a patent WO2016073853 issued.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.