
Abstract
Pexacerfont is a corticotropin-releasing factor subtype 1 receptor antagonist that was developed for the treatment of anxiety- and stress-related disorders. This report describes the results of repeat-dose oral toxicity studies in rats (3 and 6 months) and dogs (3 months and 1 year). Pexacerfont was well tolerated in all of these studies at exposures equal to or greater than areas under the curve in humans (clinical dose of 100 mg). Microscopic changes in the liver (hepatocellular hypertrophy), thyroid glands (hypertrophy/hyperplasia and adenomas of follicular cells), and pituitary (hypertrophy/hyperplasia and vacuolation of thyrotrophs) were only observed in rats and were considered adaptive changes in response to hepatic enzyme induction and subsequent alterations in serum thyroid hormone levels. Evidence for hepatic enzyme induction in dogs was limited to increased liver weights and reduced thyroxine (T4) levels. Mammary gland hyperplasia and altered female estrous cycling were only observed in rats, whereas adverse testicular effects (consistent with minimal to moderate degeneration of the germinal epithelium) were only noted following chronic dosing in dogs. The testicular effects were reversible changes with exposure margins of 8× at the no observed adverse effect level. It is not clear whether the changes in mammary gland, estrous cycling, and testes represent secondary hormonal changes due to perturbation of the hypothalamic–pituitary–adrenal axis or are off-target effects. In conclusion, the results of chronic toxicity studies in rats and dogs show that pexacerfont has an acceptable safety profile to support further clinical testing.
Introduction
Corticotropin-releasing factor (CRF) is a 41 amino acid neuronal peptide and the principle regulator of the hypothalamic–pituitary–adrenal (HPA) axis. 1 –4 One of the major sites of CRF expression in the mammalian brain is the paraventricular nucleus (PVN) of the hypothalamus. 5 –7 In response to stress, CRF is released from the PVN and activates corticotropic cells in the anterior pituitary gland, leading to the synthesis and secretion of adrenocorticotrophic hormone (ACTH) into systemic circulation. 8 –12 Adrenocorticotrophic hormone, in turn, stimulates the adrenal glands to produce glucocorticoids (eg, cortisol) which regulate a number of diverse metabolic functions. 13 –15 Glucocorticoids also act to regulate CRF secretion via a negative feedback loop. 16 Under normal physiologic conditions, the HPA axis allows animals to respond to stressful conditions through transient changes in physiology and behavior. However, persistent activation of the HPA axis can also be pathogenic, resulting in a variety of metabolic and neuropsychiatric disorders such as autoimmune disease, hypertension, affective disorders, and major depression. 2,8,9,17 –23 Therefore, modulation of the HPA axis represents a promising therapeutic approach for treating various stress-related conditions.
Corticotropin-releasing factor exerts its actions by binding to 2 types of G-protein-coupled receptors: corticotropin-releasing factor subtype 1 receptor (CRF1) and CRF2. The CRF1 receptors are highly expressed in the anterior pituitary gland, specific brain regions (eg, cerebral cortex, cerebellum, amygdala, hippocampus, and olfactory bulb), and at lower levels in peripheral tissues including adrenal glands, testis, spleen, thymus, colon, and ovaries. 2,5,11,24 –29 In contrast, CRF2 receptors are not expressed in the anterior pituitary and have a different expression pattern in the brain and periphery (eg, heart and skeletal muscle). 2,4,5,25,30 –36 Both CRF1 and CRF2 receptors also have different pharmacological properties, and the neuroendocrine activity of CRF is mediated largely through CRF1 receptor signaling.
There are numerous reports in the literature that describe the positive effects of CRF1 receptor inhibition in animal models of anxiety, depression, stress conditions, and addictive disorders, 2,20,21,37 –41 and several of these compounds have progressed into clinical trials, including pexacerfont (BMS-562086; Figure 1). The preclinical pharmacological and adsorption, distribution, metabolism, and excretion properties of pexacerfont are described in 2 published reports 42,43 and are briefly summarized as follows. Pexacerfont is a potent and selective CRF1 receptor antagonist (IC50 values of 6.1, 4.0, and 7.2 nM against rat, dog, and human CRF1 receptors, respectively) with 1,000-fold lower affinity for CRF2 receptors. In vitro, pexacerfont inhibited CRF-mediated ACTH release from primary rat pituitary cells with an IC50 value of 129 nM. In vivo, single oral doses of pexacerfont were active in rodent models of anxiolytic activity (rat defensive withdrawal model and rat elevated plus maze test) and stress-induced colonic motility. The lowest effective dose across these studies was generally 3 mg/kg; corresponding brain receptor occupancy (in the rat parietal frontal cortex) was approximately 50%. In a separate study in rats, the Cmax and AUC(INF) after a single oral dose at 5 mg/kg were 1,261 nM and 5,815 nM·h, respectively. Single oral doses as high as 100 mg/kg in rats had no statistically significant effects on spontaneous locomotor activity or rotorod performance. In preclinical pharmacokinetic (PK) studies, pexacerfont had good oral bioavailability (approximately 40%-59% in rats, dogs, and chimpanzees) and was extensively metabolized in hepatocytes from all species (rats, dogs, monkeys, and humans) with O-demethylation and hydroxylation of the N-alkyl side chain and N-dealkylation as the major metabolic pathways. Renal and biliary excretion were the major routes of elimination in bile duct–cannulated rats.
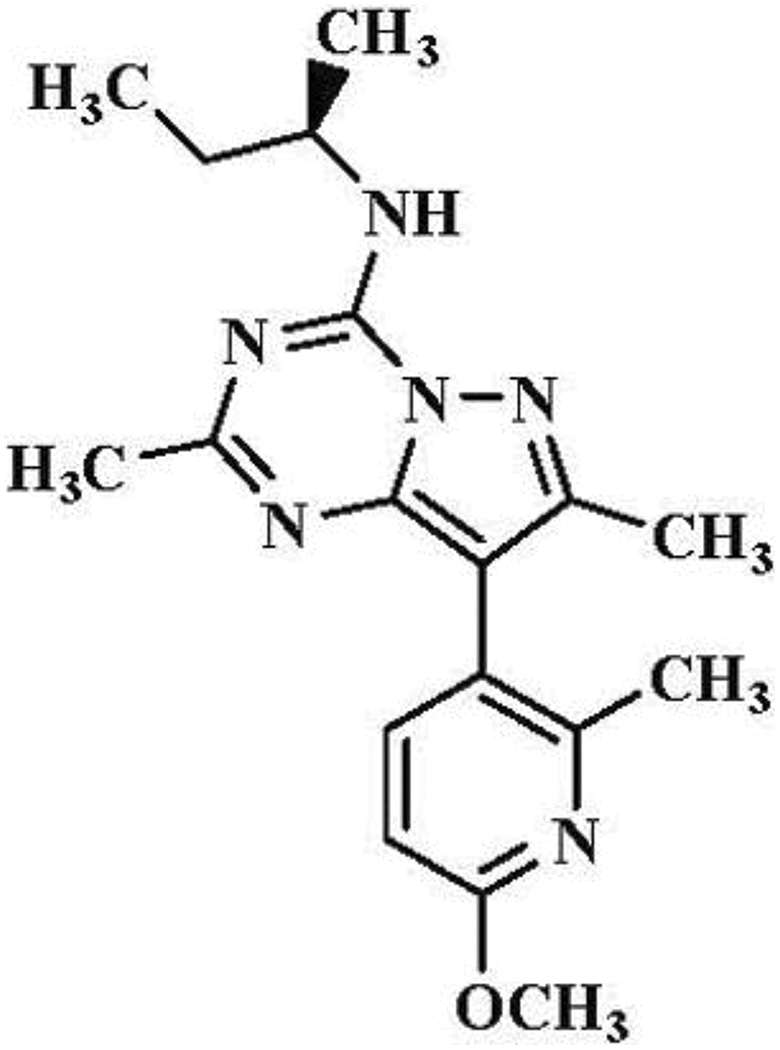
Chemical structure of BMS-562086, pexacerfont.
The toxicity of pexacerfont was evaluated in a comprehensive set of nonclinical safety studies in rats and dogs. Findings were generally limited to direct and secondary effects of hepatic enzyme induction that were most prominent in rats. Other findings included mammary gland hyperplasia and altered female estrous cycling that were only observed in rats and adverse testicular effects that were only observed in dogs after chronic dosing. The testicular effects were reversible changes with an 8× exposure margin at the no observed adverse effect level (NOAEL). It is not clear whether the changes in mammary gland, estrous cycling, and testes represent secondary hormonal changes due to perturbation of the HPA axis or are off-target effects. Therefore, based on favorable pharmacological and PK properties and an acceptable safety profile in rats and dogs, pexacerfont progressed into phase II clinical trials for the treatment of generalized anxiety disorder, irritable bowel syndrome, alcohol dependence, and withdrawal from drug dependency. 44 –47 This report describes the results from the repeat-dose toxicity studies in rats and dogs.
Materials and Methods
These studies were conducted in compliance with US Food and Drug Administration Good Laboratory Practice regulations. All animal experiments were approved by the Institutional Animal Care and Use Committee of Bristol-Myers Squibb Company and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals.
Test Article
Pexacerfont (BMS-562086) is a CRF1 receptor antagonist and is chemically described as 8-(6-methoxy-2-methyl-3-pyridinyl)-2,7-dimethyl-N-[(1R)-1-methylpropyl]pyrazolo[1,5-a]-1,3,5-triazin-4-amine. 48
Animals
The species used in the toxicology studies were Crl:CD Sprague Dawley rats (Charles River Laboratories, Inc, St-Constant, Quebec, Canada, and Kingston, New York) and beagle dogs (Marshall Bioresources, North Rose, New York). The rats were approximately 7 to 8 weeks of age and weighed between 180 and 290 g (males) or 130 and 190 g (females) at the start of dosing. The dogs were approximately 17 months of age, weighed between 10 and 12 kg (males) or 6 and 10 kg (females) in the 3-month study, and were at least 5 months of age and 5 and 11 kg in the 1-year study. All animals were housed in environmentally controlled rooms maintained on a 12-hour light–dark cycle with targeted ranges for humidity (30%-70%) and temperature (17.5 to 23 °C). Water was provided ad libitum, and certified food was provided approximately 4 hours after dosing each day: 22 g for male rats, 16 g for female rats, and 350 g for dogs.
Doses Administered
For both species, pexacerfont was dosed once daily by oral gavage. Vehicle was 0.5% (wt/vol) methylcellulose (Methocel A15C Premium methylcellulose) for rats and Labrafil M1944CS for dogs. Dose volumes were 5 mL/kg for rats and 2 mL/kg for dogs.
Dose Selection
Rats
In a 5-day oral toxicity study of pexacerfont in rats, a dose of 300 mg/kg/d was well tolerated. Clinical signs were limited to perianal staining in 2 of 6 males and soft stool in 1 of 6 males and 2 of 6 females. Based on these results, doses of 10, 30, 100, and 300 mg/kg/d were evaluated in a 3-month oral toxicity study in rats (10 or 15 per sex per group) with a 2-month recovery period.
In the 3-month study, pexacerfont was generally well tolerated at all doses. Liver, thyroid, and pituitary were identified as target organs. Hypertrophic changes in the liver and thyroid were attributed to induction of hepatic drug-metabolizing enzymes and increased biliary clearance of thyroxine. The hypertrophy/vacuolization of thyrotrophs in the pituitary pars distalis was considered secondary to prolonged thyroid-stimulating hormone (TSH) stimulation of the thyroid gland. Partial to complete recovery of all changes was noted following the 2-month postdose recovery period. Based on the low incidence and minimal changes, 100 mg/kg/d was considered the NOAEL. Based upon these data, doses of 30, 100, and 300 mg/kg/d were used in the 6-month oral toxicity study in rats (25 per sex per group) with a 2-month recovery period.
Dogs
In a single-dose study in dogs, emesis with loose/mucoid feces was observed at doses ≤300 mg/kg/d. Administration of structurally similar compounds to dogs at comparable doses in studies ranging from single dose to 3 months in duration resulted in similar dose-dependent clinical findings. Therefore, to avoid excessive clinical toxicity with extended treatment durations, pexacerfont doses of 15, 35, 70, and 140 mg/kg/d were selected for the 3-month oral toxicity study in dogs (4 or 6 per sex per group) with a 2-month recovery period. Since there were no histopathology findings in the 3-month dog study and all doses in this study were well tolerated, doses of 20, 60, and 180 mg/kg/d were chosen for the 1-year dog study.
Parameters Evaluated
Rats
Rats were observed twice daily for clinical signs, individual body weights were determined pretest and at least weekly, qualitative feed consumption was recorded once daily, and ophthalmic examinations were conducted once during pretest and once prior to study termination. Water consumption was measured during week 25 in the 6-month study. Routine hematology, serum chemistry, coagulation, and urinalysis evaluations were conducted during the dosing phase (week 5) and recovery phase (weeks 15 and 22) in the 3-month study and prior to terminal and recovery necropsies in the 6-month study. Urinary corticosterone was measured in samples collected during the dosing (week 4) and recovery phases (weeks 13 and 21) in the 3-month study and during the dosing (week 25) and recovery phases (week 33) in the 6-month study. Serum samples were collected for thyroid hormones (triiodothyronine [T3], T4, and TSH) prior to terminal and recovery necropsies. Plasma concentrations of pexacerfont were determined on day 1 (6-month study only) and on the day of study termination at 0.5, 1, 2, 4, 8, and 24 hours postdose. Blood samples were collected from 3 toxicokinetic study rats/sex in each pexacerfont-treated groups and each rat was sampled at 3 time points on each day of sampling. The mean concentration values were used to generate the composite concentration versus time profiles for each dose and sex. In the 3-month study, vaginal cytology was performed once daily during weeks 3 and 4 and 11 and 12 for all dose groups, and during the last 2 weeks of recovery for the control, 100 and 300 mg/kg/d dose groups. For both studies, all animals were necropsied at study termination (end of dose or end of recovery), selected organ weights were collected, and all animals were subjected to a thorough gross and microscopic evaluation of tissues.
Dogs
In both the 3-month and 1-year studies, dogs were examined twice daily during the dosing phase and once daily during the recovery phase for clinical signs; individual body weights were collected twice during pretest, twice weekly for the first 4 weeks of the study, and then once weekly thereafter; qualitative feed consumption was evaluated daily; and ophthalmic examinations were conducted prior to treatment and during weeks 13 and 24 (3-month study) or at predose and weeks 26 and 52 (1-year study). Routine hematology, serum chemistry, coagulation parameters, and urinalysis were evaluated on samples collected pretest, during the dosing phase (weeks 5 and 12) and recovery phase (week 21) in the 3-month study, and pretest and during the dosing phase in the 1-year study (weeks 39 and 51). Serum was collected for thyroid hormones (T3, T4, TSH) pretest, during the dosing phase (weeks 5 and 13), and during the recovery phase (week 21) in the 3-month study. Serum thyroid hormones were not evaluated in the 1-year study. In the 3-month study, urine samples were collected for urinary corticosterone and creatinine during weeks 5 and 13 of the dosing phase and during week 22 of the recovery phase. Electrocardiograms (ECGs) were performed twice prior to treatment, during the dosing phase (weeks 1 and 13), and during recovery phase (week 21) in the 3-month study, and once during weeks 26 and 52 in the 1-year study. The ECGs were taken approximately 50 to 70 minutes after dosing (approximate Tmax). Serum was collected for toxicokinetics at 0.5, 1, 2, 4, 6, 8, 12, and 24 hours after dosing on day 1 and during week 13 in the 3-month study, and on day 1 and during weeks 26 and 50 in the 1-year study. The first 4 animals/sex/group were bled for toxicokinetic evaluation. The mean concentration values were used to generate the composite concentration versus time profiles for each dose and sex. Animals were necropsied at study termination (end of dose or end of recovery), selected organ weights were collected, and all animals were subjected to a thorough gross and microscopic evaluation of tissues. See Table 1 for the study design of all rat and dog studies.
Study Design for 3-Month and 6-Month Oral Rat Toxicity Studies and 3-Month and 1-Year Oral Dog Toxicity Studies With Pexacerfont.
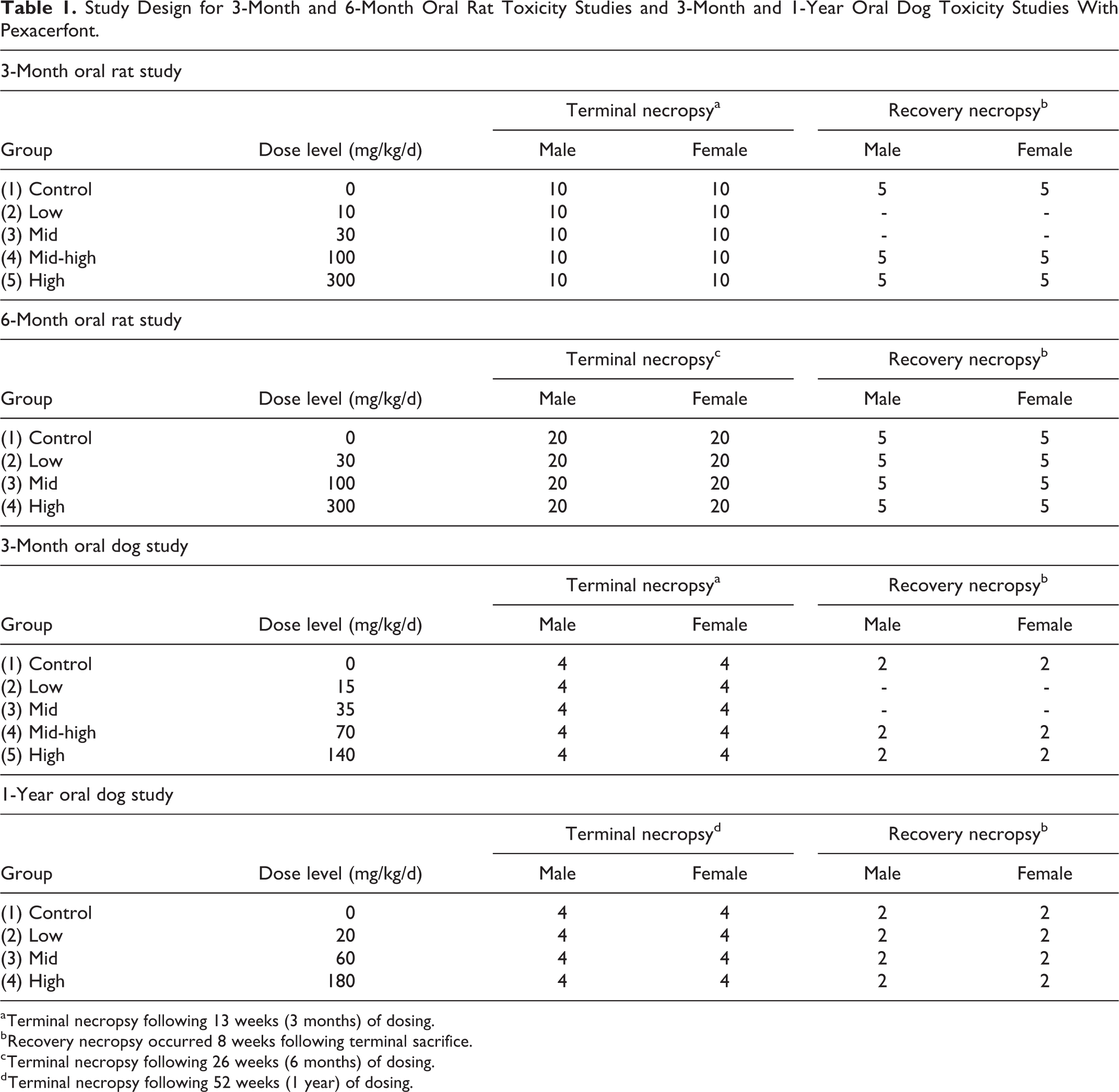
a Terminal necropsy following 13 weeks (3 months) of dosing.
b Recovery necropsy occurred 8 weeks following terminal sacrifice.
c Terminal necropsy following 26 weeks (6 months) of dosing.
d Terminal necropsy following 52 weeks (1 year) of dosing.
Results
Toxicokinetics and Exposure Multiples of Pexacerfont
The systemic exposures (AUC) and the corresponding exposure multiples relative to the mean human AUC at 100 mg/d are indicated in Table 2. In both studies in rats, AUC values generally increased in a dose-proportional manner and were slightly greater in females (<2-fold). Exposures in the 6-month study were minimally increased (<2-fold) compared to the 3-month study, indicating that steady state exposure was achieved by 3 months. The NOAEL in the 6-month rat study was 100 mg/kg/d with exposure multiples of 7× and 12× for males and females, respectively.
Systemic Exposures and Exposure Multiples of Pexacerfont in Repeat-Dose Oral Toxicity Studies in Rats and Dogs.
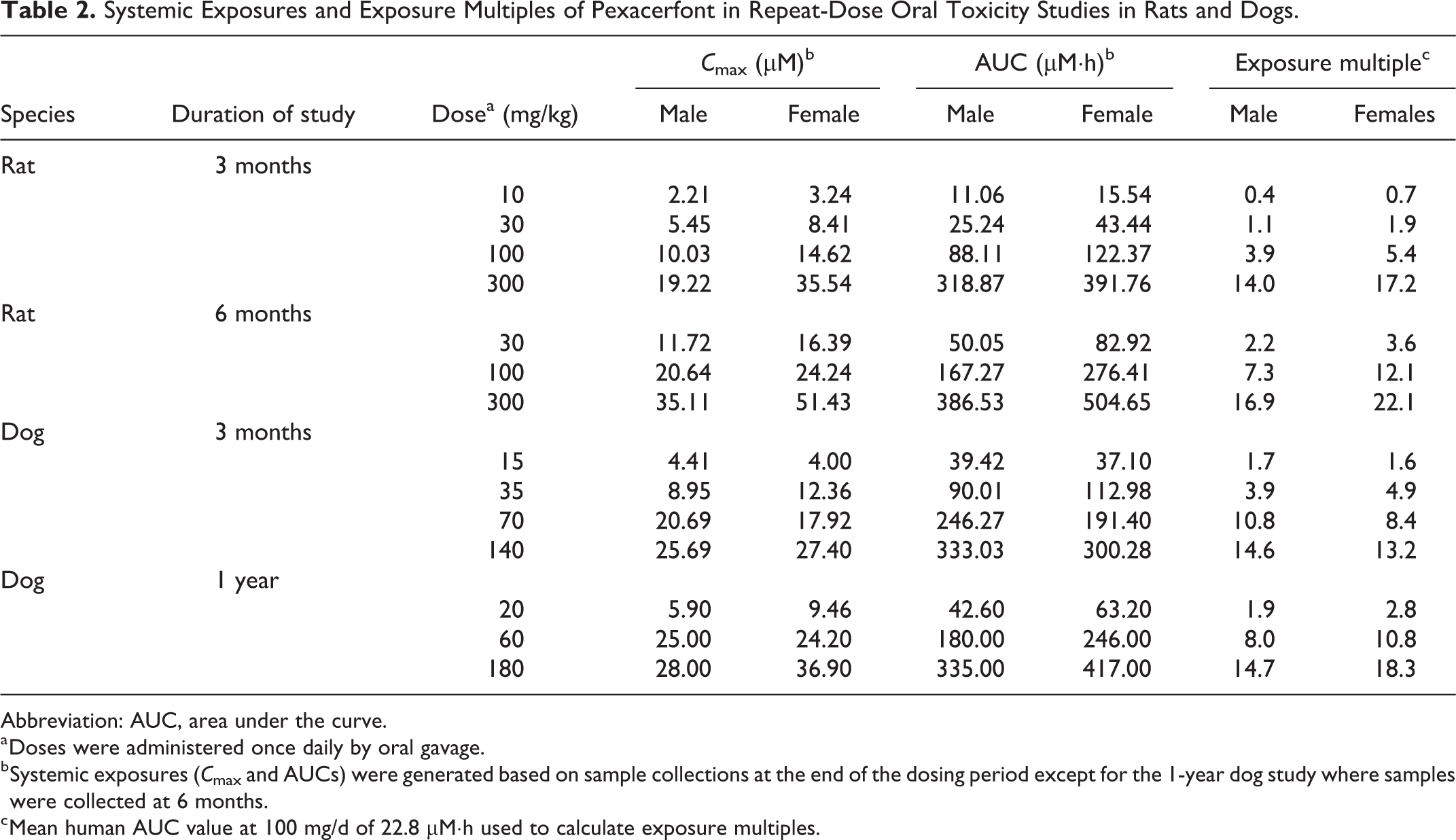
Abbreviation: AUC, area under the curve.
a Doses were administered once daily by oral gavage.
b Systemic exposures (Cmax and AUCs) were generated based on sample collections at the end of the dosing period except for the 1-year dog study where samples were collected at 6 months.
c Mean human AUC value at 100 mg/d of 22.8 µM·h used to calculate exposure multiples.
In both studies in dogs, AUC values generally increased in a dose-proportional manner up to 60 to 70 mg/kg/d but increased in a slightly less than dose-proportional manner at 140 to 180 mg/kg/d, suggesting possible saturation of absorption. There were no consistent differences in exposures between males and females in either study. There were also no apparent effects on exposures between 3 months and 1 year of dosing. The NOAEL in the 1-year dog study was 60 mg/kg/d with exposure multiples of 8× and 11× for males and females, respectively.
In-Life Observations
Rats
BMS-562086 was well tolerated at all doses in both the 3- and 6-month studies. In the 6-month study, there were minimal reductions in body weights (6%-8% lower vs controls) during the last 3 months of dosing and mildly increased (33%-35%) water consumption at study termination at 300 mg/kg/d.
Dogs
All animals survived in both studies and there were no signs of severe clinical toxicity. However, sporadic clinical findings were observed at all doses, including oily hair coats, emesis, excessive salivation, fecal changes (yellow-colored, liquid, mucoid, and/or nonformed feces), and/or foot sores. The emesis, excessive salivation, and fecal changes had a greater incidence at the 2 highest doses, while there was no clear dose–response relationship to the other findings.
Effects Associated With Hepatic Microsomal Enzyme Induction
Rats
Alterations in the liver in both rat studies included increased liver weights (up to 91% compared to controls) with a corresponding increased incidence and severity (moderate) of centrilobular hepatocellular hypertrophy at ≥30 mg/kg/d with no corresponding elevations in alanine aminotransferase or aspartate transaminase. The liver weight changes were reversible at ≤100 mg/kg/d in the 3-month study, and there was complete recovery of all liver findings in the 6-month study. Statistically significant increases in serum T4 levels at doses of 30, 100, and/or 300 mg/kg/d (Table 3) most likely reflected a compensatory response to increased clearance of T4 due to hepatic enzyme induction and increased hepatobiliary excretion. Statistically significant dose-related increases in TSH levels at 100 and 300 mg/kg/d provided additional evidence for an exaggerated compensatory feedback loop. The increases in TSH were slightly greater in males. The lack of a direct dose-related correlation between serum levels of T4 and TSH was likely due to the magnitude of the initial decreases in serum T4 (eg, lower at 30 mg/kg/d) and the corresponding dynamic range of the feedback loop. For example, large increases in TSH at 300 mg/kg/d were necessary to maintain serum T4 at or near normal levels. All changes in T4 and TSH were completely reversible. There were no changes in serum T3 levels in the 3-month study (not measured in the 6-month study).
Serum Thyroid Hormone Levels in Rats and Dogs After Repeated Dosing With Pexacerfont.a
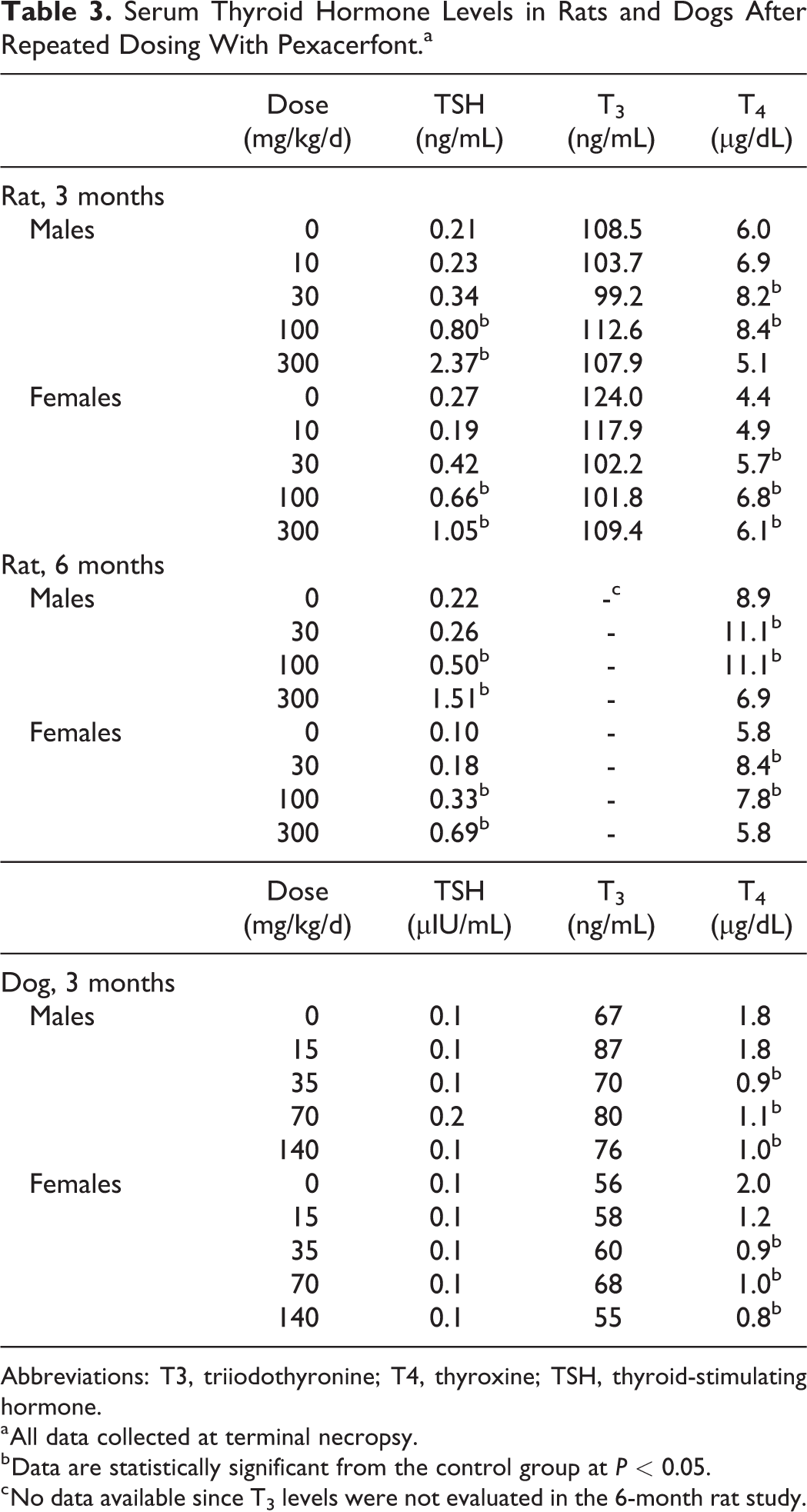
Abbreviations: T3, triiodothyronine; T4, thyroxine; TSH, thyroid-stimulating hormone.
a All data collected at terminal necropsy.
b Data are statistically significant from the control group at P < 0.05.
c No data available since T3 levels were not evaluated in the 6-month rat study.
At all doses in both studies, there was an increased incidence and severity (minimal to severe) of thyroid follicular cell hypertrophy/hyperplasia with benign thyroid follicular adenomas at 300 mg/kg/d (Figure 2). The findings at 300 mg/kg/d were considered adverse. Thyroid gland weights were also increased at 100 and 300 mg/kg/d. There was partial to complete recovery of the thyroid changes in both studies. Notably, decreased thyroid colloid with variable follicular cell attenuation was observed at ≥100 mg/kg/d at the end of recovery in the 3-month study along with follicular cell hypertrophy and follicular cell adenomas at 300 mg/kg/d. There were no thyroid findings at the end of recovery in the 6-month study.
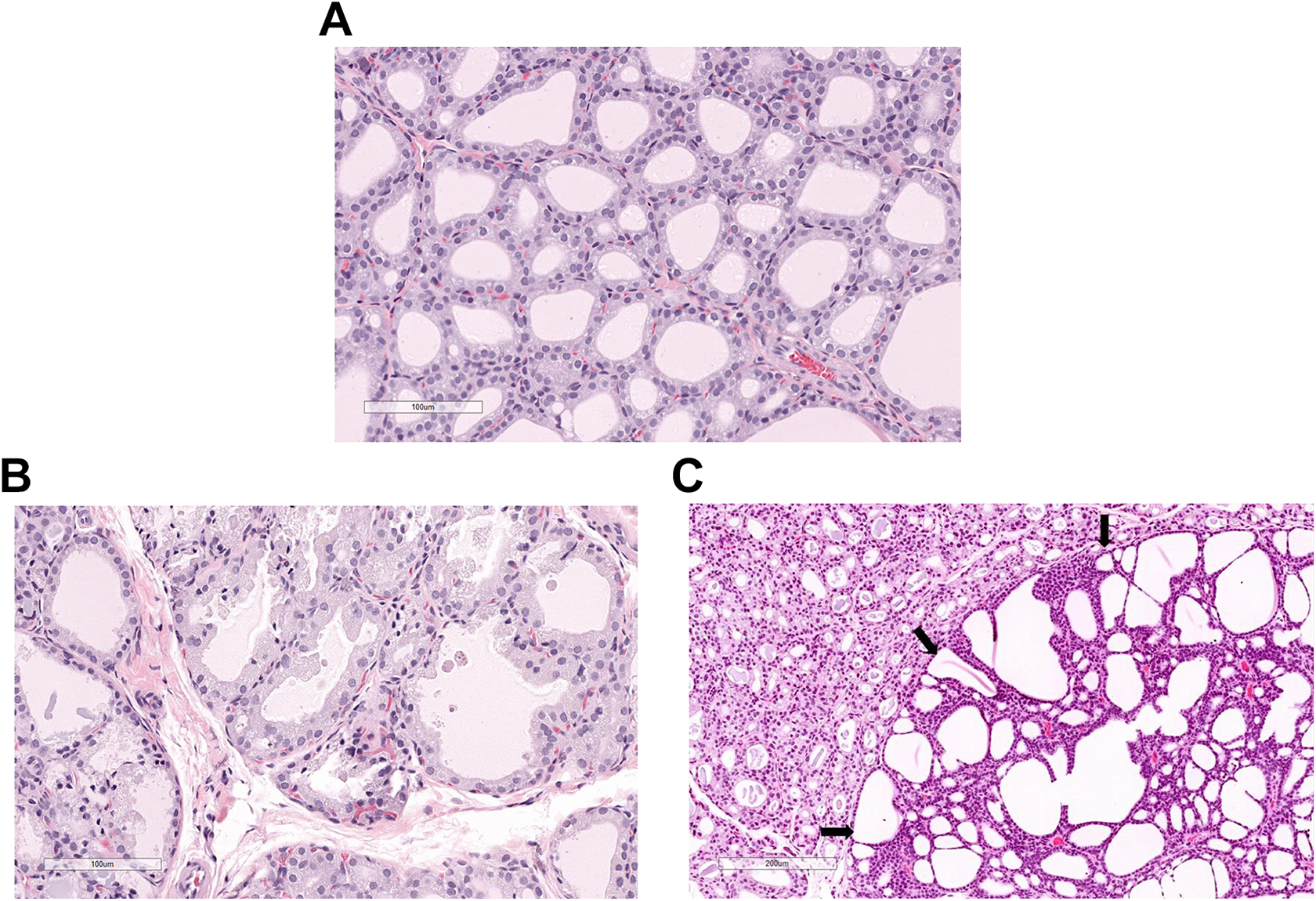
A, Male control rat with normal thyroid gland composed of numerous thyroid follicular cells that form follicles containing intraluminal colloid. B, Male rat dosed with pexacerfont at 300 mg/kg/d for 6 months. Thyroid gland exhibiting hypertrophy (larger) and hyperplasia (increased numbers) of thyroid follicular cells and decreased intraluminal colloid. C, Male rat dosed with pexacerfont at 300 mg/kg/d for 6 months. Thyroid gland containing a follicular cell adenoma (right with edge of adenoma delineated by arrows) and hypertrophic and hyperplastic thyroid follicular cells (left) with compression of the adjacent non-neoplastic tissue.
In both studies, there was an increased incidence and severity (minimal to moderately severe) of multifocal hypertrophy/vacuolization of thyrotroph cells (based on immunohistochemical staining with TSH) in the pituitary glands (pars distalis) at all doses in males only (Figure 3). These findings were partially reversible in both studies. The microscopic findings in the thyroid glands and pituitary were considered adaptive changes (not adverse) to the increased production of TSH that was somewhat greater in males.
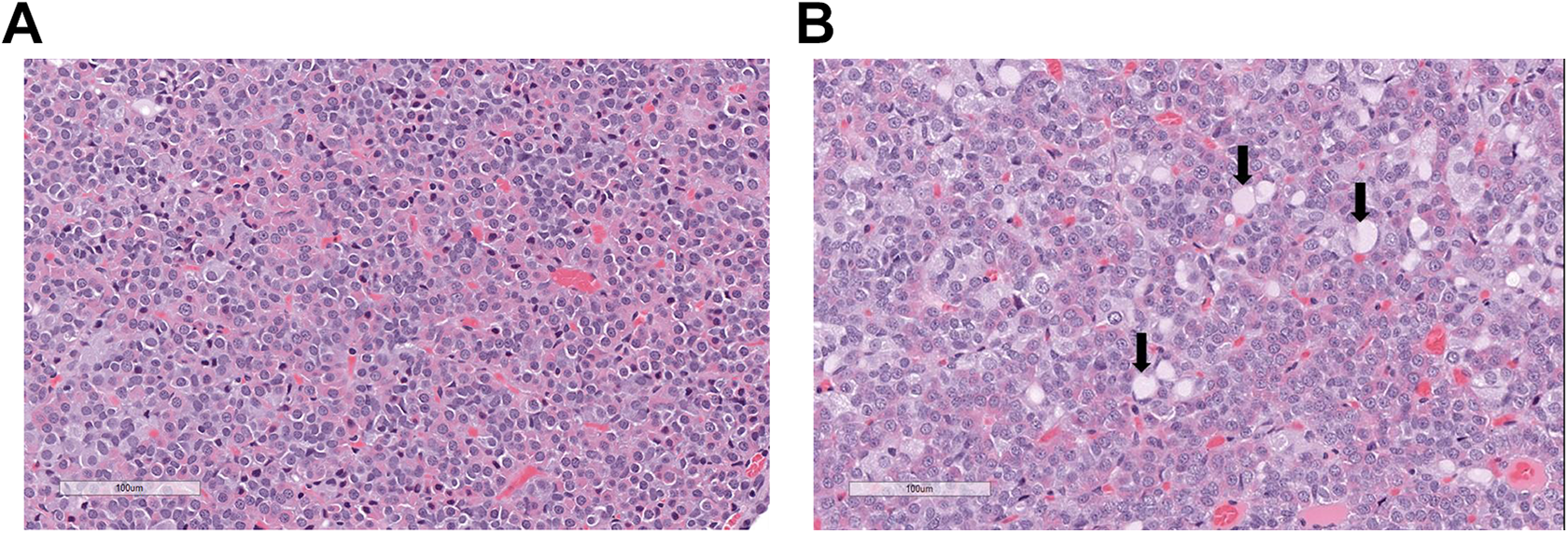
A, Male control rat with normal pituitary gland (pars distalis) containing pituicytes. B, Male rat dosed with pexacerfont at 300 mg/kg/d for 6 months. Pituitary gland with nonstaining cytoplasmic vacuoles in hypertrophic pituicytes (arrows) in the pars distalis that were identified as thyrotrophs by immunohistochemistry (not shown).
In the 6-month study, effects on several clinical biochemistry parameters (increased total cholesterol, 29% to 1.8-fold; decreased triglycerides, 25%-69%; and increased γ glutamyltransferase, 0.99- to 9.9-fold) likely reflected hepatic metabolic enzyme induction. All of these changes were fully reversible and none of these were considered adverse.
Dogs
In the 3-month study, there were mild and reversible increased liver weights at 140 mg/kg/d with no corresponding clinical chemistry, macroscopic, or microscopic findings. Additionally, mean serum T4 levels were mildly decreased in males (39%-50%) at ≥35 mg/kg/d and in females (40%-60%) at all doses; however, there were no associated changes in serum TSH or T3 levels or correlative alterations in thyroid weight or morphology (Table 3). The T4 changes were fully reversible. The reduced effects on T4 in dogs compared to rats are not surprising since rats are generally more sensitive to hepatic enzyme induction and alterations in thyroid homeostasis. 49
Other Findings
Rats
In the 3-month study, a single male at 10 mg/kg/d had minimal hyperplasia of the mammary gland. In the 6-month study, there was minimal to slight mammary glandular hyperplasia in males at all doses and an increased incidence of minimal to slight mammary gland hyperplasia in females at 300 mg/kg/d (Figure 4). There was complete recovery of the mammary gland findings in both studies. At 100 and 300 mg/kg/d in the 3-month study, there was altered estrous cycling during weeks 3 to 4 that was characterized by prolonged episodes of metestrus–diestrus and up to a 53% reduction in the mean number of complete estrous cycles, including some rats without any complete cycles. Prolonged episodes of proestrus–estrus and a 38% increase in mean cycle length were still present following recovery necropsy, indicative of partial reversibility. There were no microscopic findings associated with the altered estrous cycling noted in the female reproductive tract at either the terminal or recovery necropsy. At 300 mg/kg/d in the 6-month study, there was increased cell debris in the lumen of the epididymal tubules that were likely degenerative germinal epithelial cells, indicative of minimal testicular toxicity. This finding was not considered adverse due to the low severity and completely resolved following the 2-month recovery period.
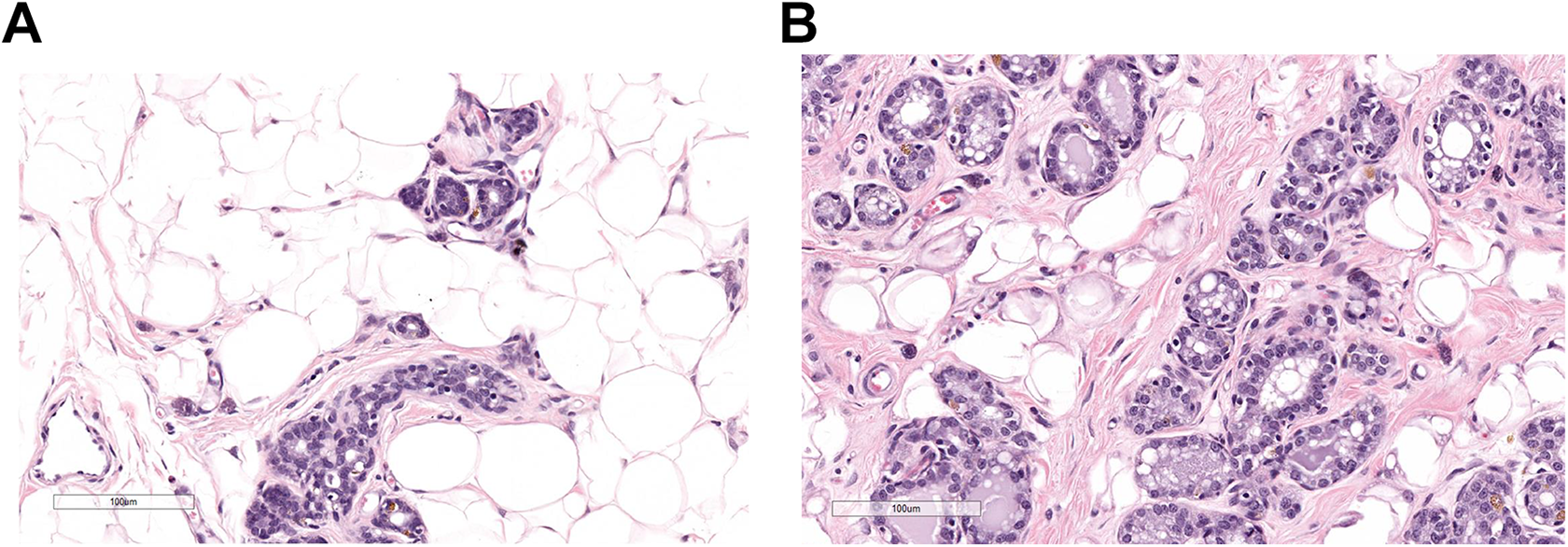
A, Female control rat with normal mammary gland tissue in the subcutaneous tissue. B, Female rat treated with pexacerfont at 300 mg/kg/d for 6 months. Hyperplastic mammary gland tissue in the subcutaneous tissue.
In both studies at ≥100 mg/kg/d, there were minimal to mild decreases in red blood cell count, and/or decreased hemoglobin, mean corpuscular hemoglobin concentration, and/or hematocrit. These changes were partially reversible with persistence of a mildly decreased red blood cell count or mean hemoglobin and hematocrit at 300 mg/kg/d in both studies. However, none of the changes in red blood cell parameters were considered adverse given the small magnitude of the reductions. There were no changes in urine corticosterone in the 3-month study, but there were mildly decreased urine corticosterone output (31%-34%) and corticosterone to creatinine ratios (27%-29%) in males at 300 mg/kg/d in the 6-month study, confirming intended pharmacology. The corticosterone changes in the 6-month study were completely reversible.
Dogs
In the 1-year study, there were microscopic findings in the male reproductive tract (testes) consisting of minimal to slight degenerative germ cells at 20 mg/kg/d and minimal to moderate degenerative germ cells at 180 mg/kg/d (Figure 5). These findings were not observed in the 3-month study (up to a 14× exposure margin). The degenerating cells were predominantly round and elongating spermatids, and in 1 of these dogs, the degeneration resulted in testicular spermatid depletion with exfoliated germ cells (spermatids) in the epididymal tubules. Although the testicular changes were considered adverse at 180 mg/kg/d (15× exposure margin), they were reversible following a 2-month recovery period.
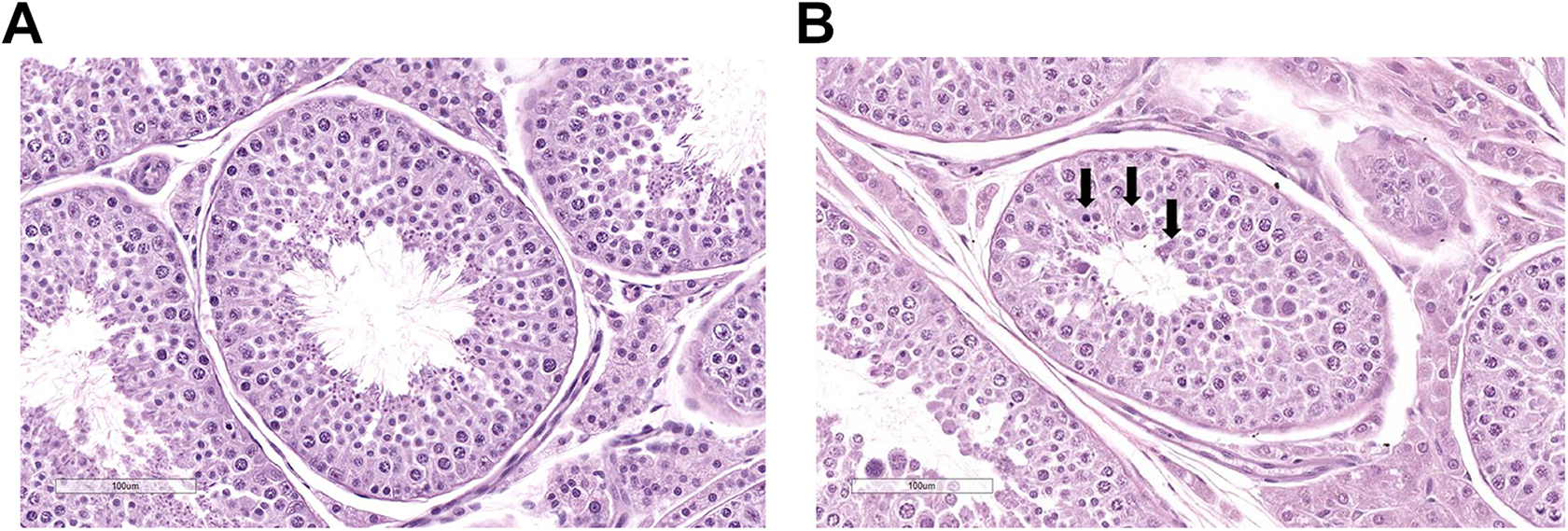
A, Control male dog with normal cross sections of seminiferous tubules in the testis containing spermatozoa. B, Treated male dog dosed orally with pexacerfont at 160 mg/kg/d for 1 year. Cross section of a seminiferous tubule in the testis containing degenerative spermatids (arrows).
In the 3-month study, transient and fully reversible increases in urinary total cortisol (1.5- to 3-fold) and cortisol to creatinine ratios (1.8- to 2.4-fold) in males and females at 140 mg/kg/d were considered to be a consequence of increased stress associated with the clinical observations (salivation, emesis, fecal alterations, foot sores).
Discussion
In both rat studies, findings of increased liver size/weights coupled with centrilobular hepatocellular hyperplasia and alterations in the thyroid gland (increased size and weight, follicular cell hypertrophy/hyperplasia, and follicular adenomas) and pituitary gland (hypertrophy/vacuolation of thyrotroph cells) were consistent with microsomal enzyme induction in the liver and subsequent alterations in thyroid homeostasis. 50 All of these findings occurred at much higher doses/exposures compared to the lowest effective dose range in the rat pharmacology models. 42 In contrast, findings in dogs associated with hepatic enzyme induction were limited to mildly increased liver weights and mildly decreased T4 levels. Microsomal enzyme induction is a well-established mechanism for hepatocellular hypertrophy in rodents. 51 Uridine diphosphate glucuronosyltransferase (UDPGT) is a microsomal enzyme that transforms bilirubin, drugs, and hormones into water-soluble metabolites that are excreted into the bile of the liver 49,50,52 and has been shown to be upregulated in the liver of rats given DMP 904—a pharmacologically and biochemically (eg, pyrazolopyrimidine) similar CRF1 receptor antagonist. 53 In studies with DMP 904, there was upregulation of UDPGT and an increased hepatobiliary clearance of thyroid hormones (principally T4) with increased TSH. Therefore, persistent elevations of TSH and associated microscopic changes in the thyroid and pituitary in rats treated with pexacerfont were also considered adaptive physiological responses due to chronic TSH stimulation in response to decreased serum levels of T4, that is, loss of negative feedback control. 54 There was no direct toxicological effects of pexacerfont on the thyroid or pituitary gland. In addition, no evidence of altered thyroid or pituitary function has been reported in humans administered pexacerfont for up to 2 months at exposures up to 22.8 µM·h. 44 –47 Finally, the type of thyroid tumorigenic response in rats (follicular cell adenoma) has no established clinical relevance to humans since rats are notably more sensitive to drugs that alter thyroid homeostasis. 54 –56
Although there were minor reductions in serum T4 levels in dogs in the 3-month study, there were no corresponding changes in TSH levels and no alterations in thyroid morphology at similar or higher systemic exposures than in rats. These results demonstrate that dogs are less sensitive to pexacerfont-induced hepatic enzyme induction.
It is not clear whether the changes in mammary gland, estrous cycling, and testes are due to off-target effects or are secondary to hormonal changes due to perturbation of the HPA axis. For example, there is some evidence to suggest that hyperprolactinemia can occur in response to CRF receptor antagonists. 57 –59 Prolactin is a peptide hormone produced in the anterior portion of the pituitary gland that is primarily responsible for lactation. Prolactin is also needed for normal cycling of the female reproductive tract and rats with hyperprolactinemia often present with prolonged or persistent diestrus. 60 Although hormone levels were not measured in the current studies, the abnormal estrous cycling in rats treated with pexacerfont is consistent with this hypothesis (hyperprolactinemia). One other possible explanation for the effects on estrous cycling is a direct effect of pexacerfont on the ovary itself. The CRF1 receptor messenger RNA has been reported in stroma cells and in the theca surrounding the ovulatory follicles in rats, suggesting an important role of CRF signaling during the ovulatory process, especially during periods of stress. 29 Estrous cycles were not monitored in dogs.
Mammary gland hyperplasia is another well-recognized response in male and female rats due to hyperprolactinemia. 59 Prolactin is the hormone primarily responsible for mammary gland differentiation, lobuloalveolar growth, and synthesis of all major milk components. 57,61,62 Increased serum levels of prolactin promote ductal branching in the mammary gland and can also lead to mammary gland hyperplasia and adenomas as was observed in rat studies. In addition to prolactin, changes in other hormones can promote increased duct development in mammary glands, including androgen, growth hormone, and epidermal growth factor. 63 –65 While none of these hormones were measured in the current studies, the testicular effects were suggestive of altered testosterone production (see below).
Testis was a target organ in dogs, but only after chronic administration. Testicular changes were characterized by degenerative spermatids in the testes and increased cell debris in the epididymal tubular lumens, which indicates the release of degenerative germinal epithelial cells. This is somewhat surprising since the literature suggests that CRF is a negative regulator of testosterone production in rats. 66 Hence, CRF1 receptor inhibitors would be expected to increase testosterone production and spermatogenesis, yet the opposite effect was observed. Although these effects on the testes are not completely understood, the spectrum of changes is consistent with reduced testosterone production by the Leydig cells to the extent that Sertoli cells were unable to support spermatogenesis. The presence of degenerative germinal epithelial cells in the lumen of the epididymal tubules in dogs and mammary gland hyperplasia in male rats are both consistent with reduced testosterone production. Since the testicular changes were minimal to moderate in severity and were fully reversible, they are not likely to have a major impact on male reproductive function. Nevertheless, possible effects on male fertility should be taken into account with long-term pexacerfont dosing in humans.
In the 6-month rat study, decreased urinary corticosterone output and corticosterone to creatinine ratios in males at 300 mg/kg/d were consistent with the known pharmacodynamic effects of pexacerfont. This finding suggests that pexacerfont was able to inhibit the actions of CRF, which resulted in decreased synthesis and secretion of ACTH in the corticotrophs of the anterior pituitary gland and subsequently decreased production of corticosterone by the cortical cells of the adrenal gland. However, it is also recognized that urinary measurements of corticosterone in rats can be variable and it is not the most sensitive biomarker for assessing alterations in the HPA axis. Therefore, the absence of effects in female rats or at lower doses in males does not necessarily indicate a lack of pharmacology, especially since single oral doses of pexacerfont at doses as low as 3 mg/kg and exposures as low as 5,815 nM·h were active in anxiolytic and stress-related models. 42,43
In summary, the results of these studies demonstrate that pexacerfont has an acceptable safety profile following chronic dosing in rats and dogs. Relative to the AUC values in humans at the clinical dose of 100 mg, exposure margins at the NOAELs in the chronic studies were 7× to 8×. Findings were generally attributed to hepatic enzyme induction, which was most prominent in rats. Other findings included mammary gland hyperplasia and altered female estrous cycling that were only observed in rats and adverse (but reversible) testicular effects that were only observed in dogs after chronic dosing. It is not clear whether the changes in mammary gland, estrous cycling, and testes are off-target effects or were secondary to hormonal changes due to perturbation of the HPA axis. However, based on favorable pharmacology and PK characteristics and an acceptable safety profile, pexacerfont progressed into clinical trials for a number of anxiety- and stress-related disorders. The results of several phase I/II studies confirmed the safety of pexacerfont in humans 44 –47
Footnotes
Author Contributions
M. R. White contributed to conception and design, contributed to acquisition, drafted the manuscript, and critically revised the manuscript. M. J. Graziano contributed to conception and design, contributed to acquisition and analysis, drafted the manuscript, and critically revised the manuscript. T. P. Sanderson contributed to conception and design, contributed to acquisition and analysis, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article: Funding for these studies was provided by Bristol-Myers Squibb.