
Abstract
The carcinogenic potential of roxadustat (FG-4592), a novel orally active, heterocyclic small molecule inhibitor of hypoxia-inducible factor prolyl hydroxylase (HIF-PH) enzymes in clinical development for treatment of anemia, was evaluated in CD-1 mice and Sprague Dawley rats. Inhibition of HIF-PH by roxadustat leads to a rapid increase in cytoplasmic HIF-α concentrations, followed by translocation of HIF-α to the nucleus and upregulation of HIF-responsive genes, including erythropoietin. Roxadustat was dosed by oral gavage 3 times weekly (TIW) for up to 104 weeks in mice at 0, 15, 30, and 60 mg/kg and in rats at 0, 2.5, 5, and 10 mg/kg. Treatment-associated changes in hematology parameters were consistent with the pharmacologic activity of roxadustat and included elevations in hematocrit in mice at 30 and 60 mg/kg TIW and elevations in erythrocyte count, hemoglobin, hematocrit, and red cell distribution width in rats at 10 mg/kg TIW. No increase in mortality or neoplastic effects compared with vehicle controls was observed after roxadustat treatment in either species. No treatment-related nonneoplastic findings were observed in mice, whereas nonneoplastic microscopic findings in rats were limited to atrial/aortic thromboses at 10 mg/kg TIW males and bone marrow hypercellularity in all treated male and female groups, consistent with the pharmacology of roxadustat. In conclusion, roxadustat administered by oral gavage to mice and rats TIW for up to 104 weeks resulted in dose-dependent exposure and hematologic effects with no effect on survival or development of neoplastic lesions at up to 60 mg/kg in mice and up to 10 mg/kg in rats.
Introduction
Roxadustat is a novel, orally bioavailable, heterocyclic small molecule that reversibly inhibits hypoxia-inducible factor prolyl hydroxylase (HIF-PH) enzymes and activates HIF and transcription of HIF-responsive genes, including erythropoietin (EPO). Roxadustat is a therapeutic modality developed/designed to elevate EPO and stimulate complete erythropoiesis, allowing correction of anemia in some settings that are underserved by recombinant human EPO therapy. Roxadustat was shown to correct anemia in phase 2 clinical trials and is currently in phase 3 clinical development for treatment of patients with anemic chronic kidney disease (CKD). 1 –4 Hypoxia-inducible factor is a key transcription factor within the body’s major oxygen-sensing mechanism. 5 Hypoxic conditions enhance EPO expression, red blood cell mass, and hemoglobin (Hb) 6 –9 via the coordinated expression of EPO and genes responsible for other aspects of erythropoiesis, including iron absorption, transport, storage, metabolism, and heme synthesis, all of which contribute to the production of new red blood cells. Many genes involved in this coordinated erythropoietic response to hypoxia are known to be regulated by HIF. 10 Genes regulated by HIF have a broad spectrum of functions, the main goal of which is to facilitate cell survival in different ways. For instance, HIF induces transcription of all enzymes involved in glycolysis and genes involved in angiogenesis, such as vascular endothelial growth factor. 5 Roxadustat inhibition of HIF-PH prevents hydroxylation and subsequent degradation of HIF-α subunits, thus activating HIF target genes under normoxic conditions. In phase 2 clinical trials conducted to date, roxadustat corrected anemia and maintained Hb levels in patients with anemia having nondialysis-dependent and dialysis-dependent CKD. 2,3
The molecular mechanisms by which HIF-PH enzymes control the activity of HIF and its target genes are well known. Hypoxia-inducible transcription factors consist of heterodimers formed between HIF-β and HIF-α subunits. Two major HIF-α isoforms are known, HIF-1α and HIF-2α, which are synthesized continuously and, under normoxic conditions, become hydroxylated by the action of HIF-PH enzymes with oxygen as a substrate in the hydroxylation reaction. The resulting hydroxylated proteins associate with the von Hippel-Lindau (VHL) E3 ubiquitin ligase complex and are further modified and ultimately degraded by the proteasome. Under hypoxic conditions, inhibition of HIF-PH leads to a rapid increase in the cytoplasmic concentration of HIF-α, which then translocates to the nucleus where it dimerizes with HIF-β and selectively upregulates HIF-responsive genes. 5,11,12
It has been postulated that pharmacologic HIF activation may increase tumorigenic potential. 13 However, evidence also exists for antitumor effects of HIF activation including studies that demonstrate HIF-induced upregulation of proapoptotic proteins 14 and studies in which genetic elevation of HIF leads to antitumor phenotypes. 15 –17
Based on the International Conference on Harmonization (ICH) compliant battery of genotoxicity (data not shown) tests, roxadustat is not mutagenic. In addition, we previously reported no neoplastic effect in rat and mouse in 2-year carcinogenicity studies conducted with an earlier generation HIF-PH compound, FG-2216. 18 In these 2 studies, FG-2216 was administered 3 times weekly (TIW) to male and female CD-1 mice and Sprague Dawley rats at pharmacologically active doses up to 80 and 25 mg/kg, respectively. Following 2 years of treatment, there was no FG-2216-related adverse effect observed for mortality and no evidence of a carcinogenic effect.
The purpose of the current investigation was to evaluate the carcinogenic potential of roxadustat in mice and rats chronically exposed to roxadustat for 2 years with an intermittent dosing regimen similar to that used in clinical studies.
Materials and Methods
All portions of the studies were performed at Charles River Laboratories, Preclinical Services, Ohio (Spencerville, Ohio) and other Charles River Laboratory sites, with the exceptions of the pathology peer review, which was conducted at Experimental Pathology Laboratories, Inc (Frederick, Maryland), and mortality and tumor statistical analyses were performed at BioSTAT Consultants, Inc (Portage, Michigan). Animal use was in accordance with Guide of the Institute for the Care and Use of Laboratory Animals. 19
The studies were conducted in accordance with US Department of Health and Human Services, Food and Drug Administration (FDA), and US Code of Federal Regulations, Title 21, Part 58: Good Laboratory Practice for Nonclinical Laboratory Studies. The design of these mouse and rat carcinogenicity studies was based on the study objective(s), the overall product development strategy, and ICH Harmonized Tripartite Guidelines S1C(R2), S1B, S1A, M3 (R2), and S3a. The study design (including dose levels) was presented and agreed upon by the FDA under a special protocol assessment.
In accordance with an FDA recommendation, a second control group (saline) was added in each study. Histopathology assessment was deemed not necessary if no differences in toxicity between the 2 controls (vehicle and saline) were noted and if tumor incidence in the vehicle control was not different from historical control values. Neither of these criteria was met in either study; therefore, histopathology of the saline control groups was not analyzed for either mice or rats. All comparisons presented in this article are between dose groups and vehicle control.
Test Article
Roxadustat (C19H16N2O5, molecular weight: 352.34) was manufactured for FibroGen, Inc by Asymchem (Tianjin, China), stored at room temperature, and protected from light. The purity of the lot used was 98.7%. Dose formulations were prepared at concentrations of 0, 1.5, 3.0, and 6.0 mg/mL for the mouse and 0, 0.25, 0.5, and 1.0 mg/mL for the rat in an aqueous vehicle with 0.5% carboxymethylcellulose sodium (CMC) and 0.1% polysorbate 80 (Tween 80). Formulations were stored refrigerated (−2°C to 8°C) and in amber bottles to protect from light. Homogeneity and stability of the test formulations were verified.
Animals and Husbandry
Male and female Crl: CD1 mice and Crl: CD(SD) rats were from Charles River Laboratories (Portage, Michigan). Animal rooms were environmentally controlled (temperature of 18°C-26°C; relative humidity of 30%-70%; 10 or greater air changes/hour; and a 12-hour light–dark cycle). Following assignment to study, each animal was individually housed in stainless steel cages. When warranted by health conditions, rats were provided with resting plates. Animals were offered Purina Mills Nutrition International Certified Rodent Chow No 5CR4 (14% protein) and water was provided ad libitum, except during specified procedures. At the beginning of the study, mice were 6 weeks old and weighed between 24.2 and 34.9 g (males) and 17.6 and 27.4 g (females) and rats were 7 weeks old and weighed between 185 and 265 g (males) and 137 and 205 g (females).
Dose Administration
The oral gavage route of administration was selected because the intended route of administration in humans is oral. Intermittent dosing of TIW was used as that is the intended clinical dosing regimen.
Roxadustat was administered by oral gavage, TIW (Monday, Wednesday, and Friday) for up to 104 weeks (dosing phase). Doses were based on the most recently recorded body weight. Animals were dosed at the volume of 10 mL/kg/dose in an aqueous vehicle with 0.5% CMC and 0.1% polysorbate 80. Dose formulations were protected from light at all times and stirred at least 30 minutes prior to use and continuously throughout the dosing procedure by using a magnetic stir bar and stir plate.
Male and female CD-1 mice (N = 60/sex/dose group for the carcinogenic evaluation) were administered 0, 15, 30, or 60 mg/kg roxadustat TIW, and Sprague Dawley rats (N = 75/sex/dose group for the carcinogenic evaluation) were administered 0, 2.5, 5, or 10 mg/kg roxadustat TIW. Additional mice and rats were included in each dose group to evaluate the toxicokinetics (TK) of chronic roxadustat administration. Satellite TK mice were administered 0, 15, 30, or 60 mg/kg TIW of roxadustat (24 animals/sex/group for 0 mg/kg and 72 animals/sex/group for the remaining groups). Satellite TK rats were administered 0, 2.5, 5, or 10 mg/kg TIW of roxadustat (6 animals/sex/group for 0 mg/kg and 24 animals/sex/group for the remaining groups). An additional 20 mice/sex and 20 rats/sex were untreated and served as sentinels to monitor for overall health of the colony.
Justification of Doses
The dose levels for this carcinogenicity study were selected in agreement with FDA recommendation based on hematologic end points (pharmacodynamics) and expected maximum tolerated doses over 2 years of chronic dosing rather than multiples of clinical exposure.
In mice, the dose selection was based on a 13-week oral toxicity study with roxadustat dosing TIW at 0, 30, 45, 60, 100, or 150 mg/kg (data not shown). Dosing for 13 weeks was well tolerated up to 60 mg/kg TIW and mortality was observed at 150 mg/kg beginning at day 59. Polycythemia was observed in mice treated with 60 mg/kg TIW for 13 weeks as evidenced by hematocrit (Hct) increases to >55% and Hb increases to approximately 17 g/dL, without other abnormalities. Based on this information and in agreement with FDA, the maximum tolerated dose was judged to be 60 mg/kg TIW and was selected as the high dose for the study, and 30 and 15 mg/kg TIW were selected as the intermediate and low doses, respectively.
In rats, dose selection was based on the results of a 6-month toxicity study of 0, 5, 15, 30, and 40 mg/kg roxadustat administered TIW to Sprague Dawley rats (data not shown). With repeated intermittent dosing, at doses ≥15 mg/kg, Hb and Hct levels increased to approximately 20 g/dL and >60%, respectively. The pharmacodynamic effect of roxadustat at doses ≥15 mg/kg TIW, reflected by polycythemia and associated adverse effects, was shown to be dose limiting for chronic treatment in rats (data not shown). Based on this information and in agreement with FDA, the high dose level of 10 mg/kg TIW was selected as it is expected to provide the desired hematological response without significant mortality, and 5 and 2.5 mg/kg TIW were selected as the intermediate and low doses, respectively.
Clinical Observations
The in-life procedures, observations, and measurements described below were performed for all carcinogenicity animals. Toxicokinetic animals were weighed and any abnormal clinical observations were recorded.
In-life end points for both mice and rats included clinical observations (twice daily) for mortality, abnormalities, or signs of pain or distress, and detailed observations were done weekly throughout the studies; abnormal findings were recorded. Beginning on week 26, detailed clinical observations included a palpable mass examination (including the occurrence, size, location, and description of palpable masses). The persistence or disappearance of these masses was documented at the next weekly clinical examination.
Body weights were recorded at baseline, once weekly to week 26, then monthly, and during the last dose week. Food consumption was recorded once weekly to week 26, then monthly through study end.
Sentinel animals were checked twice daily (
Clinical Pathology (TK Animals Only)
Blood samples for clinical pathology (hematology and clinical chemistry) were collected from TK rats at baseline and at 12 months and from TK mice and rats at termination. Mice were not fasted, whereas rats were fasted overnight prior to scheduled collections. Interim blood samples were collected via a jugular vein from rats, and terminal samples were collected via cardiocentesis from mice or from the vena cava of rats. The anticoagulant potassium EDTA was used for hematology, and a separate sample was processed without anticoagulant to provide serum for chemistry. Hematology tests conducted were for white blood cell differential, Hb, red blood cell (erythrocyte) count, mean corpuscular volume, mean corpuscular Hb, mean corpuscular Hb concentration, white blood cell (leukocyte) count, blood cell morphology, Hct, platelet count, red cell distribution width, and reticulocyte count. Clinical chemistry tests conducted were for alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, γ-glutamyltransferase, total bilirubin, urea nitrogen, creatinine, calcium, phosphorus, total protein, albumin, globulin, albumin/globulin ratio, glucose, cholesterol, triglycerides, sodium, potassium, and chloride. When possible, a blood sample for clinical pathology tests was collected from TK animals terminated at an unscheduled interval.
Necropsy and Histopathology
Individual animals were euthanized prior to scheduled termination as necessary for humane reasons. Necropsies were performed on all carcinogenicity mice or rats that died or were terminated at an unscheduled interval. Sentinel animals were euthanized after the final blood collection interval by inhalation of isoflurane followed by exsanguination. Any animals with signs of severe debility or toxicity were euthanized by carbon dioxide inhalation. Sentinel animals found dead were discarded without necropsy and no tissues were retained.
All mouse dose groups were terminated as scheduled following 104 weeks of dose administration. Male rat groups began termination on day 729 (week 105). Female rat groups began termination on day 683 (week 98) because survival in the saline control group fell to ≤20 animals. In consultation with FDA, all female rat groups were terminated to retain sufficient animals for assessment of the carcinogenetic potential as stated in the protocol. At termination, carcinogenicity animals were anesthetized with carbon dioxide, exsanguinated, and necropsied. These animals were then subjected to a gross necropsy, and tissues and lesions were saved in 10% neutral-buffered formalin, Davidson fix (eyes, optic nerve), or modified Davidson fix (testis) for possible histopathological examination (Table 1). The gross necropsy examination included evaluation of the carcass and musculoskeletal system; all external surfaces and orifices; cranial cavity and external surfaces of the brain; and thoracic, abdominal, and pelvic cavities with their associated organs and tissues. Necropsy examinations were conducted under the supervision of a board-certified veterinary pathologist. All lesions and collected tissues from all carcinogenicity animals as well as carcinogenicity animals that died at unscheduled intervals were embedded in paraffin, sectioned, mounted on glass slides, and stained with hematoxylin and eosin.
Protocol-Specified Tissue Collection at Necropsy for Histological Examination.

aBone marrow smears were collected from the femur at scheduled and unscheduled necropsies (for possible examination). Smears were not collected from animals that were found dead.
bPreserved in Davidson fixative (euthanized animals only).
cCollected from mice only.
dPreserved in modified Davidson fixative (euthanized animals only).
A histopathologic examination was conducted on the tissues listed in Table 1 and any additional lesions from all carcinogenicity animals, including animals that died or were euthanized at an unscheduled interval. A pathology peer review was conducted by a pathologist at Charles River Laboratories, Preclinical Services. A second pathology peer review was conducted by a pathologist external to the laboratory conducting the studies, from Experimental Pathology Laboratories, Inc (Seattle, Washington).
Toxicokinetics
Mouse study
Toxicokinetic information was obtained from additional mice receiving oral (gavage) administration of 15, 30 or 60, mg/kg of roxadustat (24 animals/sex/group for 0 mg/kg and 72 animals/sex/group for the remaining groups) TIW. Blood samples were collected in sodium heparin tubes predose and at 0.5, 1, 4, 12, and 24 hours after dose administration on day 1 and at 6, 12, and 18 months from n = 2 to 3 animals/sex/group per time point.
Rat study
Toxicokinetic information was obtained from additional rats receiving oral (gavage) administration of 0, 2.5, 5, or 10 mg/kg of roxadustat (6 animals/sex/group for 0 mg/kg and 24 animals/sex/group for the remaining groups) TIW. Blood samples were collected for TK analysis predose and at 0.5, 1, 2, 4, 8, 12, and 24 hours after dose administration on day 1 and at 6, 12, 18, and 24 months from up to n = 3/sex/group per time point.
Plasma roxadustat levels
After blood sample collection, all samples were placed in a Kryorack until centrifugation. Plasma was harvested and stored frozen (approximately 70°C ± 10°C) until analysis. A sparse sampling collection method was used with no more than a single sample collection per animal scheduled per collection interval.
Plasma concentrations of roxadustat were determined using a validated method by liquid chromatography–mass spectrometry at Charles River Laboratories, Preclinical Services (Montreal, Senneville, Québec, Canada).
Statistical Analysis
Statistical analyses were performed for body weights, body weight changes, and food consumption of carcinogenicity animals and for hematology and clinical chemistry of TK animals, using a protocol-specified statistical decision tree and P < 0.05 unless otherwise specified.
Survival rates of carcinogenicity animals were described (or summarized) by Kaplan-Meier estimates of group survival rates by sex. A log-rank test for survival was used to compare (1) each treated group with the control group and (2) trend test with the control utilizing ordinal coefficients. All tests were 2 sided. If an animal’s death was classified as accidental or euthanasia, the survival time was considered a censored value for the purpose of the Kaplan-Meier estimates and survival rate analyses.
Tumor incidence data for carcinogenicity animals were evaluated in accordance with the FDA draft Guidance for Industry: Statistical Aspects of the Design, Analysis, and Interpretation of Chronic Rodent Carcinogenicity Studies of Pharmaceuticals. 20 The incidence of tumors was analyzed by Peto’s mortality-prevalence method, 21 without continuity correction, incorporating the context (incidental, fatal, or mortality independent) in which tumors were observed.
Results
Mouse Carcinogenicity Study
Survival, clinical signs, body weight, and food consumption
There was no roxadustat-related effect on mortality (Figures 1 and 2). The incidence of unscheduled deaths determined at study end was slightly greater in high-dose males than in vehicle controls, but there was no dose relationship between low- and mid-dose males. Furthermore, the incidence of unscheduled deaths in mid-dose males was considerably less than for all other groups (Table 2). There were no treatment-related clinical signs or effects on mean body weight (Figures 3 and 4), body weight gain, or food consumption for either sex (data not shown).

Survival curves of male CD-1 mice over 2 years of dosing with roxadustat. All dose groups were terminated as scheduled following 104 weeks of dose administration. = 0 mg/kg (vehicle control);  = 15 mg/kg;
= 15 mg/kg;  = 30 mg/kg;
= 30 mg/kg;  = 60 mg/kg.
= 60 mg/kg.
Survival curves of female CD-1 mice over 2 years of dosing with roxadustat. All dose groups were terminated as scheduled following 104 weeks of dose administration. = 0 mg/kg (vehicle control); = 15 mg/kg; = 30 mg/kg; = 60 mg/kg.
Scheduled Euthanasia and Percentage Survival of CD-1 Mice.a
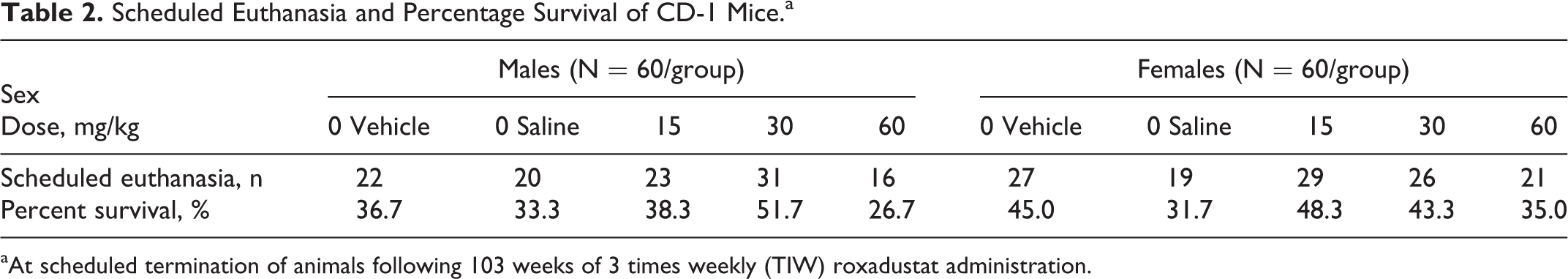
aAt scheduled termination of animals following 103 weeks of 3 times weekly (TIW) roxadustat administration.
Body weight curves of male CD-1 mice over 2 years of dosing with roxadustat. All dose groups were terminated as scheduled following 104 weeks of dose administration. = 0 mg/kg (vehicle control); = 15 mg/kg; = 30 mg/kg; = 60 mg/kg.
Body weight curves of female CD-1 mice over 2 years of dosing with roxadustat. All dose groups were terminated as scheduled following 104 weeks of dose administration. = 0 mg/kg (vehicle control); = 15 mg/kg; = 30 mg/kg; = 60 mg/kg.
Hematologic effects
The mean Hct at 18 months was slightly higher in the 30 and 60 mg/kg TIW dose groups (44.7% and 44.6%, respectively, in males [Figure 5] and 46.9% and 46.1%, respectively, in females) compared to vehicle controls (41.2% in males and 40.7% in females). These increases in Hct did not reach statistical significance, likely due to the low statistical power in the study (n = 2, 7, 11, and 10 for males and N = 3, 7, 8, and 9 for females in groups 1 to 4, respectively), but was consistent with the expected pharmacology (data not shown).

Hematocrit (Hct) data from male and female CD-1 mice at 18 months of dosing with roxadustat. Hematocrit was slightly higher at 30 and 60 mg/kg compared to controls, although the differences were not significant. = 0 mg/kg (vehicle);  = – 15 mg/kg;
= – 15 mg/kg;  = – 30 mg/kg;
= – 30 mg/kg;  = 60 mg/kg. Male: vehicle n = 2; 15 mg/kg n = 7; 30 mg/kg n = 11; 60 mg/kg n = 10; female: vehicle n = 3; 15 mg/kg n = 7; 30 mg/kg n = 8; 60 mg/kg n = 9.
= 60 mg/kg. Male: vehicle n = 2; 15 mg/kg n = 7; 30 mg/kg n = 11; 60 mg/kg n = 10; female: vehicle n = 3; 15 mg/kg n = 7; 30 mg/kg n = 8; 60 mg/kg n = 9.
Histopathology
Neoplastic lesions
There were no roxadustat-related carcinogenic findings in animals that died prior to scheduled euthanasia or in animals that completed the entire 2 years of dosing. Cause of death was determined for animals that died or were euthanized for humane reasons prior to scheduled termination; neoplastic lesions as cause of death occurred in similar numbers for different dose groups or were within historical control incidence, and there were no roxadustat-attributable deaths among these animals (Table 3). Primary neoplasm occurrence was compared between all vehicle controls and roxadustat dose group animals (both unscheduled and final terminations), and the total number of primary benign or malignant tumors and the total number of animals with tumors were similar among groups. Evaluation of all tumors within each organ revealed some differences between treated and control groups in the incidences of the following tumors: bronchioloalveolar adenoma/carcinoma, fibrosarcoma of the (sub)cutis, malignant lymphoma of hemolymphoreticular tissue, mammary adenocarcinoma, pituitary adenoma, hemangiosarcoma in various organs, and ovarian hemangioma (Table 4). However, for all of these tumor types, the differences were not considered roxadustat-related, because these were not statistically significant and the incidences were within the historical range in CD-1 mice (Historical Control Data, Charles River Laboratories 1997-2014) and/or showed a lack of dose proportionality.
Tumor-Related Causes of Unscheduled Deaths in CD-1 Mice.
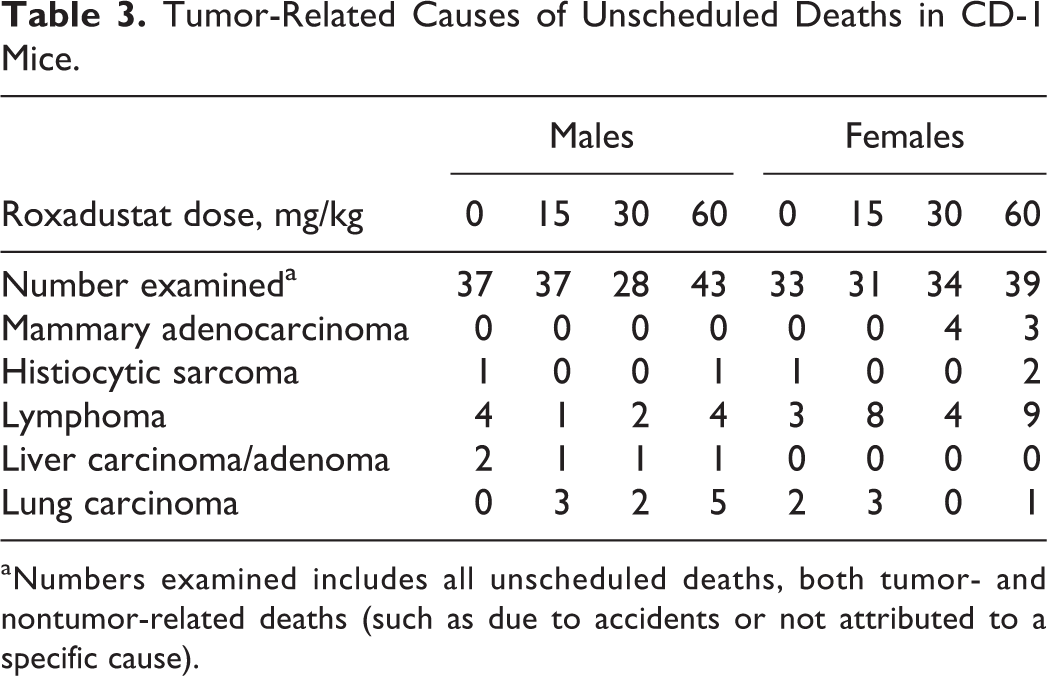
aNumbers examined includes all unscheduled deaths, both tumor- and nontumor-related deaths (such as due to accidents or not attributed to a specific cause).
Numbers of CD-1 Mice With Selected Neoplastic Tumors.a
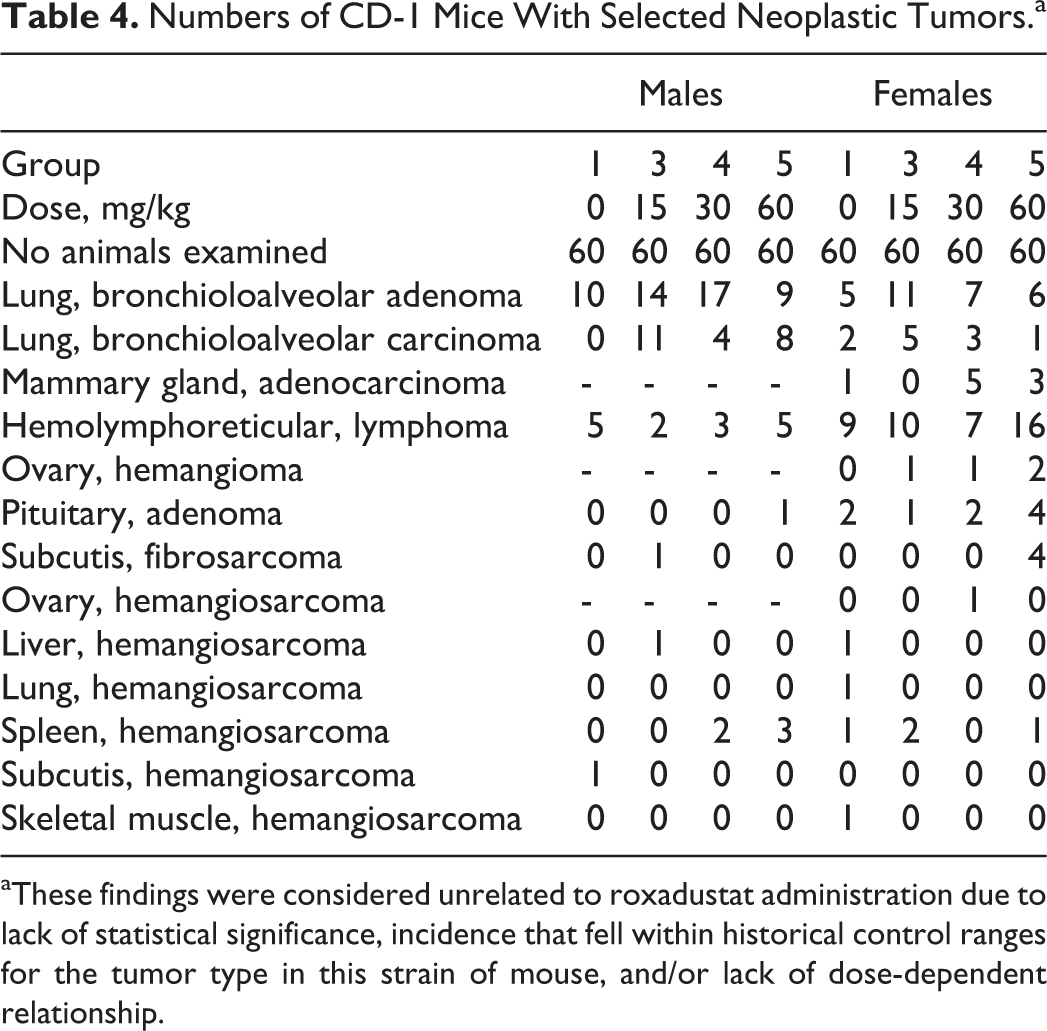
aThese findings were considered unrelated to roxadustat administration due to lack of statistical significance, incidence that fell within historical control ranges for the tumor type in this strain of mouse, and/or lack of dose-dependent relationship.
Nonneoplastic lesions
Extramedullary hematopoiesis in spleen was observed at 30 and 60 mg/kg TIW and is indicative of pharmacologic activity. Most of the other nonneoplastic microscopic findings had similar incidences among control and treated groups and/or were present in small numbers (<10% of the animals in that group). Nonneoplastic microscopic findings were considered by the study pathologist to be incidental, of a nature commonly observed in this strain and age of mice, and/or were of similar incidence and/or severity in control and treated animals and, therefore, with the exception of the extramedullary hematopoiesis in the spleen, which showed a trend of dose proportionality, were considered to be unrelated to administration of roxadustat.
Toxicokinetics
The area under the curve, AUC(0-t), and maximum observed concentrations (C max) are presented in Table 5. Due to animal deaths over time, the number of animals sampled per time point was often less than 3 leading to a decreased accuracy and increased variability of TK parameters derived after 18 months of dosing. Peak concentrations were observed at 0.5 hours postdose on day 1 and months 6 and 12. Peak concentrations ranged from 0.5 to 4 hours postdose on month 18. There were no notable or consistent sex differences for all sampling occasions and for all dose levels. The sex combined half-life (T 1/2) per dose level ranged from 2.13 to 5.51 hours and was not time or dose dependent (data not shown). The AUC(0-t) increased with dose in an approximately proportional manner on day 1 and months 6, 12, and 18. The AUC(0-t) for 15 and 30 mg/kg in males TIW on months 6, 12, and 18 was similar to the exposure on day 1, but the AUC(0-t) for the 30 and 60 mg/kg in females TIW decreased slightly compared to day 1. Oral gavage administration of roxadustat TIW to mice at the highest dose of 60 mg/kg correlated with a C max of 71 to 95 μg/mL and an AUC(0-t) of 5,434 to 633 μg·h/mL at 18 months in both sexes (combined).
Selected Toxicokinetic Parameters of Roxadustat in CD-1 Mice.a
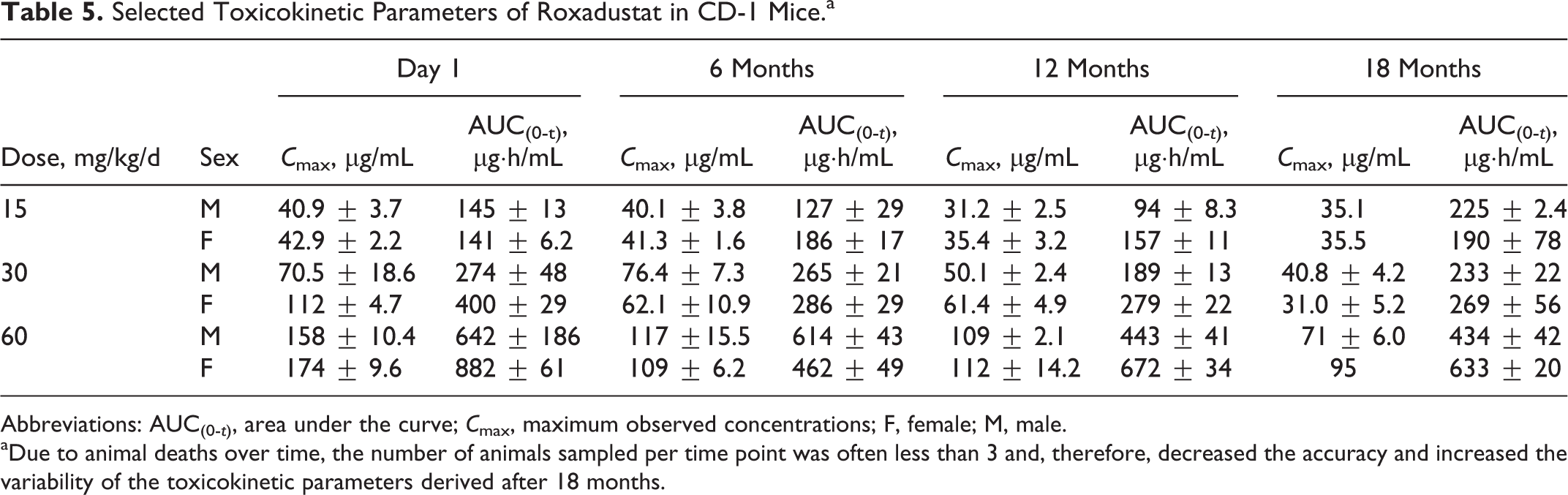
Abbreviations: AUC(0-t), area under the curve; C max, maximum observed concentrations; F, female; M, male.
aDue to animal deaths over time, the number of animals sampled per time point was often less than 3 and, therefore, decreased the accuracy and increased the variability of the toxicokinetic parameters derived after 18 months.
The exposures observed at 60 mg/kg based on steady-state average AUC/C max values are 5.7- to 4.6-fold (AUCss) and 9.1- to 6.8-fold (C max) greater than measured in a phase 1 clinical study in healthy participants using the anticipated clinical doses of 2 and 3 mg/kg and the same TIW dosing regimen.
Rat Carcinogenicity Study
Survival, clinical signs, body weight, and food consumption
There were no statistically significant differences in survival in males or females as compared to vehicle control (Table 6; Figures 6 and 7), and there were no clinical signs that were considered to be roxadustat-related. Female rat groups began termination on day 683 (week 98) because survival in the saline control group fell to ≤20 animals. In consultation with FDA, all female groups were terminated to retain sufficient animals for assessment of the carcinogenic potential as stated in the protocol. Statistical increases and decreases in mean body weight gains were noted throughout the study relative to the vehicle control, but rarely resulted in any statistical differences in absolute mean body weights during the study. Overall, mean body weights were numerically similar and within expected biological variation (Figures 8 and 9). There were statistically significant increases in mean food consumption for all treated males generally through week 64 relative to vehicle-treated males (data not shown). However, these slight increases in mean food consumption did not have impact on the overall body weights. Mean food consumption in females was generally more variable but within biological expectations. Occasionally, there were statistically significant increases in food consumption in roxadustat-treated groups relative to vehicle controls, but they were generally not dose dependent and did not have impact on overall body weights (data not shown).
Percentage Survival of Sprague Dawley Rats.a
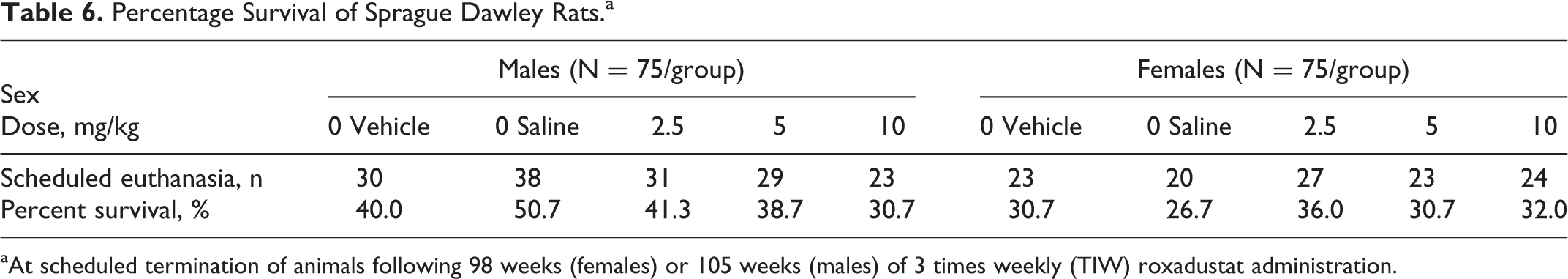
aAt scheduled termination of animals following 98 weeks (females) or 105 weeks (males) of 3 times weekly (TIW) roxadustat administration.
Survival curves of male Sprague Dawley rats over 2 years of dosing with roxadustat. All dose groups were terminated as scheduled following 104 weeks of dose administration. = 0 mg/kg (vehicle control); = 2.5 mg/kg; = 5 mg/kg; = 10 mg/kg.
Survival curves of female Sprague Dawley rats over 2 years of dosing with roxadustat. All female dose groups began termination on day 683 (week 98). = 0 mg/kg (vehicle control); = 2.5 mg/kg; = 5 mg/kg; = 10 mg/kg.
Body weight curves of male Sprague Dawley rats over 2 years of dosing with roxadustat. All dose groups were terminated as scheduled following 104 weeks of dose administration. = 0 mg/kg (vehicle control); = 2.5 mg/kg; = 5 mg/kg; = 10 mg/kg.
Body weight curves of female Sprague Dawley rats over 2 years of dosing with roxadustat. All female dose groups began termination on day 683 (week 98). = 0 mg/kg (vehicle control); = 2.5 mg/kg; = 5 mg/kg; = 10 mg/kg.
Hematologic effects
There were similar roxadustat-related statistically significant increases in erythrocytes (10.14 ×106/cm2 compared to concurrent vehicle control value of 8.79 × 106/cm2), Hb (19.1 g/dL compared to concurrent vehicle control value of 14.9 g/dL), and Hct (58.3% compared to concurrent vehicle control value of 45.4%) observed in 10 mg/kg TIW males relative to vehicle at 12 months on study (Hct data shown; Figure 10). While still increased, these parameters were no longer statistically different at the end of the study. In addition, red cell distribution width % (measured only at termination) was increased in 5 and 10 mg/kg males (16.9% and 18.1%, respectively) relative to vehicle control (14.5%) at 24 months on study. Similarly, changes in erythrocytes (8.26 × 106/cmm), Hb (16.3 g/dL), and Hct (49.2%) were observed at 12 months in 10 mg/kg TIW females compared to vehicle controls (7.72 × 106/cmm, 15.2 g/dL, and 45.5%, respectively) but were not statistically significant (Hct data shown; Figure 10).
Hematocrit (Hct) data in Sprague Dawley rats throughout 2 years of dosing with roxadustat. *P < 0.05; = 0 mg/kg (vehicle); = 2.5 mg/kg; = 5 mg/kg; = 10 mg/kg. Baseline: males; vehicle n = 6; 2.5 mg/kg n = 24; 5 mg/kg n = 24; 10 mg/kg n = 23; females: vehicle n = 6; 2.5 mg/kg n = 22; 5 mg/kg n = 24; 10 mg/kg n = 22; 12 months: males; vehicle n = 5; 2.5 mg/kg n = 23; 5 mg/kg n = 23; 10 mg/kg n = 24; females: vehicle n = 4; 2.5 mg/kg n = 24; 5 mg/kg n = 24; 10 mg/kg n = 23; 24 (males)/22 (females) months: males; vehicle n = 3; 2.5 mg/kg n = 9; 5 mg/kg n = 10; 10 mg/kg n = 10; females: vehicle n = 1; 2.5 mg/kg n = 8; 5 mg/kg n = 11; 10 mg/kg n = 7.
Histopathology
Neoplastic lesions
There were no roxadustat-related carcinogenic findings in either animals that died prior to scheduled euthanasia or animals that completed the entire dosing period. There were no increases in benign tumors, malignant tumors, or tumor-bearing animals that were considered treatment-related based on a combination of statistical significance, dose dependency, and/or normal incidence in this species. The cause of death was determined for animals that died or were euthanized for humane reasons prior to scheduled termination; neoplastic lesions as the cause of death occurred in similar numbers for different dose groups and there were no deaths attributed to roxadustat among these animals (Table 7). Primary neoplasm occurrence was also compared between all vehicle controls and roxadustat dose group animals (both unscheduled and final terminations). There were no increases in benign tumors, malignant tumors, or number of tumor-bearing animals considered treatment-related. There were 2 tumor observations that warrant discussion due to higher incidence in a dose group compared with vehicle control (Table 8). Hemangiosarcoma was present in the spleens of 2 high-dose male rats but was not noted in any other male or female rats. The incidence (2.7%) was outside the historical incidence range (0.0%-1.7%) of similar tumors in male Sprague Dawley rats at the testing facility as well as the historical incidence range (0%-2.0%) of other male Crl: CD (SD) rats (Historical Control Data, Charles River Laboratories, 2013). It is unlikely that this neoplasm was related to roxadustat, as the difference was not statistically significant. The increased incidence of mammary gland adenomas at the mid-dose of 5 mg/kg TIW (23 rats with adenomas) was statistically significantly increased when compared with the female vehicle control group (8 rats with adenomas). However, this finding was not considered to be treatment-related as there was no dose–response relationship.
Tumor-Related Causes of Unscheduled Deaths in Sprague Dawley Rats.
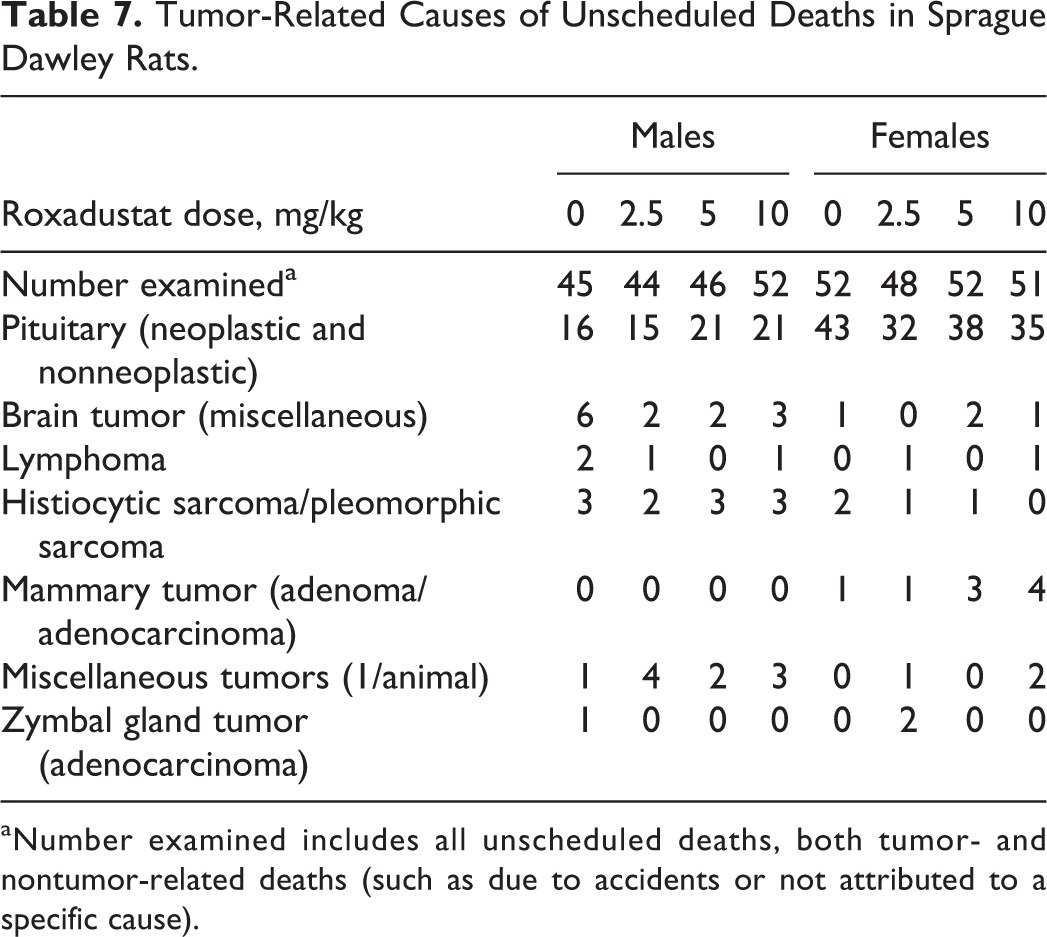
aNumber examined includes all unscheduled deaths, both tumor- and nontumor-related deaths (such as due to accidents or not attributed to a specific cause).
Numbers of Sprague Dawley Rats With Selected Neoplastic Tumors.a
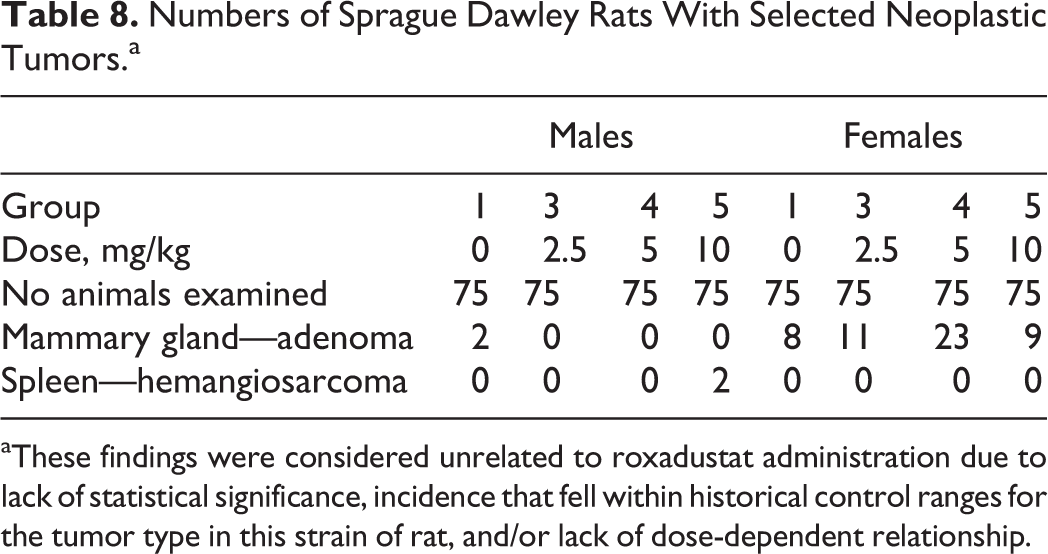
aThese findings were considered unrelated to roxadustat administration due to lack of statistical significance, incidence that fell within historical control ranges for the tumor type in this strain of rat, and/or lack of dose-dependent relationship.
Nonneoplastic lesions
Macroscopic and microscopic findings were diverse in nature, primarily of the kind generally found in normal populations of rats of this strain and age, and distributed randomly without relation to dose level. Exceptions included treatment-related nonneoplastic microscopic findings of bone marrow hypercellularity in all treated male and female groups and atrial/aortic thromboses at 10 mg/kg TIW in males, consistent with exaggerated pharmacology of roxadustat.
The cause of atrial/aortic thromboses in these animals is likely due to roxadustat increased red cell mass, leading to increased blood viscosity, decreased tissue perfusion, and thromboembolism secondary to polycythemia.
Toxicokinetics
The AUC(0-t) and C max concentrations for roxadustat are presented in Table 9. Due to animal deaths over time, the number of animals sampled per time point was often less than 3 leading to decreased accuracy and increased variability of TK parameters derived after 18 and 24 months of dosing. Peak concentrations were generally observed at 1 or 2 hours postdose on day 1 and at 0.5 or 1 hour postdose on months 6, 12, 18, and 24 (males)/22 (females). There were no notable sex differences in all sampling occasions and in all dose levels. The sex combined T 1/2 per dose level ranged from 3.4 to 5.6 hours and was not dose dependent. The AUC(0-t) increased with dose in an approximately proportional manner on day 1 and months 6, 12, 18, and 24. The exposures following repeated dosing at various doses were generally unchanged from 6 to 24 months and were approximately double the exposure values on day 1. Oral gavage administration of roxadustat TIW to rats for up to 97 weeks (females) and 104 weeks (males) at the highest dose of 10 mg/kg correlated with a C max of 56.9 μg/mL and an AUC(0-t) of 316 μg·h/mL at the end of study in both sexes combined.
Selected Toxicokinetic Parameters of Roxadustat in Sprague Dawley Rats.
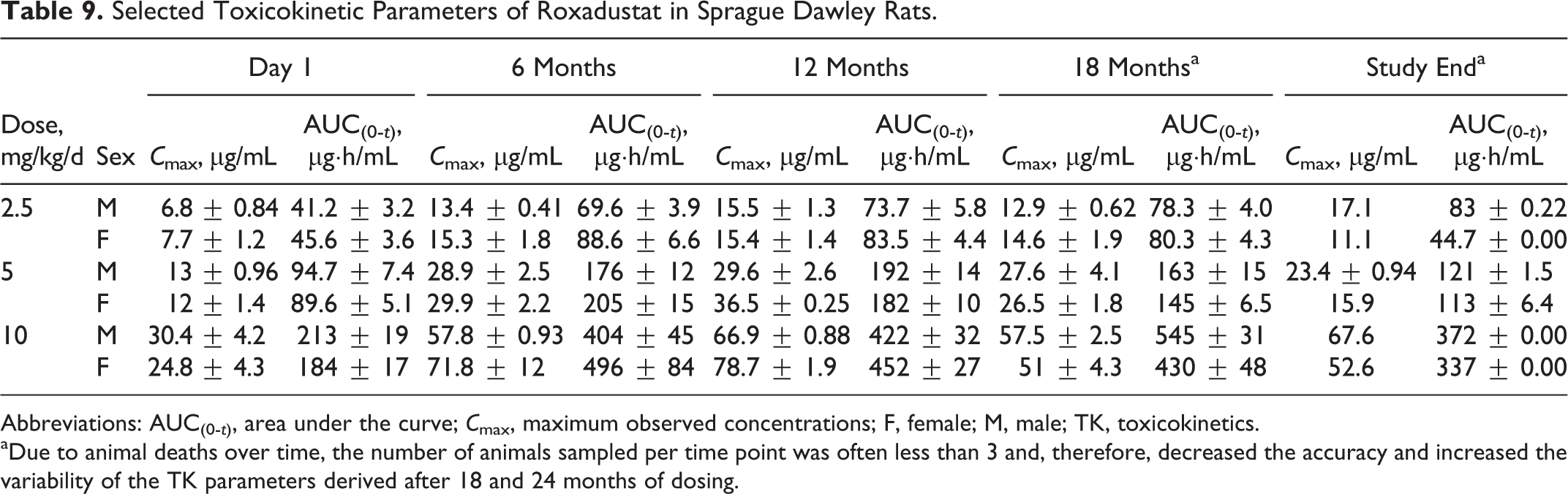
Abbreviations: AUC(0-t), area under the curve; C max, maximum observed concentrations; F, female; M, male; TK, toxicokinetics.
aDue to animal deaths over time, the number of animals sampled per time point was often less than 3 and, therefore, decreased the accuracy and increased the variability of the TK parameters derived after 18 and 24 months of dosing.
The exposures observed at 10 mg/kg, based on steady-state average AUC/C max values, are 4.5- to 3.6-fold (AUCss) and 5.4- to 4.0-fold (C max) greater than measured in a phase I clinical study in healthy participants using the anticipated clinical doses of 2 and 3 mg/kg and the same TIW dosing regimen.
Discussion
The carcinogenic potential of roxadustat was evaluated in standard mouse and rat 2-year bioassays as part of the nonclinical safety program to support clinical development. Under the conditions of these studies, there was no significant effect of roxadustat on survival or evidence of carcinogenic activity of roxadustat in male or female CD-1 mice or Sprague Dawley rats. Roxadustat was dosed TIW by oral gavage for 2 years in mice and rats at up to 60 and 10 mg/kg, respectively. The range of doses in these rodent carcinogenicity bioassays was chosen so as to not significantly reduce the life span or body weights of the treated animals unless in the event of a carcinogenic response. Chronic treatment with these doses of roxadustat had no impact on mortality or neoplastic findings in either species. The highest doses in the 2 studies, chosen based on results of 13-week and 6-month studies in mice and rats, were not expected to affect mortality but to induce pharmacologic effects based on hematologic parameters and thus enable evaluation of the carcinogenic potential of intermittent dosing with roxadustat.
No effects were noted on clinical observations, body weights, or food consumption in either species. Compared to controls, higher levels of red blood cells, Hb, and Hct were observed for the high dose in both species tested, confirming that the doses chosen were in the pharmacologically active range.
No treatment-related nonneoplastic findings were observed in mice, whereas in rats, nonneoplastic microscopic findings were limited to atrial/aortic thromboses at 10 mg/kg TIW males and bone marrow hypercellularity in all treated male and female groups, consistent with effects observed in chronic toxicity studies and an expected consequence of excessive erythropoiesis due to the exaggerated pharmacological activity of roxadustat.
The roxadustat exposures reached in the mouse and rat studies are greater than those anticipated in patients with CKD in phase 2 and 3 clinical studies. The roxadustat exposures reached at the high doses are 5.7- and 4.6-fold greater and 4.5- and 3.6-fold greater in the mouse and rat, respectively, than at the anticipated clinical doses of 2 and 3 mg/kg using the same TIW dosing regimen. The actual exposure comparisons presented are based on clinical pharmacokinetic data generated from phase 1 healthy participants since a more intensive sampling of blood collection time points was conducted in phase 1 and thus a better human to animal comparison could be made.
However, these safety margins should be good estimates since the roxadustat exposure in patients with CKD appears to be comparable to that in healthy participants based on available pharmacokinetic data from patients in phase 2 studies.
Given that hypoxia is a well-known component of the tumor microenvironment, it is not surprising that HIF-α overexpression is observed in a broad range of human cancers. 22,23 However, the fact that HIF- α overexpression has been shown to correlate with poor prognosis in many instances 24,25 has led to the oversimplified view that HIF actively drives tumor progression via the regulation of key target genes that control cell survival, cell metabolism, cell invasion, and angiogenesis. Moreover, cause and effect is difficult to infer from such clinical observations since any correlation of HIF- α expression with outcome may simply reflect the fact that more aggressive tumors are faster growing, therefore are more hypoxic, and as a result express more HIF. Over the past decade, a substantial amount of research has been carried out in numerous laboratories across the world to further our understanding of the role that HIF may play, positive or negative, in tumor growth and progression. Genetic approaches have been widely used to overexpress or delete HIF-1α and/or HIF-2α in different cell types in vivo to try and dissect the role that they may play in tumor biology. Although some of these studies have reported a pro-tumorigenic role for HIF-1α or HIF-2α, 26 –29 others have reported tumor-suppressive activity of HIF-1α or HIF-2α in certain contexts. 15 –17,29,30 Thus, the overall conclusion from this body of work currently seems to be that the relationship between HIF and tumor biology is complex, context dependent, and divergent, even opposing, with respect to HIF-1α and HIF-2α. 16,25,29 The opposing role of HIF-1α and HIF-2α is also evident in clear cell renal cell carcinoma (ccRCC), the most common form of kidney cancer. Inactivation of the VHL tumor suppressor protein (pVHL) is a common event in ccRCC, and multiple lines of evidence indicate that HIF-2 acts as a renal oncoprotein in pVHL-defective ccRCC. 31 However, evidence also suggests that HIF-1α acts as a renal tumor suppressor in this setting, 31 highlighting the opposing and complicated role that HIF plays in ccRCC.
Given the divergent effects of HIF-1α and HIF-2α overexpression on tumor growth in animal studies and the fact that in such studies HIF-α is often constitutively activated, it is difficult to predict based on these data what would be expected with a HIF-PH inhibitor such as roxadustat that only transiently and intermittently activates both HIF-1α and HIF-2α. Thus, we determined the tumorigenic potential of roxadustat in 2-year rodent carcinogenicity studies described here.
The lack of tumor effects in the carcinogenicity studies is consistent with other nonclinical safety data generated with roxadustat. Roxadustat was determined to be nongenotoxic after assessment of genotoxic potential using a battery of in vitro and in vivo assays such as bacterial reverse mutation, chromosomal aberration in human lymphocytes, and mouse micronucleus (data not shown). No pro-tumorigenic or preneoplastic effects of roxadustat were observed in chronic toxicity studies of 6 and 12 months duration in Sprague Dawley rats and cynomolgus monkeys, respectively. Furthermore, no effects on immune organs nor unexpected hyperplasia were observed and the majority of adverse effects were the result of high doses of roxadustat causing exaggerated pharmacology (excessive erythropoiesis).
Moreover, no tumor-related findings were observed with roxadustat when it was evaluated in a transgenic mouse model of breast cancer. 32 Roxadustat and another HIF-PH inhibitor, FG-4497, were evaluated in the MMTV-Neundl-YD5 (NeuYD) transgenic mouse model of breast cancer, in which mammary tumors develop spontaneously in female mice at ∼16 weeks of age. The NeuYD mice were treated with roxadustat or FG-4497 using an intermittent, erythropoietic dosing regimen beginning at ∼7 weeks of age and continuing through to study end. Treatment with either HIF-PH inhibitor led to a significant elevation in markers of erythropoiesis without any evidence of promotion of tumor initiation, progression, or metastasis. 32 Roxadustat and another HIF-PH inhibitor, FG-2216, have also been evaluated in MDA-MB-435 breast cancer and 786-O renal carcinoma xenograft tumor models. 33 Both HIF-PH inhibitors were shown to stimulate erythropoiesis, while exhibiting no effect on tumor progression. 33
The results of the carcinogenicity studies conducted with roxadustat are also consistent with results from carcinogenicity studies conducted with FG-2216. Two-year intermittent administration of FG-2216 to CD-1 mice and Sprague Dawley rats resulted in no FG-2216-related effects on mortality or carcinogenic potential. 18
In conclusion, under the conditions of these 2-year carcinogenicity studies, there was no evidence of carcinogenic activity of roxadustat in male or female CD-1 mice or Sprague Dawley rats administered oral doses of roxadustat TIW up to 60 and 10 mg/kg, respectively. The results of these studies suggest that the long-term exposure of roxadustat did not cause increased initiation, progression, or metastasis of tumors in CD-1 mice and Sprague Dawley rats. The data from these studies support the long-term intermittent clinical dosing of roxadustat without increased tumor risk.
Footnotes
Acknowledgments
The authors are grateful to Anja Slikkerveer, MD, PhD, Björn Dahl, PhD, and Gail Walkinshaw, PhD, for scientific review of this manuscript; Stefan Hemmerich, MD, for assistance in the preparation of this manuscript; Kimberly L. Bonnette, MS, LATG, and Lisa M. Diehl, BS, both of Charles River Laboratories, Inc, Spencerville, for study direction; and Gerald Long, DVM, PhD, DACVP, and Peter Mann, DVM, DACVP, FIATP, of Experimental Pathology Laboratories Inc, for peer review of the studies.
Author Contributions
James Beck and Carroll Henschel contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. James Chou contributed to analysis and interpretation, drafted the manuscript, and critically revised the manuscript. Al Lin contributed to conception and design and critically revised the manuscript. Ughetta del Balzo contributed to conception and design, contributed to analysis and interpretation, drafted the manuscript, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: James Beck, Carroll Henschel, James Chou, Al Lin, and Ughetta del Balzo are employees of FibroGen and hold stock and/or stock options in FibroGen.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.