
Abstract
Clenbuterol, a β2-selective adrenergic receptor agonist, is illicitly used in weight loss and performance enhancement and animal production. Increasing evidence demonstrates that clenbuterol induces various kinds of arrhythmias and QTc interval prolongation. However, little is known about the underlying mechanism. Most drugs are associated with QTc prolongation through interfering with human ether-a-go-go-related gene (hERG) K+ channels. The present study aims to investigate the effects and underlying mechanisms of clenbuterol on the hERG channel. HEK 293 cells were transfected with wild type and Y652A or F656A mutants of the hERG channel and treated with clenbuterol. The hERG current was recorded using whole-cell patch-clamp technique, and protein level was evaluated by Western blot. We found that clenbuterol decreases the mature form of the hERG protein at the cell membrane in a concentration- and time-dependent manner, without affecting the immature form. Correspondingly, clenbuterol chronic treatment reduced hERG current to a greater extent compared to acute treatment. In the presence of Brefeldin A (BFA), which was used to block hERG channel trafficking to cell membrane, clenbuterol reduced hERG on plasma membrane to a greater extent than BFA alone. In addition, the hERG channel’s drug binding sites mutant Y652A and F656A abolished clenbuterol-mediated hERG reduction and current blockade. In conclusion, clenbuterol reduces hERG channel expression and current by promoting the channel degradation. The effect of clenbuterol on the hERG channel is related to the drug-binding sites, Tyr-652 and Phe-656, located on the S6 domain. This biophysical mechanism may underlie clenbuterol-induced QTc prolongation or arrhythmia.
Introduction
Clenbuterol is a β2-selective adrenergic receptor agonist, which was previously used as a bronchodilator in the treatment of asthma and chronic obstructive pulmonary disease. 1 However, due to cardiovascular toxicity demonstrated by increasing evidence, US Food and Drug Administration has banned the use of clenbuterol as a therapeutic drug. 2 In recent decades, clenbuterol was shown to induce skeletal muscle and cardiac hypertrophy via agonizing β2-adrenoceptor and stimulate lipolysis via the β3-adrenoceptor in adipocytes. 3 Therefore, clenbuterol was used to induce a mild myocardial reverse remodeling and improve cardiac function in end-stage heart failure following left ventricular-assist device support. 4,5 In addition, due to the alleged anabolic and lipolytic properties and protein synthesis stimulating effects, clenbuterol is illicitly used by bodybuilders or athletes and in livestock breeding to increase muscle mass. 6,7 Consequently, outbreaks of clenbuterol intoxication for consuming contaminated meat occurred in some areas of the world. 8,9 Human case reports of clenbuterol intoxication in weight-losing and performance-enhancing bodybuilders or athletes also appear occasionally. 10,11 The main manifestations of clenbuterol intoxication include neurologic, cardiovascular, and gastroenteric damages. 12 The cardiovascular toxic effects, such as various kinds of arrhythmias (tachycardia, supraventricular tachycardia, atrial fibrillation), hypertension, and hypokalemia have been reported in patients after clenbuterol intake. 9 Clenbuterol was recently reported to increase heart rate and prolong QTc interval in rat, 13 which may predispose to ventricular arrhythmias and sudden cardiac death, but the underlying molecular mechanism has not been fully elucidated.
The hallmark mechanism of acquired QTc interval prolongation is the blockade of I Kr by specific drugs. I Kr, the rapidly activating delayed rectifier K+ current channel, mainly composed of the pore-forming subunit encoded by human ether-a-go-go-related gene (hERG), also known as KCNH2. 14 I Kr is the most important component of phase 3 repolarization and plays an important role in normal human cardiac electrical activity. 7,13 Reduction of I Kr, resulting from mutations in hERG or drug blockade, delays ventricular repolarization and consequently induces arrhythmia. The presence of aromatic amino acids, Tyr652 and Phe656, in hERG protein with side chains directed toward the large central cavity of the pore region, provides high-affinity binding sites for many of drugs, and accounts for the unusual susceptibility of I Kr channels to various drugs. 15 We, therefore, hypothesized that QTc interval prolongation induced by clenbuterol is related to the change of hERG channel expression and I Kr. In the present study, electrophysiology and Western blot analysis were used to investigate the effect and potential mechanism of clenbuterol on hERG channel expression and I Kr in HEK 293 cell lines.
Methods
Cell Culture and Drug Treatment
hERG complementary DNA (cDNA) was provided by Dr Gail Robertson (University of Wisconsin-Madison). HEK 293 cells were cultured in minimum essential medium (MEM, Invitrogen (CA, USA)) supplemented with 10% fetal bovine serum (Invitrogen) at 37°C in a humidified 5% CO2 incubator. A stable cell line expressing hERG (hERG-HEK) was created by transfecting hERG cDNA into HEK 293 cells using lipofectamine 2000 according to the manufacturer’s instructions. After transfection, the cells were cultured in 10% FBS-supplemented MEM containing 1 mg/mL G418 for selection of transfected cells. The single colony with characteristic I Kr current and hERG protein expression was selected to establish the hERG-HEK stable cell line. To maintain the stable cell lines, the selected cells were cultured in MEM supplemented with 10% fetal bovine serum and 0.4 mg/mL G418. The hERG mutants, F656A and Y652A, were generated using the site-directed PCR mutagenesis and verified by sequencing. For electrophysiological studies with transiently expressed channels, 1.5 μg hERG and 0.5 μg pEGFP-N3 (Clontech) were cotransfected into HEK 293 cells. Twenty-four to 36 hours after transfection, the cells were cultured under different treatments for various periods. And then the cells were harvested for Western blot, immunocytochemistry, and electrophysiological analysis. For electrophysiological studies, the cells were harvested from the culture dish by trypsinization with 0.05% trypsin (Invitrogen) and stored in standard MEM-based medium at room temperature. The cells were studied within 8 hours of harvest.
Western Blot Analysis
Twenty-four hours after clenbuterol treatment, the cells were scraped in ice-cold PBS (phosphate buffer saline) and were collected by centrifuging at 3,000g for 5 minutes. And then the pellet was added with RIPA (radio immunoprecipitation assay) buffer (containing 1% PMSF (phenylmethanesulfonyl fluoride), 10% protease inhibitor cocktail) and incubated on ice for 30 minutes, centrifuged at 15,000g for 10 minutes. The supernatant was collected and measured with a protein assay. The equal amounts of lysate (15 μg/lane) were subjected to SDS (sodium dodecyl sulfate)-polyacrylamide gel electrophoresis (8% gel) followed by transfer onto PVDF (polyvinylidene fluoride) membranes, and then the membranes were blocked for 1 hour with blocking solution (5% nonfat dry milk powder and 0.1% Tween-20 in TBS (tris-buffered saline)). After that, the membranes were incubated with goat polyclonal anti-HERG (1:500 dilution) primary antibody at 4 C overnight or 1 hour at room temperature, after 3 washings with TBST, the membrane was blotted with donkey anti-goat horseradish peroxidase-conjugated secondary antibody, and signals were visualized with an ECL (electrochemiluminescence) detection kit (Bio-Rad, Hercules, CA, USA). To quantify the Western blot data, the band intensities of proteins of interest in each gel are first normalized to their respective actin intensities; then normalized intensities are compared with the band intensity from control cells and expressed as relative values to their controls.
Whole-Cell Patch-Clamp Recording
Cells were collected from the culture dish by trypsinization and stored in standard MEM at room temperature. Cells were studied within 8 hours of harvest. Whole-cell currents were measured using a MultiClamp 700B patch-clamp amplifier (Axon Instruments, Union City, CA, USA). pCLAMP 9.2 software was used to generate voltage clamp protocols and acquire data. Pipettes were pulled from borosilicate glass with a P-97 micropipette puller (Sutter, Novato, CA) and had a resistance of 2 to 3 MΩ. The extracellular solution was (in mM): 137 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.4 with NaOH). The pipette solution was (in mM): 130 KCl, 1 MgCl2, 5 EGTA, 5 MgATP, and 10 HEPES (pH 7.2 with KOH). All experiments were performed at room temperature (22°C-24°C).
Reagents and Antibodies
Rabbit anti-Kv11.1 (HERG) antibody was purchased from Alomone Labs. Goat anti-HERG (C-20), goat anti-GAPDH, rabbit anti-GAPDH, mouse anti-actin, donkey anti-goat IgG, goat anti-rabbit IgG, and Protein A/G PLUS-Agarose for immunoprecipitation assays were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Clenbuterol hydrochloride (clenbuterol), G418, and Brefeldin A (BFA) were purchased from Sigma Aldrich. Clenbuterol was solubilized in distilled water to prepare the stock solution with a concentration of 30 mmol/L. The stock solution of BFA (20 mM) was prepared by dissolving BFA in dimethyl sulfoxide (DMSO). The drugs’ stock solution was diluted in DMEM medium for cell treatment. The highest concentration of DMSO used in this study was 0.05%.
Statistical Analysis
Data are expressed as the mean ± standard error of the mean (SEM). A 1-way ANOVA (analysis of variance) or 2-tailed student t test was used to determine statistical significance between the control and test groups. A P value of 0.05 or less was considered significant.
Results
Clenbuterol Decreases hERG Channel at Cell Membrane
To assess the effect of clenbuterol on hERG channel expression, hERG-HEK cells were exposed to different concentrations of clenbuterol (0, 3, 30, and 300) for 24 hours or to 300 μM clenbuterol for different times (0, 12, 24, and 48), then the cells were harvested for Western blotting analysis. Both the control group and the clenbuterol treatment group show typical hERG signature of 2 single bands at approximately 135 and 155 kDa. The band at 155 kDa represents mature, fully glycosylated form of hERG protein on the plasma membrane, and the band at 135 kDa represents immature, core-glycosylated form of the hERG protein retaining in endoplasmic reticulum. As shown in Figure 1A, treatment of hERG-HEK cells with 3, 30, and 300 μM clenbuterol for 24 hours selectively decreased the 155 kDa, but not the 135 kDa hERG band in a concentration-dependent manner. A total of 300 μM clenbuterol shows a maximum inhibition of hERG channel protein. Then, 300 μM clenbuterol was used to treat hERG-HEK cells for various periods. Figure 1B shows that clenbuterol-induced reduction of hERG expression became obvious at 24 hours and peaked at 48 hours. Then, 300 μM clenbuterol incubation for 24 hours was used for mechanistic investigations in the present study. Furthermore, our data show that clenbuterol exerts no effect on 135 kDa hERG band level, indicating that the reduced hERG protein at cell membrane may result from accelerated retrograde degradation or slowed anterograde trafficking.
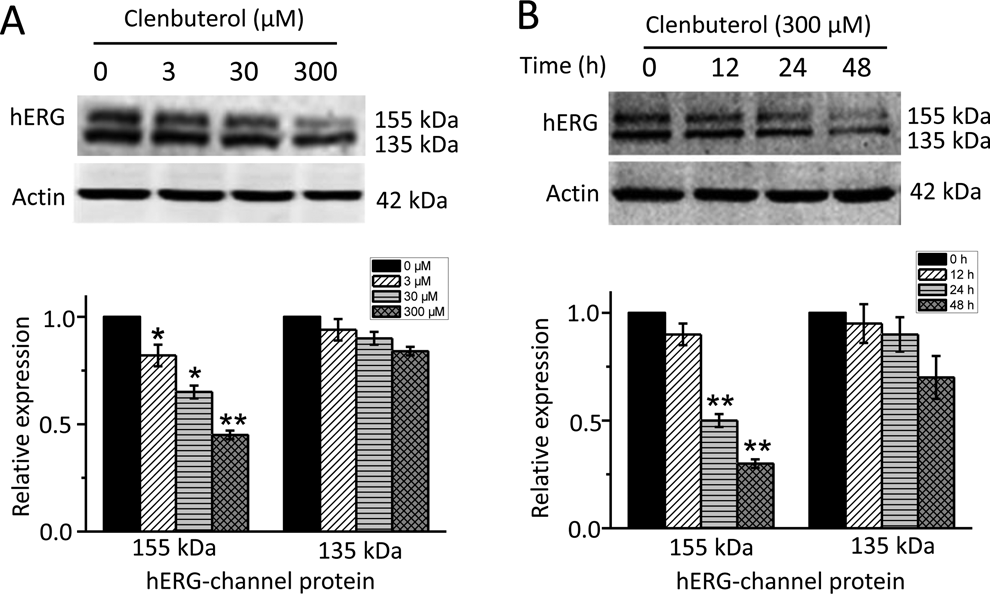
Clenbuterol reduces surface membrane expression of the human ether-a-go-go-related gene (hERG) channel in a concentration- and time-dependent manner. hERG-HEK cells were incubated with clenbuterol at different concentrations (0, 3, 30, 300 μM) for 24 hours as indicated. A, Representative immunoblot and densitometric measurements of fully glycosylated 155 kDa or core-glycosylated 135 kDa hERG proteins normalized to actin. n = 3, *P < 0.05 versus 0 μM, **P < 0.01 versus 0 μM. B, Representative immunoblot and densitometric measurements (normalized to actin) of 155 or 135 kDa hERG proteins in HERG-HEK293 treated without (0 hour) or with 300 μM clenbuterol for 12, 24, and 48 hours as indicated. n = 3, *P < 0.05 versus 0 hours, **P < 0.01 versus 0 hour.
Clenbuterol Treatment Effectively Reduces hERG Current
To examine the effect of clenbuterol on hERG current (I hERG), whole-cell patch clamp was used to record the current in hERG-HEK cells treated with or without clenbuterol (as a control, Ctrl). Cells were depolarized to test voltages between −70 and +70 mV in 10 mV increments for 5 seconds from a −80 mV holding potential, followed by a step to a level of −50 mV for 5 seconds to elicit tail current. The protocol was applied at a frequency of 0.07 Hz. Representative hERG currents were recorded under control conditions and the incubation of clenbuterol (300 μM) for 24 hours. As shown in Figure 2, clenbuterol significantly reduces hERG peak steady-state current amplitude by 45% (Figure 2D, P < 0.05) and peak tail current by 56% (Figure 2F, P < 0.05). The I/V curve of tail currents (Figure 2C) and current measured at the end of depolarizing steps (Figure 2E) shows that clenbuterol reduces hERG current at different test voltages.
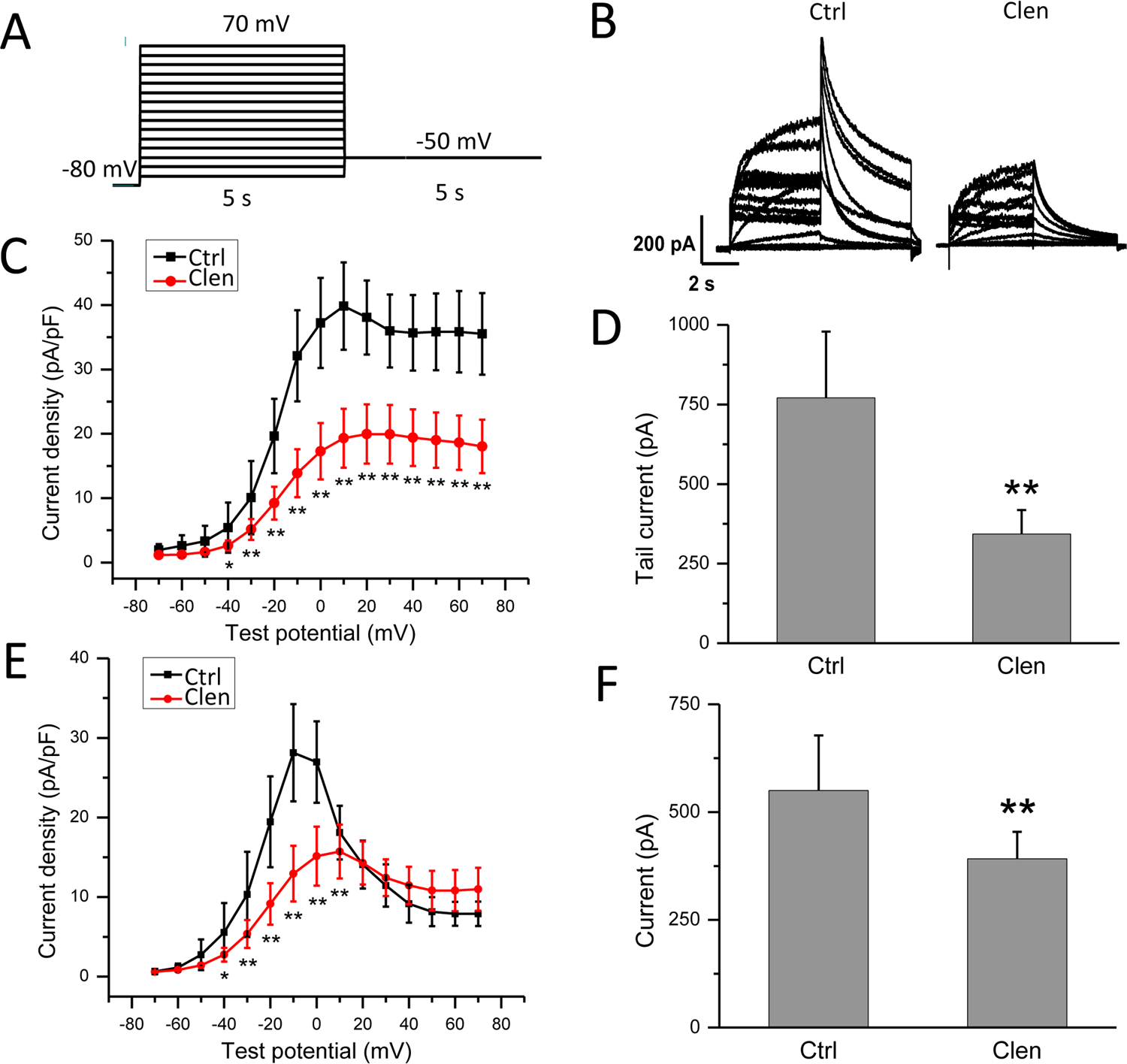
Chronic clenbuterol treatment reduces human ether-a-go-go-related gene (hERG) current in hERG-HEK cells. A, The voltage-clamp protocol used to record whole cell hERG current. Cells were depolarized to test voltages between −70 and +70 mV in 10 mV increments for 5 seconds from a −80 mV holding potential, followed by steps to a level of −50 mV for 5 seconds to elicit tail current. B, Representative hERG current traces in hERG-HEK cells control (Ctrl) and clenbuterol (Clen) exposure. I–V relationships (C) for tail currents and bar graphs (D) of summarized peak tail currents (n = 8, **P < 0.01 vs Ctrl). I–V relationships (E) and bar graphs (F) for current measured at the end of depolarizing steps in control cells and in cells exposed to clenbuterol (n = 8, *P < 0.05 vs Ctrl, **P < 0.01 vs Ctrl).
The Acute Clenbuterol Treatment Reduces hERG Current
The hERG currents were elicited using a protocol the same as Figure 2 and also presented in Figure 3A. After the control measurement had been obtained, hERG-HEK cells were incubated with clenbuterol for 30 minutes in the bath solution. The original recordings of hERG current are presented in Figure 3B. Application of clenbuterol causes a reduction in hERG peak tail current by 18% (current density: Ctrl 55.90 ± 5.28 vs clenbuterol, Clen 45.63 ± 5.86 pA/pF, P < 0.05, Figure 3D) and peak steady-state current amplitude by 19% (current density: Ctrl 36.15 ± 2.72 vs Clen 29.62 ± 3.48 pA/pF, P < 0.05, Figure 3F). The current–voltage relationship (I–V curve) of hERG tail currents and current measured at the end of depolarizing steps by clenbuterol treatment is depicted in Figures 3C and E, respectively. The acute treatment of clenbuterol reduces hERG current slightly and shows direct blocking of wild-type hERG channel.
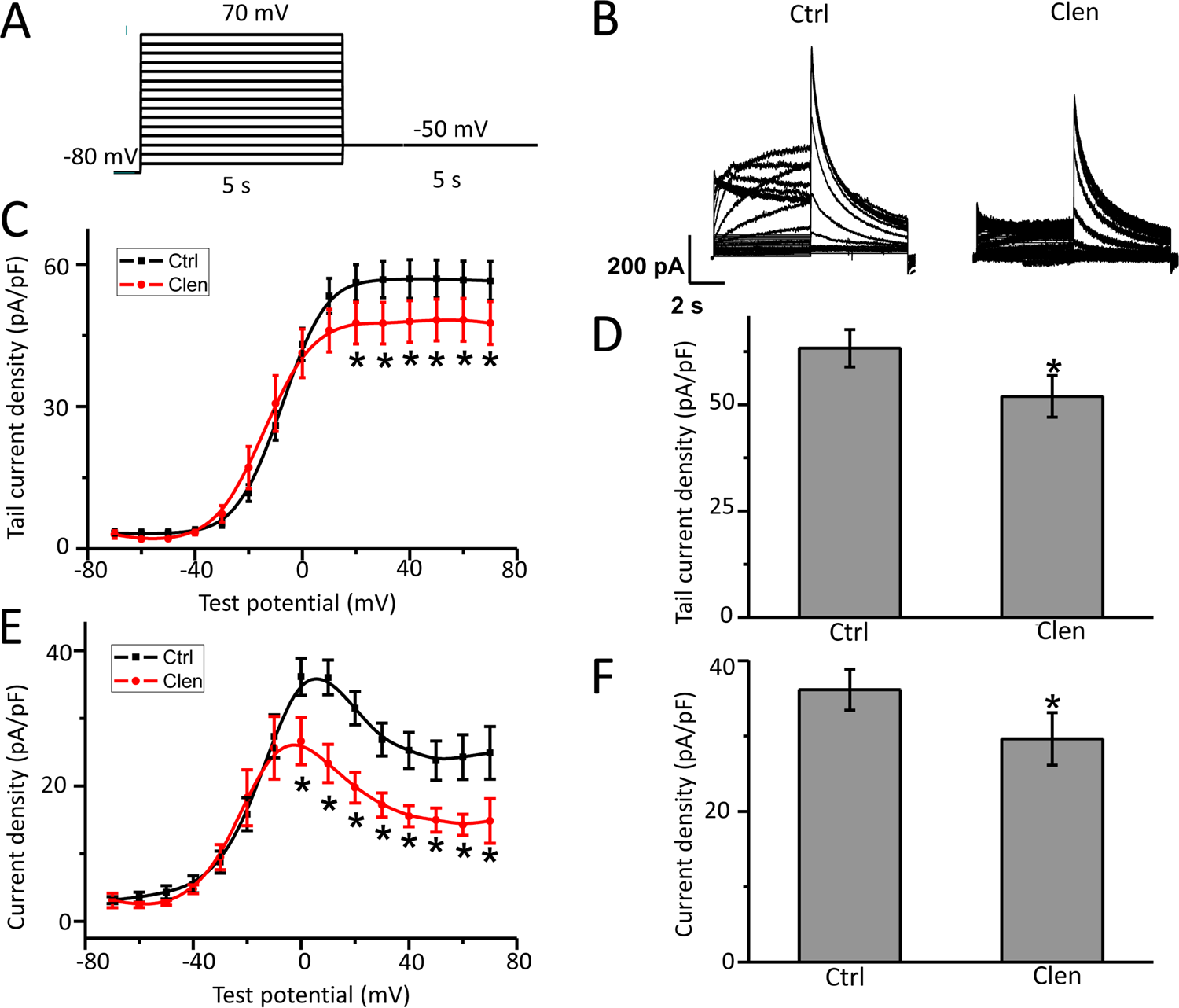
Acute clenbuterol treatment reduces human ether-a-go-go-related gene (hERG) current in hERG-HEK cells. A, The voltage-clamp protocol used to record whole cell hERG current (see also in Figure 2A). B, Representative hERG current traces in hERG-HEK cells control (Ctrl) and clenbuterol (Clen) exposure. I–V relationships (C) for tail currents and bar graphs (D) of summarized peak tail currents (n = 9-12, *P < 0.05 vs Ctrl). I–V relationships (E) and bar graphs (F) for current measured at the end of depolarizing steps in control cells and in cells exposed to clenbuterol (n = 9-12, *P < 0.05 vs Ctrl).
Frequency Dependence of Clenbuterol-Induced hERG Channel Block
The repetitive pulsing at frequencies of 1 and 0.33 Hz was used to test the frequency dependence of hERG channel block by clenbuterol for 60 seconds. The protocol for hERG current recording is presented in Figure 4A, namely depolarizing voltage step was +40 mV (300 milliseconds) and followed by a pulse to −50 mV (300 milliseconds). Having obtained the control current with repetitive pulsing over 60 seconds, the hERG-HEK cells were incubated with clenbuterol for 30 minutes and the protocol was conducted again. Figure 4B shows the representative hERG current for the first and last repetitions recorded at a frequency of 1 Hz. The hERG peak tail current by clenbuterol treatment was normalized to control (relative current) at each pulse (Figure 4C). We find that inhibition of hERG current by clenbuterol is not frequency dependent.
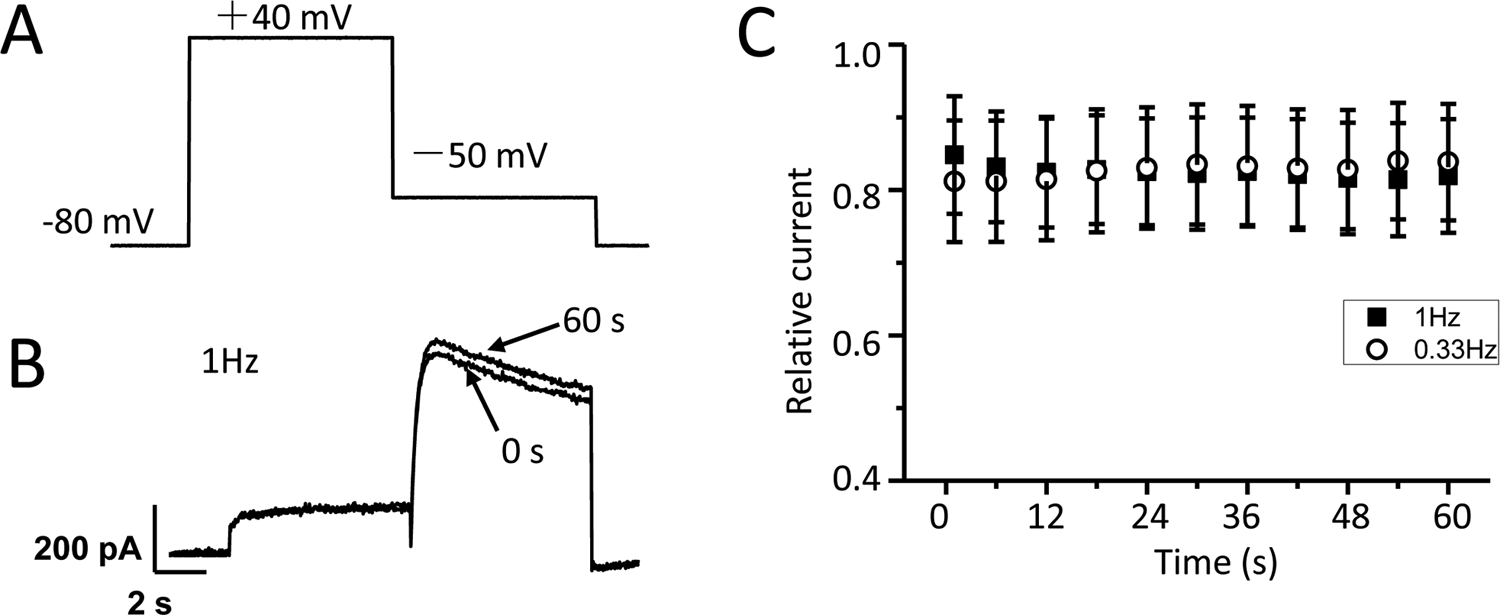
Inhibition of human ether-a-go-go-related gene (hERG) channel by clenbuterol is not frequency dependent. The test pulses were applied at frequencies of 1 and 0.33 Hz under control (Ctrl) conditions and clenbuterol incubation (Clen). A, Test protocol: from a holding potential of −80 mV, test pulses to +40 mV for 300 milliseconds were used to activate hERG currents, then tail currents were elicited by a pulse to −50 mV for 300 milliseconds. B, Representative current traces for the first and last traces at a frequency of 1 Hz. C, Relative peak tail current density are presented as a function of time. For 1 Hz recording (1-second interval), only every sixth and for 0.33 Hz (3-second intervals), only every second measurements are displayed.
Clenbuterol Decreases hERG Channel by Promoting Mature Channel Degradation
Whether clenbuterol reduces the mature hERG band by facilitating hERG degradation is next evaluated. The functional number of hERG channels at cell membrane is balanced between forward trafficking and retrograde internalization/degradation. In the present and previous study, BFA was used to test the degradation rate of hERG and other membrane proteins. 16 –18 Brefeldin A (10 μM), the fungal metabolite that causes disassembly of the Golgi apparatus and inhibits transport of newly synthesized proteins from the endoplasmic reticulum to Golgi, was used to block the transforming of hERG from its immature (135 kDa) form to its mature (155 kDa) form. 17,18 Thus, the level of 155 kDa of hERG protein with BFA incubation represents the degradation rate of the preexisting mature channels. The degradation rate of hERG was evaluated by detecting the 155 kDa band (mature form of hERG at the plasma membrane) expression level at different time points (0, 6, 12, and 24 hours) with (BFA + Clen) or without clenbuterol (BFA) incubation in the presence of BFA in hERG-HEK cells. As shown in Figure 5, 10 μM BFA alone leads to a time-dependent reduction of the 155 kDa hERG band, reflecting the degradation rate of the remaining hERG channels at the cell surface. Clenbuterol treatment significantly reduced the 155 kDa hERG band to a greater extent than BFA alone at 12-hour and 24-hour treatment, respectively. As shown in Figure 5B, following a 12-hour BFA incubation alone, the 155 kDa hERG band intensity decreased by about 40%, whereas it decreased by 60% in BFA plus Clen-treated cells. A 24-hour BFA incubation alone reduced 155 kDa hERG band intensity for 64%, while together with clenbuterol treatment reduced about 93% (n = 3, P < 0.05 vs BFA). These data suggest that clenbuterol reduces hERG channel expression and current by promoting the degradation of mature channel on the cell membrane.
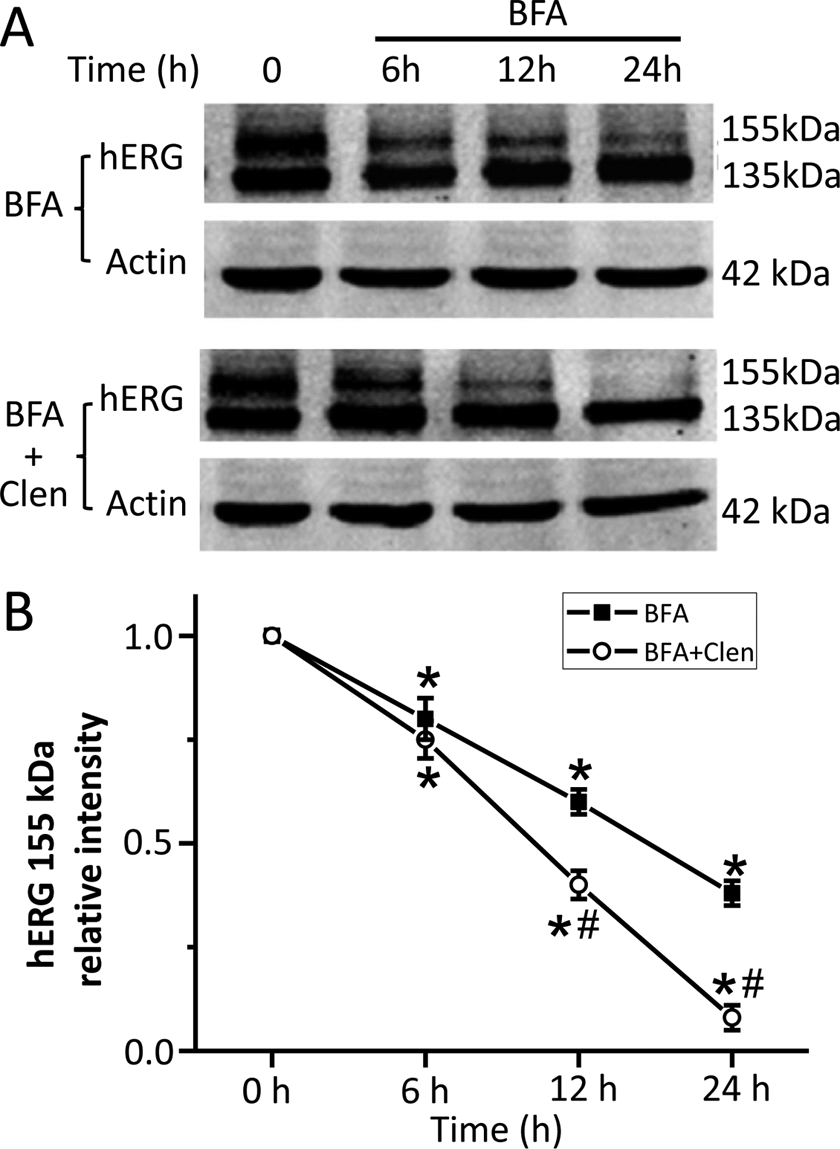
Clenbuterol accelerates the degradation rate of human ether-a-go-go-related gene (hERG) channels at the cell membrane. Post 2 hour BFA (10 μM) pretreatment to block hERG transport from the ER to the Golgi, hERG-HEK cells were incubated together without (BFA) or with clenbuterol (300 μM, BFA + Clen). Cells were harvested at different time points (0, 6, 12, and 24 hours) for Western blot analysis. (A) Representative immunoblot and (B) the intensities of mature hERG bands (155 kDa) at various time points normalized to their initial value (time point 0 hour) and summarized in the line chart, (n = 3, *P < 0.05 vs 0 hour,
Abolish of Clenbuterol Blockage by Y652A and F656A Mutations
The mechanism of clenbuterol on hERG channel reduction was further evaluated. The amino acids of Tyr-652 and Phe-656 on the transmembrane S6 segments are important for binding of most hERG channel blockers. 19 –21 Therefore, Y652A and F656A hERG mutants are commonly used to attenuate I hERG block by a number of drugs. To study whether clenbuterol reduced hERG protein expression and I hERG by binding with Tyr-652 or Phe-656, the Y652A or F656A mutant plasmids were constructed by site-directed mutagenesis and transiently transfected into HEK 293 cells, and then treated with clenbuterol to record I hERG and protein expression. As shown in Figure 6, clenbuterol exerts no significant effect on the mature or immature form of hERG protein expression (Figure 6A and B, n = 4, P > 0.05) and the tail current of Y652A-hERG mutants. Similar results were observed in F656A-hERG mutant plasmid transfected HEK 293 cells, which are shown in Figure 7. These results show that the aromatic residues Tyr-652 or Phe-656 in S6 of hERG channels are critical in clenbuterol-induced hERG reduction.
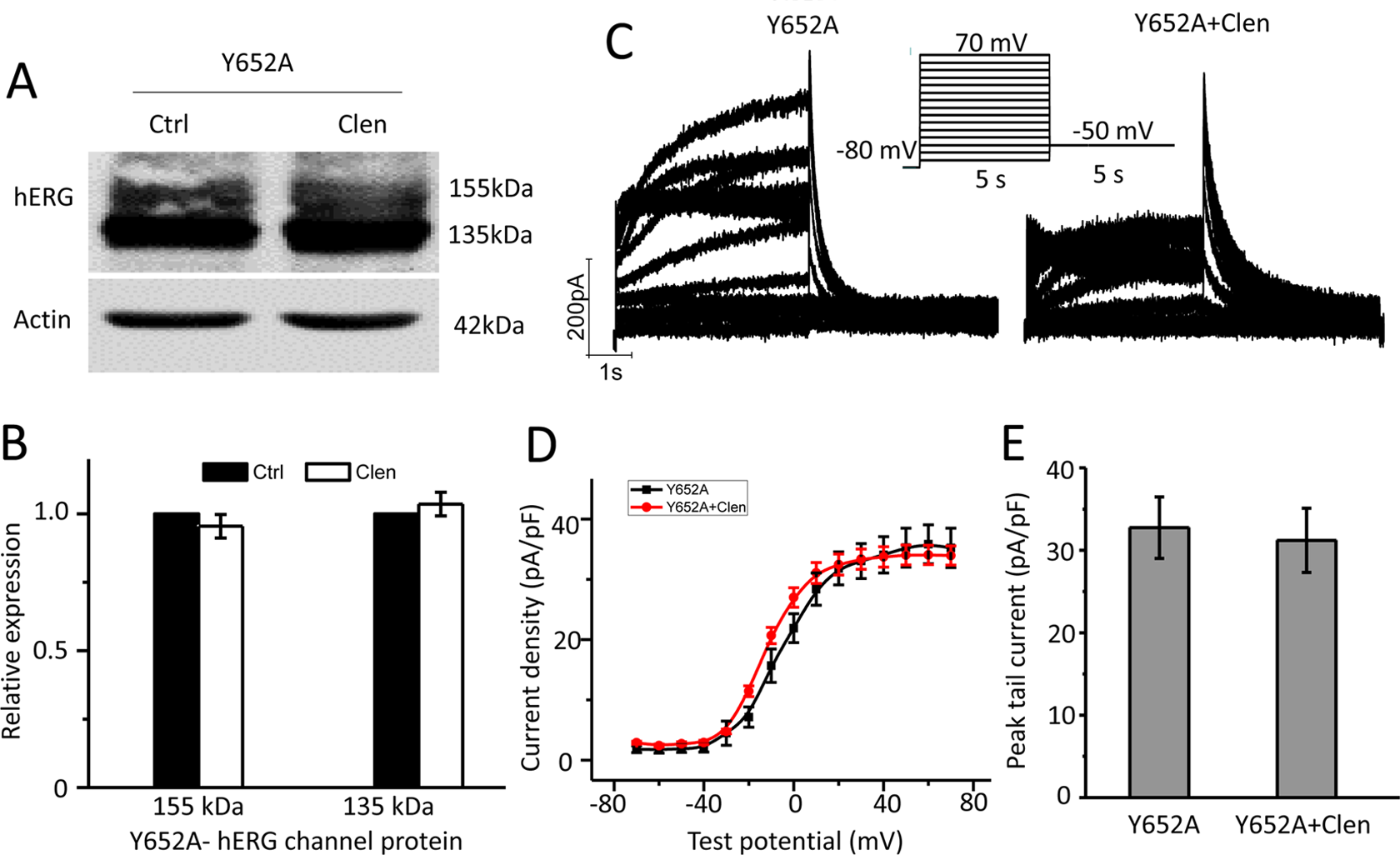
Y652A mutation abolished clenbuterol-mediated wild-type human ether-a-go-go-related gene (WT-hERG) inhibition. Y652A-hERG cDNA was transiently transfected into HEK 293 cells and incubated without (Ctrl) or with clenbuterol (Clen, 300 μM) for 24 hours to measure hERG protein expression or to record hERG current. (A) Representative immunoblot and (B) densitometric measurements (normalized to actin) of mature (155 kDa) or immature (135 kDa) hERG bands (n = 4). (C) Representative current traces for Y652A-hERG channels in the absence (Y652A) and presence of clenbuterol (Y652A + Clen). The protocols used were presented as the inset (see also in Figure 2A). I–V relationships for tail current (D) and summarized peak tail current (E; n = 9-10 cells).
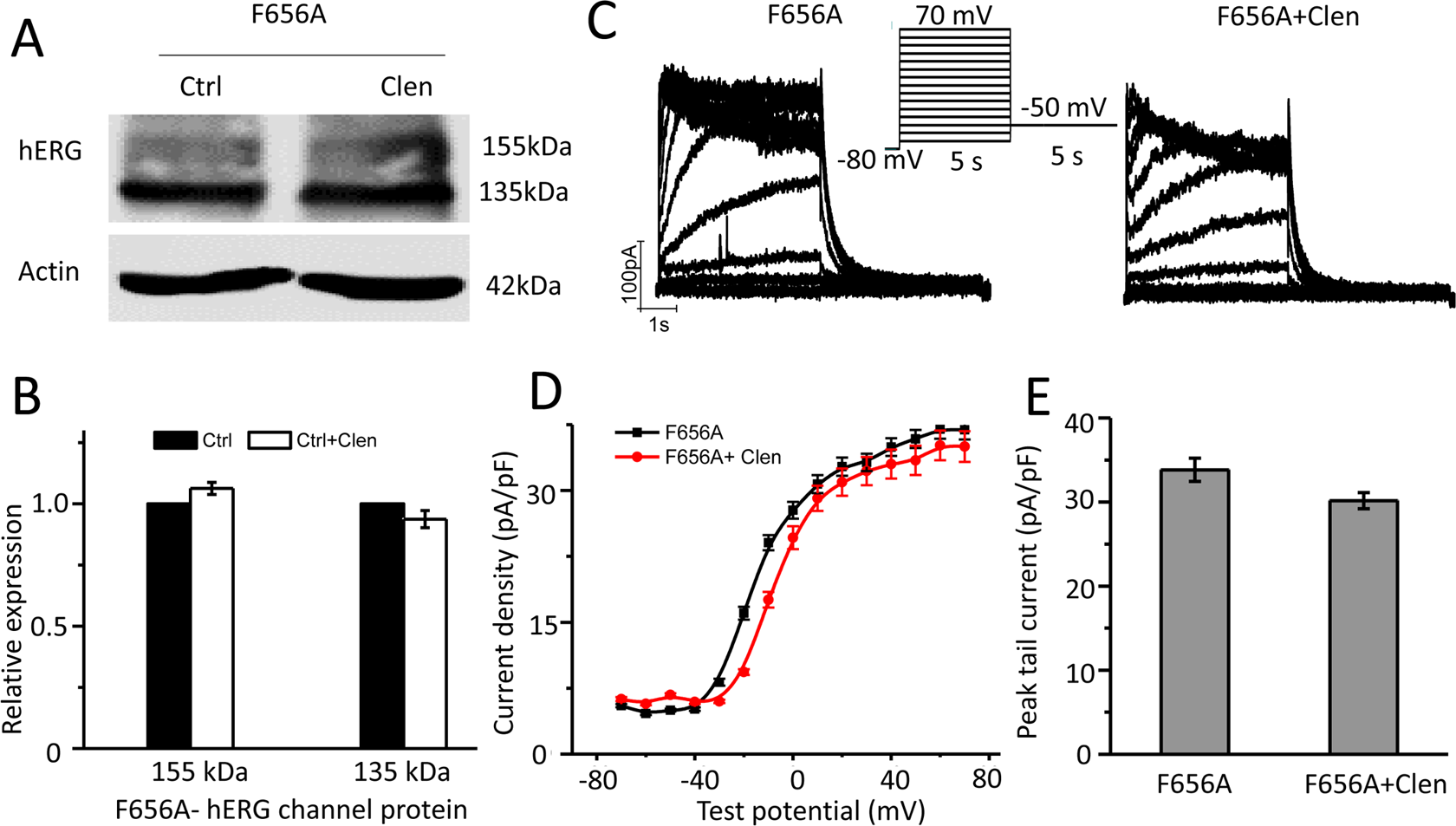
F656A mutation abolished clenbuterol mediated wild-type human ether-a-go-go-related gene (WT-hERG) inhibition. F656A-hERG cDNA was transiently transfected into HEK 293 cells and incubated without (Ctrl) or with clenbuterol (Clen, 300 μM) to measure hERG protein expression or record hERG current. (A) Representative immunoblot and (B) densitometric measurements (normalized to actin) of mature (155 kDa) or immature (135 kDa) hERG bands (n = 4). (C) Representative current traces for F656A-hERG channels in the absence (F656A) and presence of clenbuterol (F656A + Clen). The protocols used were presented as the inset (see also in Figure 2A). I–V relationships for tail current (D) and summarized peak tail current (E; n = 8-10 cells).
Discussion
The present report is the first to study the effects and mechanisms of clenbuterol on the hERG channel. Ke et al 2 demonstrated that acute intravenous administration of clenbuterol causes serious, dose-dependent cardiovascular toxicities and is even life-threatening in rabbit and they found that clenbuterol induced various arrhythmias including atrioventricular block, ventricular capture, and progressive prolongations of P–R intervals and QRS duration. Another study conducted by Paul J. R. Barton group showed that clenbuterol was able to increase the QTc interval in rats. 13 These data indicate that clenbuterol may affect the function of ion channels contributing to action potential in cardiomyocytes. The outward currents, carried predominantly by K+ (making the cell more negative intracellularly), exceeding inward currents, carried by Na+ and Ca2+, contribute to the repolarization process. 22 The loss of function of K+ channel or gain of function of inward current channels by gene mutations or acquired factors (such as drugs, electrolytes, ischemic heart disease) result in QTc interval prolongation. 23,24 I Kr is the most important component of the third phase in repolarization of the action potential in human ventricular myocytes. 14,25 The blockade of I Kr by specific drugs is the hallmark of acquired QTc interval prolongation. Long QTc interval may lead to delay after depolarization and consequently result in various arrhythmias. 26 Various kinds of drugs were demonstrated to induce arrhythmias by interfering with the hERG channel. 27 –29 It is reasonable to assume that the arrhythmia induced by clenbuterol intoxication is related to the change of hERG channel expression and I hERG. In the present study, clenbuterol-reduced hERG channel expression in a dose- and time-dependent manner. In humans, clenbuterol has been used as a bronchodilator by inhalation at doses of 20 to 40 μg daily. In healthy volunteers 8 days after commencing oral dosing of 20 μg clenbuterol twice daily, mean peak serum concentration of clenbuterol was 293 pg/mL (≈1 nM). 8 However, there is no standardized oral dose for bodybuilders. The actual dose is highly variable with a median dose of 0.8 mg (range, 0.08-5,000 mg) according to the literature. In rats administered with clenbuterol, the peak blood concentration of protein-unbound form will rise to 35 μM. 30 In our research, the clenbuterol inhibited the hERG channel protein in a concentration-dependent manner (range, 3-300 μM), which is within the blood concentration range of clenbuterol intoxication.
However, not all cases who received clenbuterol presented with QTc prolongation and arrhythmia, and it may be partially explained by the homeostatic regulation of ion channels. Homeostatic regulation of ion channels refers to different processes that are interplayed to maintain a relatively stable state under constant physiological conditions, providing a degree of plasticity for myocytes and allowing the myocytes to respond appropriately to the changing physiological demands. 31 In most physiological systems, if one component declines in function or abundance, another compensates with similar function by promoting itself to higher activity levels to sustain a relative stable state. 32 For instance, K+ currents were the major components in cardiac repolarization. The loss of one K+ channel function impairs cardiac repolarization, but there is a redundancy of the K+ channel so that when one K+ current is reduced other K+ currents increase to compensate, a phenomenon called “repolarization reserve.” 22 Besides I Kr, the I Ks which is conducted by both KCNQ1 (also known as KvLQT1, encodes the pore-forming α subunit of I Ks) and KCNE1 (also known as mink, encodes the regulatory β subunit of I Ks), are also critical for cardiac repolarization. 33 In our study, we demonstrated that clenbuterol reduced I Kr, however, the effect of clenbuterol on I Ks is unknown. There are reports, which demonstrated that β-adrenergic stimulation increased the L-type calcium channel current (I Ca, L) density in adult cardiomyocytes. 34,35 However, in adult rat received an osmotic minipump containing clenbuterol, the I Ca,L values were not changed. 4 Another study reported that acute clenbuterol treatment (30 μM) reduced Ca2+ transient amplitude and I Ca,L via the inhibitory G protein pathway in rat ventricle cardiomyocytes. 36 This difference may be due to a different way and concentration of clenbuterol treatment. Moreover, other channel currents including I K1 and I to also participate in cardiac action potential repolarization, 15 so further experiments are necessary to confirm the effect of clenbuterol on those currents.
There are 2 mechanisms for drug-induced hERG channel inhibition: (1) directly block channel conduction 37 and (2) indirectly disrupt channel trafficking to the cell surface or accelerate the degradation/internalization rate. 38 –40 In the present study, clenbuterol acute treatment reduces hERG current by about 20%; however, the chronic clenbuterol incubation for 24 hours reduces hERG protein expression and hERG current by 49% and 56%, respectively. In addition, clenbuterol reduces hERG protein in a concentration- and time-dependent manner. These data indicate that besides direct blocking effects, there are other mechanism mediated hERG channel reduction by clenbuterol. Previous research found that a variety of compounds including ketoconazole, fluoxetine, norfluoxetine, and citalopram cause long QT syndrome by directly blocking the hERG channel or reducing the channel protein expression on cell membrane. 27,39,41,42 Some drugs such as pentamidine, geldalamicin, arsenic trioxide, and digoxin interfere with the trafficking of channels from the endoplasmic reticulum to the cell membrane. 43 Nascent hERG channels are synthesized and undergo N-linked glycosylation in ER to an apparent molecular mass of 135 kDa. Upon proper folding and assembly, hERG traffics to the Golgi and is subject to additional glycosylation, increasing the apparent molecular mass to 155 kDa before export to the plasma membrane. Cell surface-resident hERG is then internalized for degradation caused by Nedd4-2-mediated ubiquitination. 44 As a result, the membrane density of the hERG channel is balanced between forward trafficking and backward internalization. The defect of hERG forward trafficking always leads to an increase in the immature protein level in the endoplasmic reticulum and a decrease in the mature form. 28 In this study, we found that clenbuterol reduced the mature form of hERG (the band of 155 kDa), but did not increase the immature protein (band of 135 kDa), indicating that clenbuterol does not block the hERG channel forward trafficking. Recently, probucol was demonstrated to reduce hERG protein and current via enhancing hERG channel degradation. 17,45 In our research, in the presence of BFA, which blocks hERG protein forward trafficking, clenbuterol reduced mature hERG protein on the cell surface to a greater extent than BFA alone. At 24 hours, BFA incubation alone reduced 155 kDa hERG band intensity by 64%, while together with clenbuterol treatment reduced about 93%. These data demonstrate that clenbuterol reduced hERG channel protein by accelerating its degradation of the mature form on the cell surface.
There are 4 known drug-binding sites within the hERG channel, including Thr-623 and Ser-624, locating at the base of the pore helix, and 2 aromatic residues, Tyr-652 and Phe-656 locating on the S6 domain. 15 Among the 4 binding sites, 2 pore helix residues, Thr-623 and Ser-624, are highly conserved in the Kv channel family. However, the other 2 S6 residues, Tyr-652 and Phe-656, are not conserved. Most Kv channels have an Ile and a Val in homologous positions. 17 This finding may help explain the astonishing sensitivity of hERG channels to sheer number and diversity of compounds. 46 Previous research demonstrated that Tyr-652 is required for cisapride and terfanadine blockade, suggesting the cation-π interactions or π-stacking interactions is important for hERG channel blockade. 17 More studies identified that 2 aromatic residues, Tyr-652 and Phe-656, are important for direct binding of most hERG channel blockers. 19 –21,47 However, whether the binding sites mediate drug-induced indirect mechanism is less elucidated. Berberine was reported to cause hERG channel trafficking deficiency which was abolished by the absence of Tyr-652 and Phe-656 on the S6 domain. 48 To determine whether Tyr-652 and Phe-656 were required for clenbuterol-mediated hERG channel inhibition, the effects of clenbuterol on Y652A and F656A mutants’ expression and function were tested. Our results showed that Y652A and F656A hampered clenbuterol-induced inhibition of hERG protein and current after overnight incubation. These data manifested that binding to the 2 residues accounts for clenbuterol-induced hERG channel inhibition. Y652A and F656A abolished clenbuterol-induced hERG protein reduction, we propose that the interaction between clenbuterol and hERG protein might affect the stability of channels on the cell membrane, thus accelerating its degradation.
There is a limitation in the present study that all patch-clamp experiments were performed at room temperature, but not at the physiological temperature. Temperature affects hERG channel function by at least 2 ways. First, the acute altering in temperature affects the channel’s gating properties. Second, the chronic changes in temperature affects channel’s density. Previous studies has demonstrated that hERG current density recorded at near physiological temperature (35°C) compared to room temperature (23°C) was increased and the gating properties including activation, inactivation, recovery from inactivation, and deactivation kinetics were rapid at 35°C. 49,50 The chronic increasing in temperature accelerates hERG degradation through altered K+ concentration. 51 Previous studies indicated that the hERG channel at higher temperature poise channels more sensitive to some hERG blockers. 52,53 However, some research studies also demonstrated that, by changing temperature from 22°C to 35°C, the alteration in potency of drugs on the hERG channel was diversified: for E-4031 the potency was unchanged, for ketoconazole it was slightly increased, and for astemizole it was decreased. 54,55 Obviously, testing the effects of drugs at 35°C gives the superiority of revealing drug actions in physiological environments. However, due to the difficulty in maintaining appropriate temperature in current recording, researchers have demonstrated that, in most cases, the potency of hERG channel blockers did not significantly change at near-physiological temperature (short pulse protocols) compared to the potency obtained at room temperature (long pulse protocols). 54 The present study found that the chronic treatment of clenbuterol reduces hERG channel density at cell surface indicated by patch clamp and Western blot assay. All of our patch-clamp studies in this study were performed at room temperature (22°C-24°C) using long pulse protocols, which is likely to provide reliable data for drug screening purposes.
In conclusion, our study suggests that clenbuterol reduces the expression of hERG channel in time- and concentration-dependent manner and also decreases I hERG, which may be one mechanism of clenbuterol-induced QTc prolongation and arrhythmia. Furthermore, our study shows that clenbuterol reduces hERG channel membrane expression by promoting mature channel degradation. The effect of clenbuterol on the hERG channel is related to the drug-binding sites of Tyr-652 and Phe-656 located on the S6 domain. This study is the first to demonstrate the biophysical mechanism of clenbuterol-induced arrhythmia, in addition to expanding and complementing the mechanisms of drugs-induced disruption of hERG channel function.
Footnotes
Authors Contributions
L. Ling contributed to design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript; H. Peijing contributed to acquisition, analysis, and interpretation and drafted the manuscript; M. Changqing contributed to analysis and interpretation and critically revised the manuscript; M. Aiqun contributed to analysis and interpretation and critically revised the manuscript; W. Tingzhong contributed to conception and design, contributed to analysis and interpretation, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (No 81570672) and Key Program of International Cooperation and Exchanges of Shaanxi, China (No 2014KW23-05).