
Abstract
Inflammation and oxidative stress are important risk factors affecting various cells in the formation of atherosclerosis. MicroRNAs (miRs) are regulators of inflammation and atherogenesis. The expressions of endothelial cell (EC)-specific miR-10a and miR-21 and monocyte-specific miR-33a and miR-221 were investigated using coculture of the ECs and monocytes upon exposure to H2O2 as an oxidative stressor, and endotoxin/lipopolysaccharide (LPS) as a microbial stressor. Human umbilical endothelial cells (HUVECs) and peripheral blood mononuclear cells (or monocytes) were cocultured in M199 complete medium and were incubated with LPS (20 ng/mL) or H2O2 (1%) for 8 hours at 37°C. The HUVECs and monocytes were then separated from the cellular mix using a magnetic bead negative selection technique. The relative expression of miRs was determined by real-time polymerase chain reaction. In both cell types, H2O2 induced miR10a (P = 0.05) and LPS induced miR21 (P = 0.0003) compared to the untreated controls. Coculture increased miR-10a and miR-21 expression in monocytes (P = 0.0008 and <0.0001); however when cultured alone, HUVECs expressed higher levels of miR-10a and miR-21 (P < 0.0001 and <0.0001). Coculture decreased the expression of miR-33a in monocytes (P < 0.0001) while increasing miR221 in HUVECs and monocytes (P < 0.0001 and <0.0001). The expression pattern of miRs in HUVECs and monocytes changes in the coculture compared to culturing alone in response to oxidative and microbial toxic compounds. Moreover, different cellular stressors induce different athero-miRs, which may affect the course of inflammation.
Introduction
Atherosclerosis is a chronic and progressive disease characterized by the fatty degeneration and vessel stiffening. It commonly affects medium and large-sized arteries in intima especially where the vessels divide. 1 Atherosclerosis is the potent cause of cardiovascular diseases such as myocardial infarction, stroke, and heart failure. 2
Inflammatory cells are abundant in the atherosclerotic lesion and are likely to play a major role in the pathogenesis of atherosclerosis. 3 Early in the process of atherogenesis, endothelial cells (ECs) become activated due to the effect of biochemical substances, such as oxidized low-density lipoprotein (LDL) and growth factors. Reactive oxygen species (ROS) are known to play a major role in the lipid oxidation. 4 Upon encountering these stimuli, ECs overexpress adhesion molecules, which lead to the recruitment of inflammatory cells. Monocytes and T cells attach to the endothelium and penetrate into the intima, where they continue to release pro-inflammatory cytokines and chemokines. Upon continuous exposure to stimulants and lipids, macrophages appear more as foamy cells and generate fatty streaks. 4 Smooth muscle cells (SMCs) migrate to the intimal layer and proliferate, and with time, fatty streak evolves to fibrous cap, which is a hallmark of the established atherosclerosis. Finally, the lesion evolves to enclose large amounts of lipid, and upon destabilization, the plaque ruptures and thrombosis or vessels occlusion occur. 5
Although the initiation of the atherosclerotic lesion is poorly described, the toxicity of infectious agents and oxidants in the pathology of atherosclerotic inflammation has been thoroughly discussed. Several trials have investigated the infection hypothesis of atherogenesis. 2,6 Pathogens can exhibit proatherogenic effects either directly or indirectly. Pathogens can infect ECs and SMCs and persist in them while replicating in low levels. Thus via releasing pro-inflammatory cytokines and continued low-level immune stimulation, chronic inflammation will prevail. 7 In such a condition, endotoxin/lipopolysaccharide (LPS) has a major contribution to the inflammatory process. 8 Several reports have shown a correlation between the incidence of atherosclerosis and the presence of infectious microorganisms, such as Cytomegalovirus, Helicobacter pylori, and Chlamydia pneumonia. 9
On the other hand, extensive ROS production occurs in atherosclerosis via chronic activation of ECs and the components of the immune system. According to the oxidative stress hypothesis, atherosclerosis begins with the oxidative modification of LDL. Previous experiments have suggested that atherosclerosis risk factors lead to ROS production from ECs, SMCs, and macrophages. 10 Further studies clearly demonstrated that ROS production from both monocytes and macrophages played a crucial role in the atherosclerosis. Indeed, ROS cause damage to the lipids, proteins, DNA, and membranes due to their toxic effects. 4 It has been shown that oxidative stress, especially as a result of exposure to H2O2, increases tyrosine kinase phosphorylation that causes further binding of neutrophils to the endothelium affecting endothelium permeability. 11 Another mechanism by which oxidative stress may lead to atherosclerosis is nuclear factor κB (NFκB) and activation protein-1 (AP-1) that impacts the expression of vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1, E-selectin adhesion molecules, and production of pro-inflammatory cytokines. 12
Reactive oxygen species molecules are also major signaling molecules that can affect several signaling pathways in the cells. 13 In addition to ROS-dependent activation of different transcription factors, 14,15 other gene-regulatory molecules can be targeted by ROS signaling among which microRNAs (miRs) can be named. 16 MicroRNAs are a class of highly conserved, small (18-24 nucleotide), noncoding RNA that bind to target mRNA. 17 MicroRNAs regulate gene expression at the posttranscriptional level and also control cellular processes such as growth, survival, and differentiation. 18
Recent evidence has shown that ECs function, innate and adaptive immune responses, and lipid metabolism are among the key regulatory processes in atherosclerosis that can be controlled by miRs. 16,19,20 Based on their role in atherogenesis, miRs are divided into athero-miRs and atheroprotective miRs. Athero-miRs such as miR-92, miR-21, miR-633, and miR-33a are a class of miRs that lead to atherosclerosis by induction of inflammatory conditions. Oscillatory shear stress, as an atherogenic mechanism, stimulates the expression of miR-21 in cultured ECs and leads to an inflammatory response during peroxisome proliferator-activated receptor α by 3′-UTR targeting. 21 MiR-33a restrains cellular cholesterol efflux to ApoA1 and mature high-density lipoprotein (HDL) and decreases circulating HDL-cholesterol (HDL-C) levels in mice and nonhuman primates. 22 Previous studies showed that genetic deletion of miR-33a in mice increased plasma HDL-C levels and reduced the progression of atherosclerosis. 22
Atheroprotective miRs such as miR10a, miR-221, miR-19, and miR-126 are a class of miRs that regulate genes of inflammatory component by which inhibit atherosclerosis. 17 Decreased miR10a in atherosusceptible regions of swine inner aortic arch and aorta–renal branches of the vessels is already shown. 3 In miR-10a knockdown models, IκB/NFκB-mediated inflammation is suggested to be the main player, where it leads to significant upregulation of inflammatory biomarkers such as MCP-1, VCAM-1, E-selectin, interleukin-6 (IL-6), and IL-8. 3 Moreover, miR-221 is recognized as an antiangiogenic miR through inhibiting c-kit–mediated migration in ECs. 21 MiR-221 targets c-kit in ECs and suppresses ECs migration and proliferation and reduces nitric oxide synthase (NOS) activity and angiogenesis. 18 It is shown that miR-221 levels are higher in endothelial progenitor cells in coronary artery disease associated with better prognosis. 21
In previous studies, an altered pattern of miRs expression in ECs and blood cells has been shown. 23, 24 However, not much information is available on the miRs expression or dysregulation in the more physiologic condition of ECs and blood cells encounters to stress. In this study, we aimed to provide new insight on the miRs expression in the coculture of ECs and monocytes under oxidative and nonoxidative stressors. Therefore, we examined the expression of EC-specific miR-10a and miR-21 and macrophage-specific miR33a and miR-221 in the coculture exposed to microbial or oxidative stress as compared to the monoculture of ECs and monocytes.
Materials and Methods
The methods and sampling used in this study were approved by the Clinical Research Ethics Committee of the Shiraz University of Medical Sciences, Shiraz, Iran. Informed consent was given by the participants for entering to the study.
Endothelial Cell Isolation and Culture
Human umbilical endothelial cells (HUVECs) were isolated using collagenase digestion method. Briefly, the 2 ends of the cord were cut with a scalpel and a cannula was introduced at each extremity of the vein and tightly maintained with a string. The cord was washed 2 times with sterile phosphate-buffered saline using the 50-mL syringe. Then, the collagenase type II (1 mg/mL) was injected at 1 end of the vein using the 30-mL syringe. Then, the ECs were detached from the vein basal membrane using collagenase type II by incubation at 37°C for 15 minutes. The cells were collected in a 50-mL falcon tube containing medium 199 (M199; Sigma), 20% fetal calf serum (FCS; Gibco, Germany). The cells were washed at 377 g for 5 minutes and suspended in 10 mL of M199 supplemented with 20% FCS, penicillin (200 U/mL; Biosera, UK), streptomycin (200 µg/mL; Biosera, United Kingdom), and ECs growth supplement (15 μg/mL, Sigma). Two milliliter of the cell suspension was added in each collagen-coated 12-well culture plates. Cultures were placed in a humidified 95% atmosphere containing 5% CO2 at 37°C. The following day, nonadherent cells were removed by changing the culture medium. The culture medium was changed every 2 days and cells were subcultured at 80% confluency. For this purpose, ECs were harvested with trypsin-EDTA 0.5% (Gibco, Germany). At passage 2, the purity of ECs was confirmed by FACS analysis of CD31 expression as a marker of almost all ECs. For this purpose, a sample of ECs was extracellularly stained with FITC-conjugated anti-CD31 after each separation. The average purity of ECs was 98.4% ± 0.96% (Figure 1).
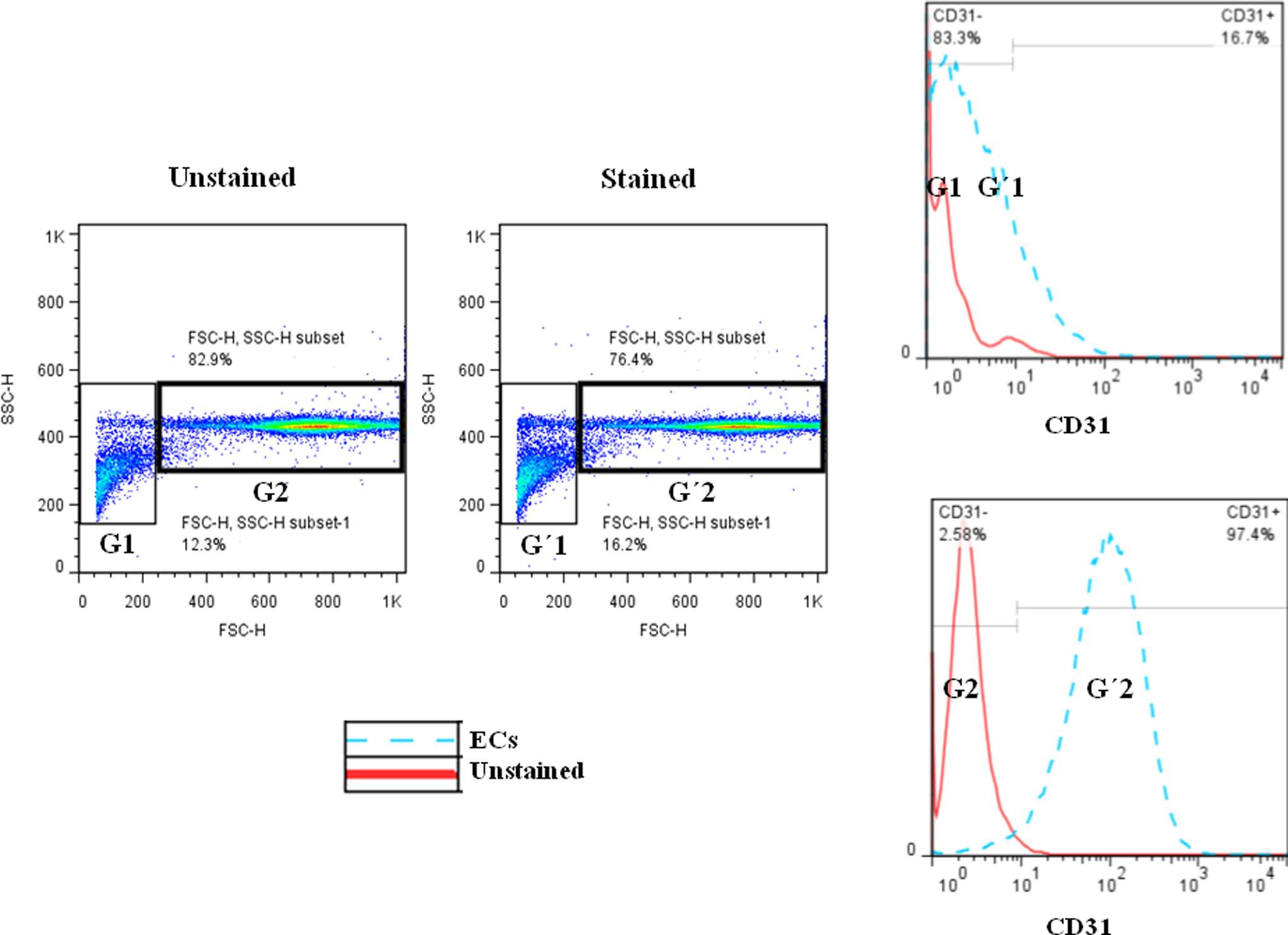
The purity of separated HUVECs. The cells were stained with FITC-conjugated anti-CD31 after each separation and the percentage of CD31 positive cells was determined by flowcytometry. HUVECs indicate human umblical endothelial cells.
Monocyte Isolation
For monocyte isolation, peripheral blood mononuclear cells (PBMCs) were isolated from 100 mL of peripheral blood of each of the 3 normal individuals (1 female and 2 males) by ficoll density gradient centrifugation. Then using the pan monocyte isolation kit (Miltenyi Biotec), human monocytes were isolated by depletion of nonmonocytes (negative selection). Nonmonocytes were indirectly magnetically labeled with a cocktail of biotin-conjugated monoclonal antibodies, as primary labeling reagent, and anti-biotin monoclonal antibodies conjugated to microbeads, as secondary labeling reagent. The magnetically labeled nonmonocytes were depleted by retaining them on a magnetic antibody cell sorter (MACS) column in the magnetic field of an MACS separator while the unlabeled monocytes passed through the column. The purity of the isolated monocytes was evaluated by a flow cytometry method. To evaluate the purity of isolated monocytes from MACS column, we stained the monocytes with anti-CD14 and anti-CD45 that were conjugated to PE and FITC, respectively. Our results showed that the average purity of the isolated monocytes from MACS column was 95.5% ± 1.15% (Figure 2).
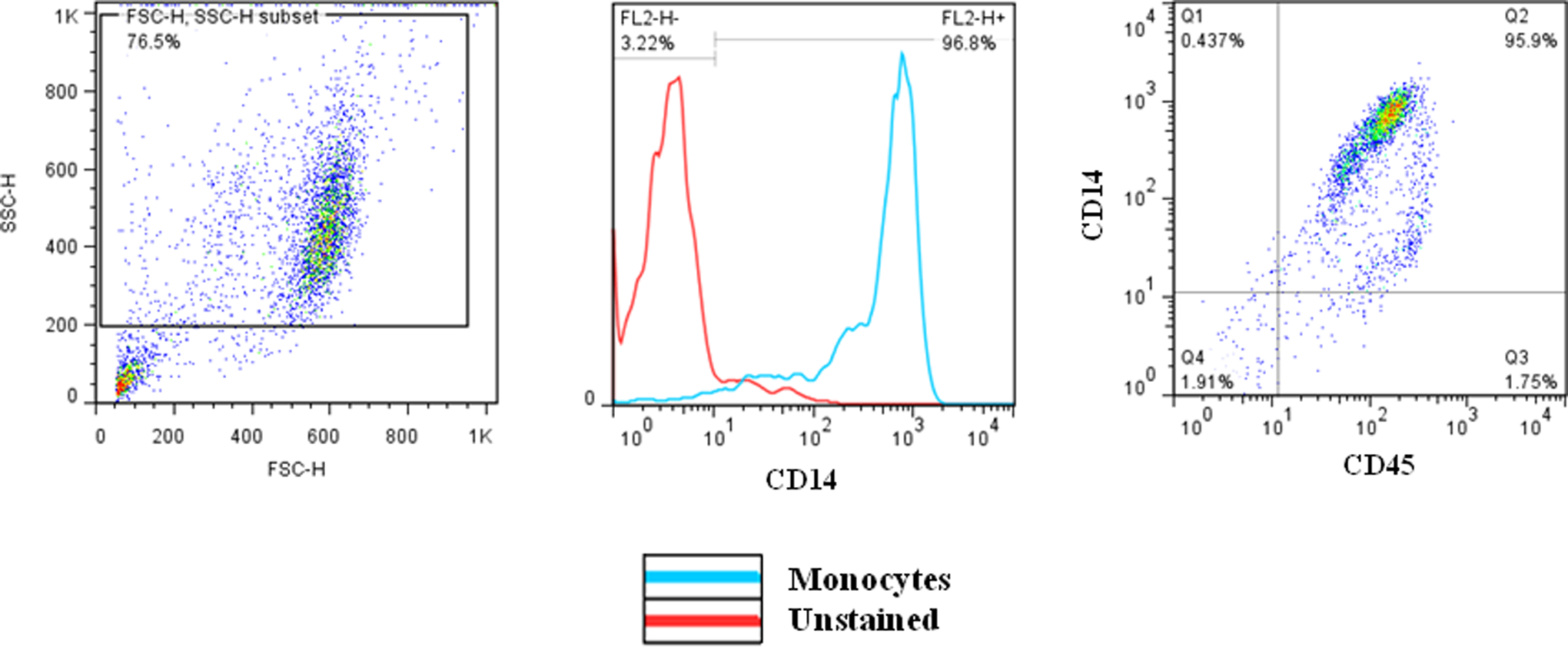
The purity of isolated monocytes. The cells were stained with PE-conjugated anti-CD14 and FITC-conjugated anti CD45 antibodies. The percentage of positive cells was determined by flow cytometry method.
Lipopolysaccharide Extraction
Lipopolysaccharide from Escherichia coli was extracted using the LPS extraction kit, and extracted LPS was treated with proteinase K (75 µg per 30 µg LPS) to digest the contaminating proteins. Then, LPS concentration was determined using the Pierce limulus amebocyte lysate chromogenic endotoxin quantitation kit (Thermo Scientific) according to the manufactures’ instructions. To obtain appropriate LPS concentration for ECs stimulation, we stimulated ECs with different concentrations of LPS (20, 40, and 160 ng/mL), and apoptosis was evaluated by Annexin V, 7-AAD, using the flow cytometry method.
Endothelial Cell Isolation by MACS
After the coculture of ECs and PBMCs, we used human CD45 microbeads (Miltenyi Biotec) to isolate ECs from PBMCs. The CD45+ cells (PBMCs) were magnetically labeled with CD45 microbeads. Then, the cell suspension was loaded onto an MACS column which was placed in the magnetic field of an MACS separator. The magnetically labeled CD45+ cells were retained within the column, whereas the unlabeled ECs which were depleted of CD45+ cells ran through. The purity of cocultured ECs after MACS separation from PBMCs was assessed by immunostaining. The FITC-conjugated anti-CD31 was used to stain the cells, and the purity of cells was determined by the flow cytometry method to be 96.3% ± 1.1% (Supplementary Figure 1).
Endothelial Cells and Monocytes Stimulation
Endothelial cells at passage 3 and freshly isolated monocytes were stimulated with 20 ng/mL LPS or 1% H2O2 for 8 hours as described elsewhere. 25,26 For the coculture experiments, ECs were originally cocultured with PBMCs and exposed to the LPS or H2O2. The ECs and monocytes were then isolated by MACS separator as explained above. The purity of cocultured monocytes after MACS separation from ECs was assessed by immunostaining. The FITC-conjugated anti-CD45 and PE-conjugated anti-CD14 were used to stain the cells, and the purity of cells was determined by the flow cytometry method to be 93.8% ± 0.7% (Supplementary Figure 2). The purity of ECs after separation from monocytes was determined by CD31 staining of the cells (Supplementary Figure 1).
Total RNA Extraction and Real-Time Polymerase Chain Reaction
Total RNA was extracted from monocytes and HUVECs treated with LPS and H2O2 as well as monocytes that were cocultured with HUVECs in the presence of LPS and H2O2, using Trizol reagent (Invitrogen). Poly.A tail was synthesized by 2 μL of 10× reaction buffer, 0.5 μL poly.A polymerase enzyme, 1 μL adenosine triphosphate (ATP; 10 mM), and 2 μg RNA as instructed by the manufacturer (Pars-genome, Tehran, Iran). Then complementary DNA was synthesized by 2 μL of 5× reaction buffer, 1 μL dNTPs, 0.5 μL RT enzyme, 0.5 μL of specific primers (15 pmol), and RNA poly.A tail (Pars-genome). The relative expression of miRs was measured by real-time polymerase chain reaction (RT-PCR) method, and miR levels were normalized to U6 expression. Amplification was carried out using the 7500 Real-Time PCR system (Applied Biosystems, Massachusetts). Cycling conditions for RT-PCR consisted of an initial step at 95°C for 5 minutes and 95°C for 5 seconds, followed by 40 cycles of 63°C for 20 seconds and 72°C for 31 seconds. Product amplification was measured during the extension phase, and all samples were run in triplicate. Fold changes relative to the untreated controls were calculated using the following formula: that is fold change = 2−▵▵CT, where ▵CT is the expression difference between target miR and internal control (U6), and ▵▵CT is the difference of ▵CT between treated and untreated conditions.
Results
Analysis of Apoptosis in ECs Stimulated by Different Concentrations of LPS
Different concentrations of LPS (20, 40, and 160 ng/mL) resulted in different levels of apoptosis in ECs. It was found that 20 ng/mL (0.5 μL) of LPS solution caused minimal necrosis (8.91%) and minimal apoptosis (9.59%) rate and was selected for treatment of ECs and monocytes in the culture (Figure 3). In the next parts, we compared the effects of a well-known toxin, H2O2, to a less-appreciated toxin in atherosclerosis, that is LPS, to see if the effects of the 2 toxins in provoking athero-miRs are different. Moreover, many toxicity experiments are performed on separate cells or cell lines, we also asked whether the presence of 2 cell types, as it happens in the body, would change the dynamic of miR expressions and thereby the outcome.
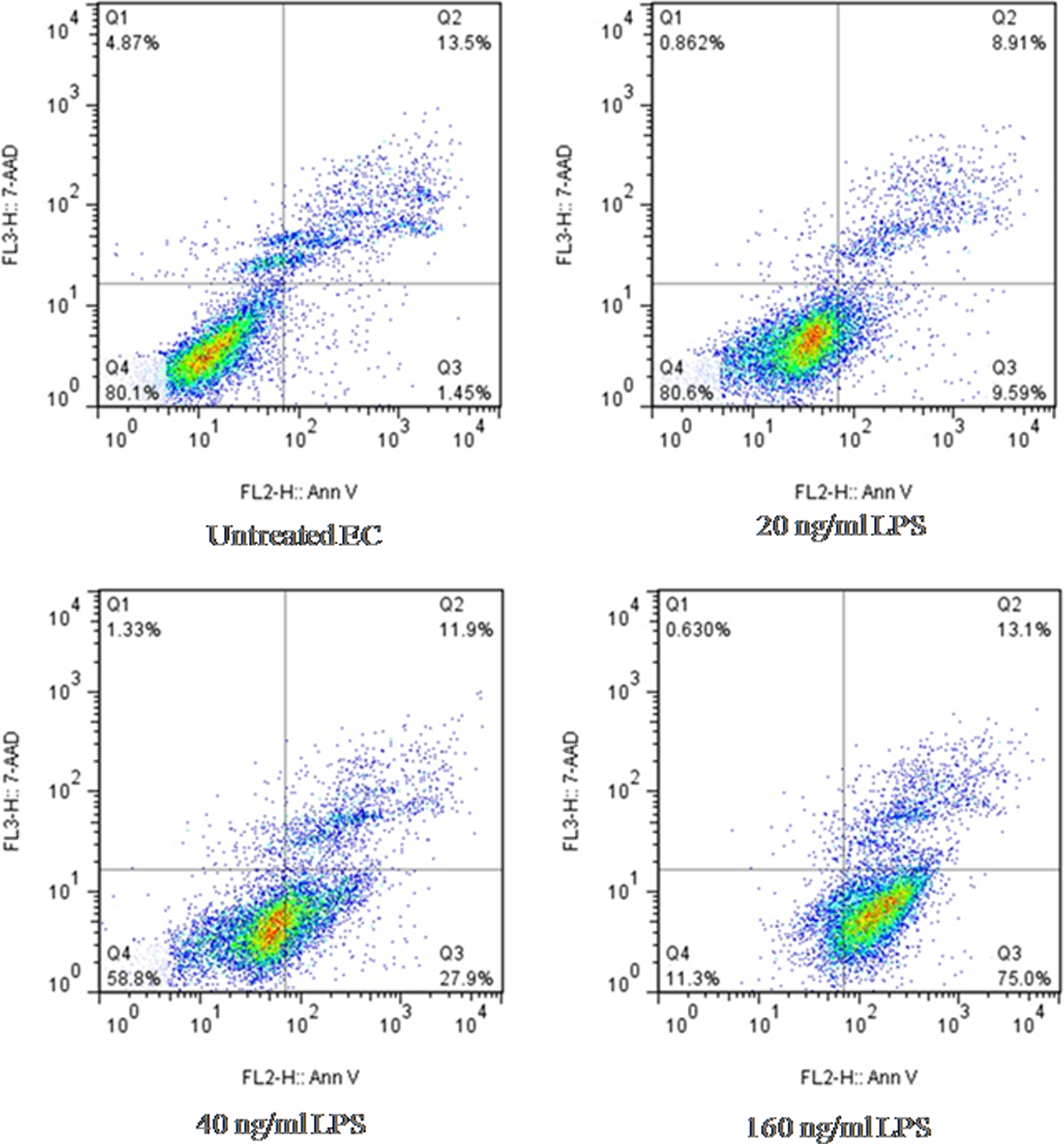
Rate of apoptosis in ECs that were stimulated with LPS. Different concentrations of the extracted LPS (20, 40, and 160 ng/mL) in 0.5, 1, and 4 μL volumes were added to the HUVEC cultures, and the rate of apoptosis and necrosis was determined after 8 hours by flow cytometry method. ECs indicate endothelial cells; HUVECs, human umblical endothelial cells; LPS, lipopolysaccharide.
The Effects of LPS and H2O2 on miR-10a Expression in ECs and Monocytes
As shown in Figure 4, both ECs and monocytes expressed higher level of miR-10a in the presence of H2O2 (8.77% ± 1.31% and 10.83% ± 2.30%, respectively) compared to LPS (5.92% ± 1.08% and 6.35% ± 2.01%, respectively, P = 0.05). Our data showed that the expression of miR-10a in ECs that were cultured alone under H2O2 and LPS stimulation was higher than ECs cocultured with monocytes (4.34% ± 1.72% and 2.45% ± 0.81%, respectively, P < 0.0001, Figure 4A). On the other hand, miR-10a, an EC-specific miR, had no expression in monocytes and the expression of miR-10a was increased in monocytes cocultured with ECs under H2O2 and LPS stimulation (P = 0.0008 and <0.0001, respectively; Figure 4B). Analysis of fold changes confirmed above results as well (Supplementary Figure 3).
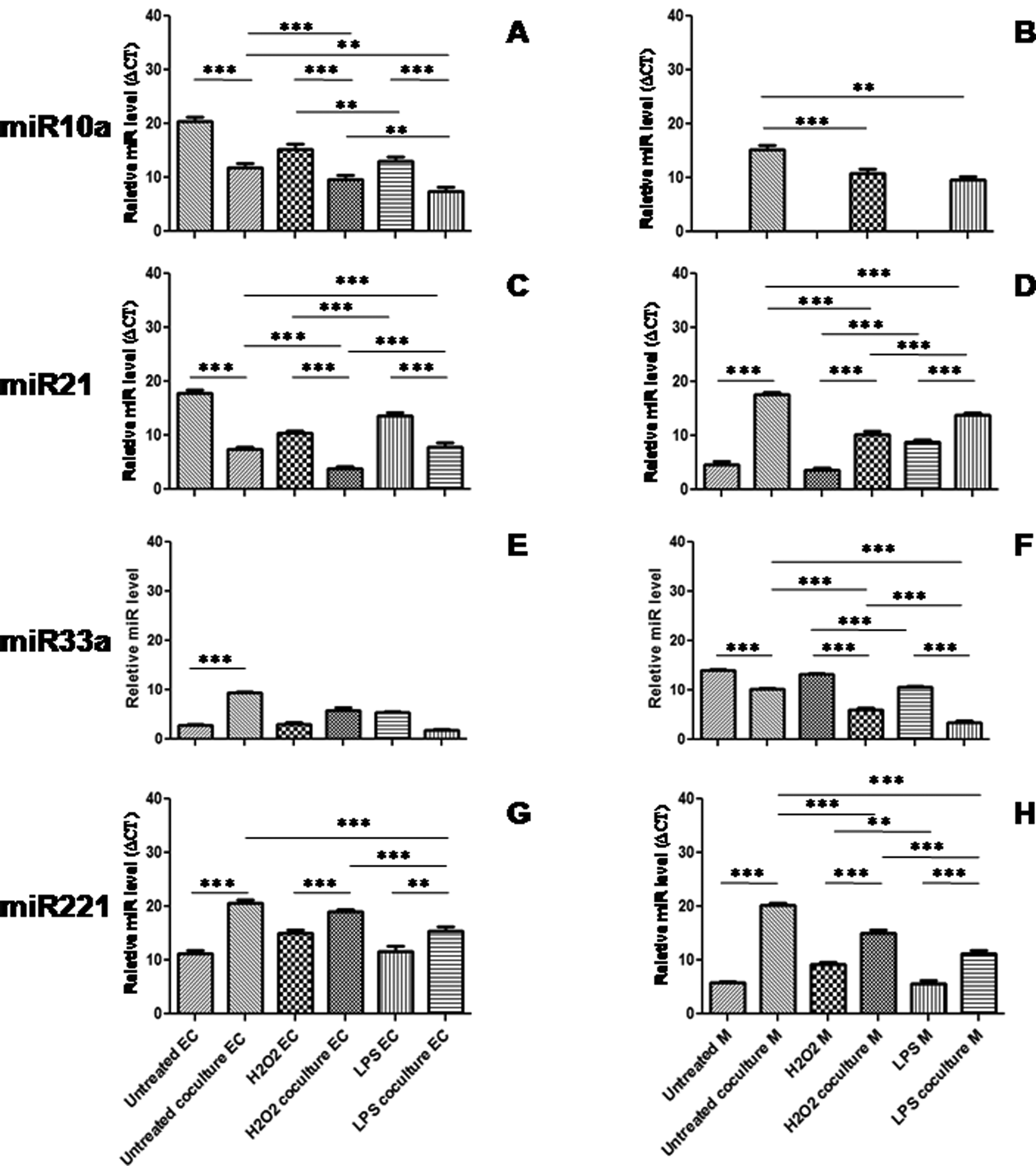
The level of miR-10a (A, B), miR-21 (C, D), miR-33a (E, F), and miR-221(G, H) expression in ECs and monocytes. MiR expression levels in ECs and monocytes are shown relative to internal control U6. *P < 0.05, **P < 0.001, ***P < 0.0001. ECs indicate endothelial cells; miR, microRNA.
The Effects of LPS and H2O2 on miR-21 Expression in ECs and Monocytes
The expression of miR-21 in ECs and monocytes was more responsive to LPS stimulation than H2O2 (18.97% ± 2.63% vs 13.81% ± 2.54%, P = 0.0003, 6.84% ± 1.66% vs 3.20% ± 1.69%, P = 0.0004, respectively; Figure 4C). Similar to miR-10a, monocytes expressed higher levels of miR-21 while in coculture with ECs (P < 0.0001, Figure 4D), and ECs expressed higher levels of miR-21 while cultured alone (P < 0.0001, Figure 4C). Analysis of fold changes confirmed above results as well (Supplementary Figure 3).
The Effects of LPS and H2O2 on miR-33a Expression in ECs and Monocytes
MiR-33a, a monocyte-specific miR, showed no significant expression in ECs in the presence of either of the stimuli (P = 0.01; Figure 4E). However, the expression of miR-33a was more responsive to H2O2 when monocytes were cultured without ECs (35.61% ± 3.93%, P < 0.0001, Figure 4F). Analysis of fold changes confirmed above results as well (Supplementary Figure 3).
The Effects of LPS and H2O2 on miR-221 Expression in ECs and Monocytes
The expression of miR-221 by ECs was noticeable while the cells were in the coculture with monocytes under H2O2 stress (4.16% ± 1.88%, P < 0.0001, Figure 4G). In addition, we observed that monocytes expressed higher levels of miR-221 in the coculture as well (22.2% ± 2.85%, P < 0.0001, Figure 4), and both ECs and monocytes were more responsive to H2O2 stress than LPS (0.76% ± 27.7%, P = 0.001 and 9.18% ± 2.90%, P = 0.003, respectively). Analysis of fold changes confirmed above results as well (Supplementary Figure 3).
Discussion
We found that LPS, as a major constituent of endotoxin and H2O2 and as an oxidant, affects the expression of miR-10a, miR-21, miR-33a, and miR-221 in different fashions. In this regard, ECs responded to H2O2 by miR-10a expression and LPS by miR-21 expression. According to the previous studies, the major targets of miR-10a are phosphatase and tensin homology (PTEN) deleted on chromosome 10, mitogen-activated kinase kinase kinase 7 (MAP3K7), and β-transducin repeat containing (bTRC) gene. The PTEN acts as a tumor-suppressor protein and regulator of cellular growth and apoptosis. In general, PTEN regulates cell growth, survival, migration, invasion, and differentiation. It also affects ECs proliferation and permeability through vascular endothelial growth factor (VEGF) regulation. The VEGF is a critical stimulator of inflammation and recruitment of neutrophils and monocytes. The PTEN inhibition leads to decreased VEGF production and ECs proliferation, 26 as well as decreased ROS production in pathological conditions such as Parkinson disease and stroke. 27 Moreover, binding of miR-10a to 3′-UTR of PTEN and suppression of its gene is already shown. 28 Other target genes of miR-10a are MAP3K7 and bTRC. MiR-10a binds to 3′-UTR of these 2 genes and inhibits NFκB activation. Therefore, the release of pro-inflammatory cytokines, such as IL-1β, IL-6, IL-8, and MCP-1, and the expression of E-selectin and VCAM-1 are suppressed. 29 It is therefore interesting to see whether the expression of miR-10a in response to H2O2 is a mechanism to subside inflammation to protect EC damage or has other consequences.
In the coculture of ECs and monocytes and in the absence of cell contact, monocytes engulf miR-10a exosomes from ECs and suppress NFκB activation. 30 In our study, miR-10a expression level in ECs was lower in the coculture with monocytes than isolated culture. On the other hand, miR-10a expression level was higher in monocytes cocultured with ECs compared to those cultured alone. Currently, it is not clear if miR-10a exosomes can affect miR-10a expression in monocytes. Moreover, the nature of miR-10a–based cross talk between the 2 cell types remains to be investigated.
We found that miR-21 expression was more responsive to LPS compared to H2O2. MiR-21 expression is time dependent in different cell types. 31 MiR-21 leads to increased JAG-1 expression and activation of NOTCH-1 signaling pathway and inflammation. 32 In addition, miR-21 activates AP-1 which results in pro-inflammatory cytokines production, overexpression of adhesion molecules, and immune cells infiltration. 32 On the other hand, miR-21 can target PTEN, decrease its activation, and induce immunosuppressive conditions which, in association with prolonged survival of damaged cells, delays the resolution of inflammation. 32 In addition, miR-21 targets the superoxide dismutase 2 (SOD-2) and leads to increased oxidative stress. The SOD-2 is an important mitochondrial protein that defends the cell against oxidative components. 33 Therefore, ECs respond to LPS by increasing an inflammation promoting miRs.
In contrast to ECs, miR-21 expression level in cocultured monocytes was higher than monocytes alone. Despite being an EC-specific miR, miR-21 affects the AP-1 gene in monocytes and leads to excessive miR-21 expression in monocytes, as well. 34 It is possible that cocultured monocytes received miR-21 from ECs, thereby targeting AP-1 gene and increased miR-21 expression in a positive feedback.
Our data showed that monocytes responded to H2O2 by the expression of miR-33a and miR-221 to a greater extent than LPS. MiR-33a is an athero-miR that is specifically expressed in monocytes and macrophages. The critical target gene of miR-33a is ATP binding cassette gene (ATBC). In physiological conditions, ATBCA1 and ATBCG1 are expressed on monocyte surface and play important roles in cholesterol metabolism. The ATBCA1 mediates efflux of cholesterol from cell to ApoA1 and its reverse transport to liver. 35 Previous studies have demonstrated that the 3′-UTR of ATBCA1 contains 3 binding sites for miR33a, by binding to which it inhibits cholesterol efflux from the cell and results in cholesterol accumulation and foam cell formation. 36 MiR-33a also targets genes that are associated with autophagy. Dysfunctional autophagy leads to inflammasome activation, dysfunctional efferocytosis in macrophages, and atherosclerosis. Efferocytosis refers to the process by which different phagocytic cells engulf the apoptotic cells 37 by MiR-33a and inhibits autophagy due to the suppression of ATG5, ATG7, lysosomal acid lipase, and lysosomal-associated membrane protein-1 genes. 38,39 In our study, miR-33a expression in ECs was cultured alone and was not significant. In contrast, miR-33a expression in monocytes cultured alone was higher than monocytes that were cocultured with ECs. It is likely that when ECs are cocultured with monocytes, ECs exhibit a protective effect and inhibit miR-33a expression in monocytes. However, this assumption needs further proof.
MiR-221 was expressed to a higher level in both ECs and monocytes in response to H2O2 than LPS. MiR-221 targets c-kit in PBMCs and suppresses their proliferation. In addition, miR-221 targets c-kit in ECs and suppresses ECs migration and proliferation and reduces NOS activity and angiogenesis. 17 Also miR-221 targets ETS-1 transcription factor, affects ETS-1 and downstream genes involved in inflammation, and thus reduces the expression of adhesion molecules and thereby atherosclerosis. 17 MiR-221 leads to reduced PTEN expression and reduced trail activities and suppresses cellular apoptosis. 34
We also observed that miR-221 expression in cocultured monocytes and ECs increased. It is shown that miR-221 expression from monocytes results in a feedback loop by which miR-221 expression in ECs increases. 40 It means that in the coculture conditions, miR-221 expression in both ECs and monocytes maybe in an attempt to extinguish inflammation.
Conclusion
Our results suggest a cross talk between ECs and monocytes by which the pattern of miRs expression in ECs and monocytes changes in the coculture in response to the atherosclerotic stressors. Moreover, both atheroprotective miRs (miR-10a and miR-221) increased in response to oxidative stress. Of the 2 athero-miRs, only miR-21 increased in response to LPS in ECs and monocytes. Since H2O2 can induce both athero-miRs and atheroprotective miRs in monocytes, it would be interesting to study the kinetics of miRs expression after oxidative stress. Finally, our findings can help us to better understand the atherosclerosis inflammatory process under different stimuli and differentiate the microbial and oxidative pathways of atherogenesis, which has implications in the therapeutic designs.
Footnotes
Authors’ Note
This work was performed as a part of Atefe Ghamar Talepoor dissertation as a requirement for graduation in MSc of Immunology from Shiraz School of Medicine (Shiraz, Iran). No writing assistance was utilized in the production of this manuscript.
Author Contributions
Ghamar Talepoor contributed to acquisition, analysis, and interpretation of data; drafted manuscript; gave final approval; and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Kalani contributed to acquisition and interpretation, critically revised manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Samsami Dehaghani contributed to acquisition, drafted manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Doroudchi contributed to conception and design, critically revised manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was financially supported by a grant (93-7072) from Shiraz University of Medical Sciences, Shiraz, Iran.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.