
Abstract
The intent of cancer immunotherapy (CIT) is to generate and enhance T-cell responses against tumors. The tumor microenvironment establishes several inhibitory pathways that lead to suppression of the local immune response, which is permissive for tumor growth. The efficacy of different CITs, alone and in combination, stems from reinvigorating the tumor immune response via several mechanisms, including costimulatory agonists, checkpoint inhibitors, and vaccines. However, immune responses to other antigens (self and foreign) may also be enhanced, resulting in potentially undesired effects. In outbred mammalian pregnancies, the fetus expresses paternally derived alloantigens that are recognized as foreign by the maternal immune system. If unchecked or enhanced, maternal immunity to these alloantigens represents a developmental and reproductive risk and thus is a general liability for cancer immunotherapeutic molecules. We propose a tiered approach to confirm this mechanistic reproductive liability for CIT molecules. A rodent allopregnancy model is based on breeding 2 different strains of mice so that paternally derived alloantigens are expressed by the fetus. When tested with a cross-reactive biotherapeutic, small molecule drug, or surrogate molecule, this model should reveal on-target reproductive liabilities if the pathway is involved in maintaining pregnancy. Alternatively, allopregnancy models with genetically modified mice can be interrogated for exquisitely specific biotherapeutics with restricted species reactivity. The allopregnancy model represents a relatively straightforward approach to confirm an expected on-target reproductive risk for CIT molecules. For biotherapeutics, it could potentially replace more complex developmental and reproductive toxicity testing in nonhuman primates when a pregnancy hazard is confirmed or expected.
Background
A functional and balanced maternal immune system is essential to establish and maintain a successful pregnancy. This occurs locally through production of soluble factors such as cytokines, chemokines, and angiogenic molecules important for implantation and placentation as well as the highly regulated activity of specific immune cell populations throughout pregnancy. 1,2 While the maternal immune system is critical for protecting the fetus from invading pathogens, it is also constantly exposed to paternally derived alloantigens expressed by the developing fetus at the maternal–fetal interface. Therefore, antigen-specific tolerance mechanisms have evolved to protect the fetus from immune-mediated rejection. While the exact mechanisms are not completely understood, several local pathways and mechanisms are involved in maintaining feto–maternal tolerance, including expression of nonclassical major histocompatibility complex (MHC) molecules by trophoblasts, presence of uterine natural killer cells, expansion of regulatory T cells (Tregs), increased T cell apoptosis, inhibition of complement factors, and immune tolerance mediated by the programed death-ligand 1 (PD-L1)/programed cell death 1 (PD-1) pathway and tryptophan catabolism by indoleamine 2,3-dioxygenase (IDO). 1,2
With the recent approvals of several biotherapeutic molecules designed to enhance patients’ own immune system to fight and eliminate tumors, cancer immunotherapy (CIT) has established itself as a clinically validated approach for treating cancer. 3 –5 Based on current understanding, an effective anticancer immune response involves several steps, including release of cancer cell antigens, presentation of these antigens to antigen-specific T cells, generation of protective CD8+ T cell responses with cytotoxic potential, trafficking to the tumor, penetration of T cells into the tumor, recognition of cancer cells by T cells, and killing of cancer cells; collectively this has been referred to as the “cancer immunity cycle.” 6 Throughout this cycle, there is a coordinated balance between stimulatory and inhibitory factors that promote or suppress tumor immunity and also prevent autoimmunity. 6,7 Enhancement of immune responses to other antigens (self and foreign) can result in unintended consequences such as exacerbated inflammatory responses or autoimmunity, both of which have been reported clinically. 4,6 –8 Importantly, several of the factors thought or known to actively suppress the local antitumor T-cell response, such as cytotoxic T lymphocyte antigen-4, PD-L1, Tregs, and IDO also prevent the developing fetus from being recognized by the maternal immune system. 1,3,6,9 –16
Since the primary pharmacology of cancer immunotherapeutics is intended to increase the activity of the maternal immune system, rather than on developmental pathways intrinsic to the developing fetus, the traditional reproductive and developmental toxicity test systems may have limited utility as they have been primarily designed to unmask direct effects on the fetus. Since the primary goal of developmental and reproductive toxicity (DART) testing is to understand the potential hazards to mother and fetus due to drug exposure during pregnancy, and for the most part, controlled clinical trials in pregnant women are rarely warranted, the nonclinical reproductive toxicity testing conducted during drug development may contribute pivotal data used for characterization of reproductive risk. Therefore, alternative models such as allopregnancy models described subsequently may represent a more relevant nonclinical pregnancy model in which to test CIT molecules for pharmacologically mediated reproductive liabilities.
Reproductive toxicity studies are meant to reveal any potential effects on mammalian reproduction at various stages but are not necessarily intended to support intentional administration of the drug during pregnancy. Typical components of DART testing include (1) fertility and early embryonic development (ie, dose administration before mating and continuing through mating and implantation, also known as Segment 1 and typically conducted in rats), (2) embryofetal development (EFD) during the period of organogenesis (ie, dose administration to the dams between implantation and closure of the hard palate, also known as Segment 2 and typically conducted in 2 species, often rat and rabbit), and (3) prenatal and postnatal development (PPND, includes dose administration to dams during pregnancy and through weaning, also known as Segment 3 and typically conducted in rats). 17 –20
Embryofetal development and PPND studies primarily identify direct effects of the new molecular entity on the developing fetus, especially when the molecular targets are represented by intrinsic pathways that regulate growth, proliferation, differentiation, and/or angiogenesis. Standard readouts for an EFD study include assessment of implantation success and both fetal and placental development through late pregnancy, as based on a timed cesarean-section end point. The PPND studies focus on pregnancy outcomes through weaning, including general growth and development. Both study types capture pregnancy losses and should include monitoring for maternal toxicity. 18,21 Therefore, when testing a new molecular entity, as long as the fetus is exposed to pharmacological levels of the investigational molecule during critical times of development, the pregnancy outcomes of these models should enable one to identify and characterize potential maternal and fetal risk. 18 –20
For biopharmaceuticals, the principles for reproductive toxicity testing are similar to those for small molecule pharmaceuticals and generally follow the regulatory guidance outlined in International Conference on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use document S5(R2). 18,22 However, because many biopharmaceuticals are designed to be highly specific for human targets, they often have limited cross-reactivity in nonclinical species traditionally used to test reproductive toxicity for small molecule pharmaceutics (eg, rodents and rabbits) and may only cross-react with nonhuman primates (NHPs; ICH S6(R1)). Furthermore, there are also instances where the biopharmaceutical only reacts with the human target. Therefore, alternative approaches such as use of surrogate molecules that cross-react in rodent species or the examination of transgenic animals that are genetically modified with regard to the intended pharmacological target may be considered if scientifically justified. 19,20,22 –25
The pregnancy outcome or phenotype of the developing fetus from genetically modified animals (e.g., knockout/knockin) often provides useful information of the specific role of the targeted pathway on embryo–fetal development. For example, the hepatocyte growth factor HGF/c-MET pathway has been demonstrated to have a critical role in embryogenesis based on findings from HGF and c-MET knockout mice. The absence of c-MET signaling in these genetically engineered mice revealed severe developmental abnormalities of the placenta, liver, neurons, and muscle in the limb, diaphragm, and tongue, suggesting an on target liability to the developing fetus. 26 –30 Administration of a one-armed monoclonal antibody (mAb) that inhibits HGF/c-MET signaling pathway to pregnant cynomolgus monkeys resulted in severe compromise of the placenta and resultant adverse pregnancy outcomes in the absence of overt toxicity to the pregnant or lactating female monkeys (manuscript in preparation).
Secondary effects on the fetus can also occur as a result of maternal toxicity or direct effects on maternal reproductive organs. 31,32 For example, administration of insulin or insulin analogues to pregnant animals at dose levels that exceeds clinically relevant doses results in maternal hypoglycemia, which in turn leads to fetal malnutrition, pre- and postimplantation losses, and skeletal and visceral abnormalities. 33,34 These effects can be considered secondary to maternal toxicity as a result of exaggerated pharmacology.
Limitations of Current DART Models for CIT
In situations where a biotherapeutic is only pharmacologically active in humans and NHPs, the nonclinical safety testing, including DART studies, is also limited to NHPs. 22 –24,35 Nonhuman primates offer several advantages over rodents and rabbits with regard to DART testing because of the similarity to humans in the physiology, placental morphology, and rates of embryonic development. Furthermore, since NHPs maintain high genetic variation within the species, they more closely represent the alloantigenic recognition that occurs with human pregnancy. However, NHPs have limitations in that they generally only bear a single offspring and have a relatively high spontaneous pregnancy failure rate of up to 30%. 22,35 Consequently, large numbers of pregnant cynomolgus monkeys must be treated with the biotherapeutic in order to ensure an adequate number of fetuses/offspring can be evaluated and to provide an assessment of effects on pregnancy outcomes. As a result, even if the biopharmaceutical does cross-react in NHPs, alternative approaches/models may be considered if they are appropriate and scientifically justified in order to reduce the use of NHPs. 22 Importantly, both ICHS5(R2) and ICHS6 guidelines emphasize the need for flexibility in DART testing strategies to this end.
CD1 mice and Sprague-Dawley (SD) rats are commonly used rodent species in DART studies for small molecule therapeutics. They may also represent appropriate species for biotherapeutics if the clinical candidate is shown to cross-react and possess similar pharmacological effects to humans. For both species, the animals are derived from genetically outbred colonies; however, the degree of genetic heterogeneity within these closed-colony stocks can vary greatly depending on the history of the stock and is expected to have more limited genetic variation between breeding animals when compared to cross-strain breeding (ie, allogeneic pregnancy) in rodents or that seen with NHPs. 36 Consistent with this idea, both rodent stocks commonly used for DART testing have a relatively low spontaneous resorption rate of approximately 10%. This is in contrast to the previously mentioned high spontaneous abortion rate of NHPs but is comparable to the spontaneous resorption rate of approximately <5% in the syngeneic pregnancies of inbred strains of mice. This suggests that induction of robust immune-tolerance mechanisms may not be required for pregnancies in the rodent models commonly used in toxicology testing. 37 –40 Alternative rodent models in which robust immune tolerance mechanisms to the fetus are induced to sustain the pregnancy may be more representative of human pregnancy, and therefore more appropriate than the currently used outbred stock colonies of rodents. We propose that the murine allopregnancy model described subsequently may represent an alternative murine system to test potential immunologic reproductive liabilities of both small molecules and biotherapeutic CIT drugs. In cases where the on-target immune-modulating reproductive liability is confirmed, further reproductive testing in other pregnancy models, including NHPs, may not be warranted since the on-target hazard can inform the clinical liability. Furthermore, the ability to detect teratogenic and developmental effects is substantially compromised when the pregnancy failure rate is high due to failed implantations, early and late resorptions, and abortions. Conversely, if an overt liability is not identified in this model, one could then progress with confidence in the study design of a more traditional DART model, as outlined in the ICH guidance documents (ICH S5(R2); ICH S6(R1)).
Mouse Allopregnancy Model for CIT
The mating of 2 different strains of mice (eg, C57/Bl6 × CBA mice) results in a fetus that expresses paternally derived alloantigens (allogeneic pregnancy) and leads to the establishment of maternal/fetal tolerance mechanisms, which is more representative of human pregnancy. 1,2,12,14,41 Similar pathways and/or cells, such as Tregs, PD-L1, FasL, and IDO, have been reported to be involved in maternal/fetal tolerance in both mouse and humans. 1 Interestingly, the spontaneous resorption rate increases as the level of genetic diversity between the mother and father increases. For example, in syngeneic pregnancies, the spontaneous resorption rate is less than 5%, whereas in outbred stock colonies and in allopregnancy models, the spontaneous resorption rate is approximately 10% and 20%, respectively, suggesting that the level of genetic diversity between parents can impact the maternal immune system in response to the developing fetus in the absence of pharmacologic immunostimulation (Figure 1). 37,38,42 To further support this concept, it has been reported that pregnancies resulting from fertilized egg donation from unrelated human donors, where the complete fetal genome is fully allogeneic, are associated with a higher incidence of pregnancy-induced hypertension and placental pathology when compared to spontaneously conceived human pregnancies or in vitro fertilization pregnancies, where the semi-allogeneic fetus only expresses paternal alloantigens. 2
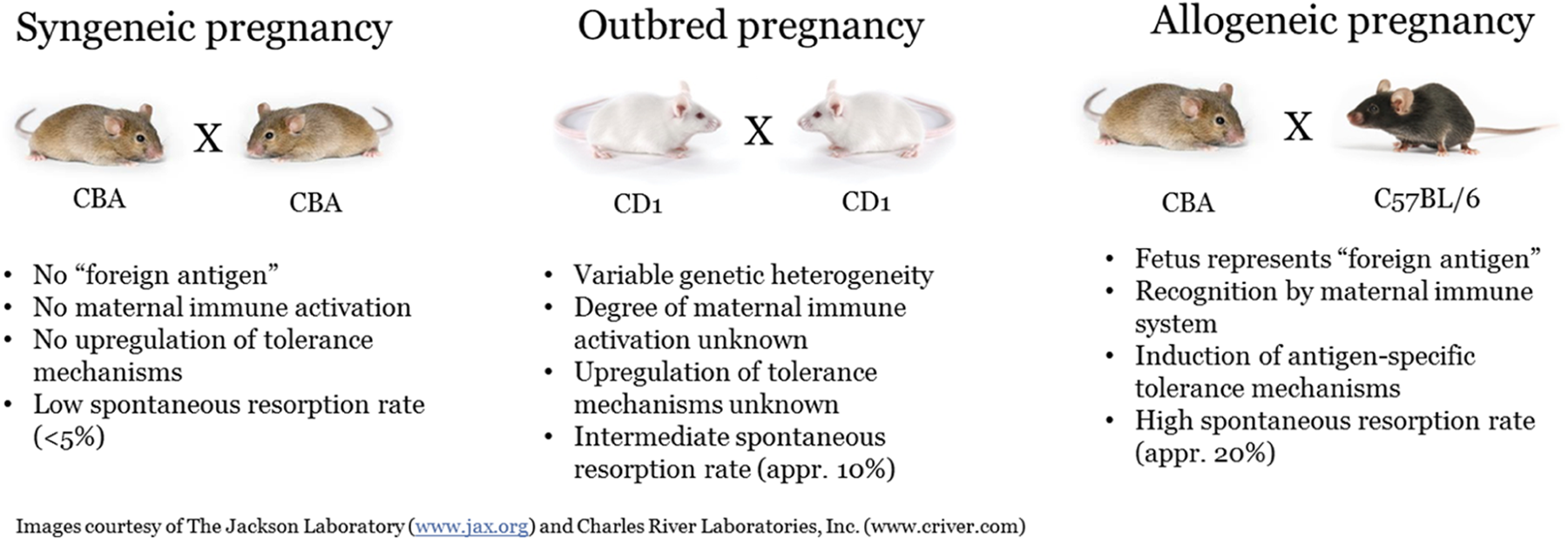
Comparison of syngeneic, outbred, and allogeneic pregnancies.
Below are 2 specific examples where the murine allopregnancy model clearly demonstrated on-target, pharmacology-dependent reproductive liabilities, whereas the syngeneic pregnancy did not identify these reproductive risks. In the first case study, a mAb directed against PD-L1 was used to demonstrate the critical role of the PD-L1/PD-1 pathway in maintaining maternal–fetal tolerance. In the second case study, a small molecule inhibitor of IDO, 1-methyltryptophan (1-MT), was used to demonstrate the involvement of the IDO pathway in maternal–fetal tolerance.
Case Study 1: PD-L1
Programed death ligand 1 is one of the 2 ligands that regulate the activity of PD-1, an inhibitory receptor that modulates T cell signaling and whose expression is induced on T cells following activation and sustained in sites of chronic stimulation. 43 –45 Ligation of PD-1 impairs the capacity of chronically activated T cells to proliferate, produce cytokines, or effectively kill target cells in response to their cognate antigen. PD-L1 is elevated in many human tumors, and its overexpression is associated with poor prognosis for patients with several epithelial cancers. 46 –50 Elevated expression of PD-L1 on tumor cells has been reported to impede antitumor immunity, resulting in immune evasion by tumor cells. PD-L1/PD-1 interactions regulate and maintain peripheral tolerance via 2 proposed mechanisms. 43,50 First, PD-1 ligands expressed on antigen presenting cells can contribute to suppression of autoreactive T cells to induce peripheral tolerance. Second, PD-1 ligands on parenchymal cells prevent tissue destruction by inhibiting T cell effector functions at peripheral sites. Conversely, inhibition of the PD-L1/PD-1 pathway enhances T cell function and increases the lethality in graft-versus-host disease and other autoimmune disease mouse models.
The critical role that the PD-L1/PD-1 pathway plays in the maintenance of feto–maternal tolerance is likely due to the unique features of the placental architecture, which is comprised of cells from both maternal and fetal origin. The placenta has a primary role in maintaining immune regulation at the maternal–fetal interface. In the placenta, PD-L1 is expressed by villous syncytiotrophoblasts and cytotrophoblasts and the fetal cells that are in close contact with the maternal blood and tissues. 14,51 –55 In humans, PD-L1 expression is low in the first trimester but rises around the onset of the second trimester when the maternal blood supply to the placenta is established. Concomitant with this placental development, the maternal immune system is constantly exposed to fetal-derived, paternally inherited alloantigens, which can be detected in the maternal spleen and lymph nodes. 12,56 The temporal and spacial expression of PD-L1 during human pregnancy suggests that this pathway plays a role in establishing antigen specific tolerance of maternal T cells to paternally derived antigens and is key in maintaining pregnancy. 14,15
Several nonclinical studies have also demonstrated that the PD-L1/PD-1 signaling pathway is essential in establishing maternal–fetal tolerance, which is essential for embryo fetal survival during gestation. 38,57 –62 Using an allogeneic pregnancy model (CBA/CaJ × C57Bl/6), Guleria and colleagues demonstrated that inhibition of the PD-L1/PD-1 pathway by administration of an anti-PD-L1 mAb increased fetal rejection rates when compared to syngeneic (CBA/CaJ × CBA/CaJ) pregnancies treated with the same anti-PD-L1 mAb. 38 The spontaneous resorption rate in CBA × CBA (syngeneic) mated mice was < 10% and was not increased following anti-PD-L1 administration. This was also reflected by the comparable fetal survival rate in both control and anti-PD-L1−treated mice (8−9 pups per female). In contrast, the spontaneous resorption rate in CBA × C57 (allogeneic) mated animals was 18% in control treated mice and was significantly increased following anti-PD-L1 administration (resorption rate increased to 86%). The increase in resorption rate was also reflected by the decreased overall fetal survival, which was approximately 5.5 and 1.2 pups per female from control or anti-PD-L1−treated CBA × C57 mice, respectively (Table 1). In contrast, anti-PD-L2 or anti-B7.2 monoclonal antibodies had no effect on the spontaneous resorption rate in the allogeneic pregnancy model, suggesting the model is sensitive to targets specifically involved in maternal–fetal tolerance. 38
Effect of Anti-PD-L1 Treatment on Fetal Survival in Syngeneic and Allogeneic Pregnancy Models.a
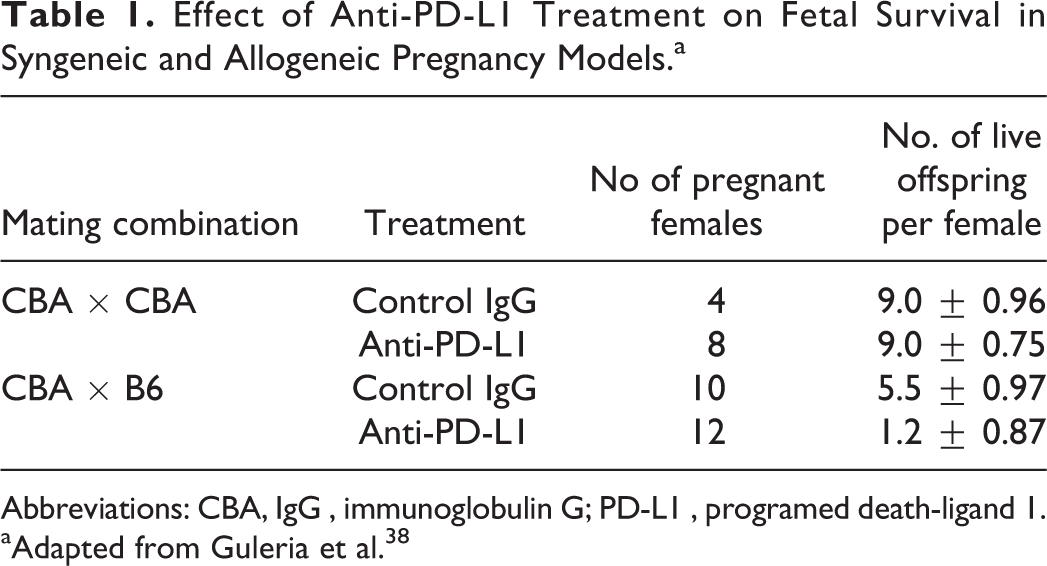
Abbreviation: CBA, IgG , immunoglobulin G; PD-L1 , programed death-ligand 1.
aAdapted from Guleria et al. 38
A negative impact on pregnancy outcomes was also noted using PD-L1-deficient females when mated to wild-type (WT) allogeneic male mice. In this instance, the number of surviving pups from allogeneic mating was 2.7 pups per female compared to 8 pups per female from syngeneic matings (Table 2). Furthermore, in a mixed leukocyte reaction assay, an increase in the expansion/frequency of interferon γ (IFN-γ)-producing lymphocytes responding to paternal alloantigens was detected in splenocytes from anti-PD-L1–treated pregnant WT mice or pregnant PD-L1-deficient females mated with allogeneic male mice. A similar increase in IFN γ production was detected in placental homogenates from PD-L1-treated or PD-L1-deficient pregnant female mice. Additional studies determined that fetal rejection was mediated via a T cell- but not B cell-dependent mechanism. 38 In the allogeneic pregnancy model, anti-PD-L1 administration caused approximately 35% to 40% fetal rejection in B cell-deficient animals, whereas PD-L1 inhibition had no effect in B cell-deficient female mice that were mated with syngeneic males. In contrast, administration of anti-PD-L1 to female RAG 1-deficient animals, which lack T and B cells, did not lead to an increase in fetal rejection when compared to RAG 1-sufficient females given anti-PD-L1. 38
Effect of PD-L1 Deficiency on Fetal Survival in Syngeneic and Allogeneic Pregnancy Models.a
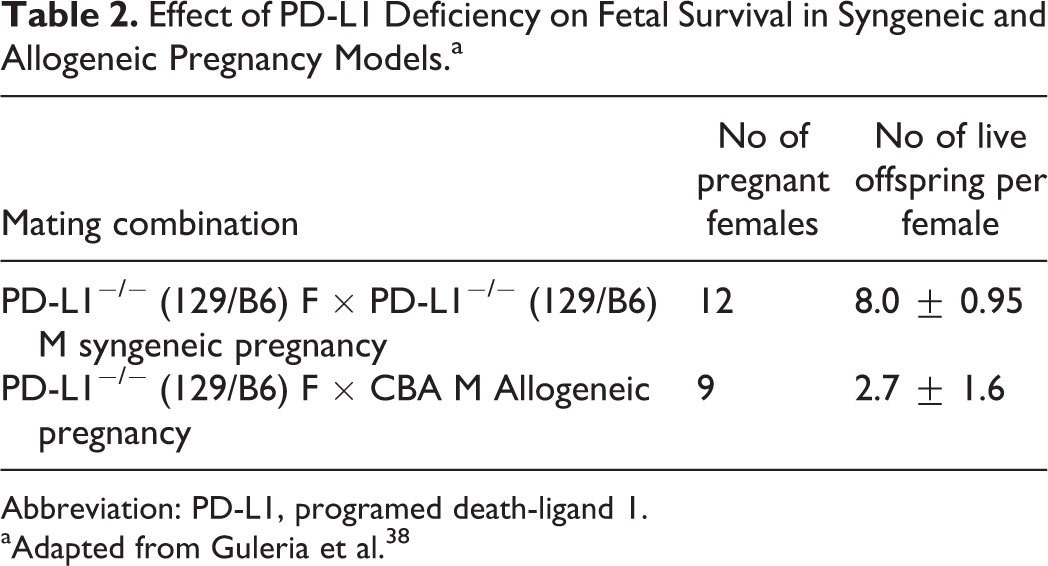
Abbreviation: PD-L1, programed death-ligand 1.
aAdapted from Guleria et al. 38
Recent studies in a murine model in which ovalbumin (OVA) is used as a model paternally derived antigen demonstrated that inhibition of the PD-L1/PD-1 axis, by means of PD-L1 or PD-1-deficient animals, resulted in an increase in activated maternal T cells that recognized paternally derived antigens expressed on the fetus. 63 In this model, male mice, which express ovalbumin under the actin promoter (ACT-mOVA Tg), are mated with WT C57Bl/6 female mice. When mated, the fetus thus inherits and expresses ovalbumin from the male, which represents a novel, fetus specific antigen in the pregnant WT female animal. To track the maternal T cell responses to fetal expressed ovalbumin, fluorescently labeled CD8+ TCR transgenic T cells (OT I T cells), which recognize a specific 8 amino acid peptide (SIINFEKL; amino acids 257-264) of ovalbumin as the cognate antigen when presented by the class I MHC molecule H 2Kb, were adoptively transferred into pregnant female mice on gestation day 10.5. Three days after adoptive transfer (gestation day 13.5), accumulation of fetal antigen specific T cells (ie, OT I T cells) was measured in the uterus draining lymph nodes and spleens of ACT-mOVA Tg mated females and compared to the frequency of OT I T cell in WT mated females. In the uterine draining lymph nodes of females mated with WT males, OT I cells were rare (< 3% of the total lymphocytes). In contrast, OT I T cells were readily detected (approximately 10% of total lymphocytes) in the uterine draining lymph nodes of females mated with ACT-mOVA Tg male mice. Furthermore, PD-1 deficient ovalbumin specific T cells (ie, PD-1−/− OT I cells) were more abundant than WT OT I T cells when transferred into females mated with ACT-mOVA Tg male mice. Accumulation of PD-1−/− OT I cells was not noted in WT female mice mated with WT (ie, lacking ovalbumin expression) male mice. Further characterization of the PD-1−/− OT-I cells from ACT-mOVA Tg mated females demonstrated that PD-1 limits accumulation of paternal antigen specific T cells through induction of apoptosis. 63 These data demonstrate that a functional PD-L1/PD-1 pathway limits maternal T cell responses to fetal antigens.
Similarly, D’Addio et al demonstrated in a separate allogeneic pregnancy model (Thy1.1 B6 female mice mated with Bm12 male mice) that PD-L1 blockade increased fetal resorption and reduced litter size. 57 In this model, anti-PD-L1 administration to previously mated Thy1.1 B6 females increased fetal resorption from 1.4% ± 10% in control treated mice up to 34% ± 10% in anti-PD-L1–treated mice. The higher resorption rate was associated with a concomitant reduction in litter size from 8.5 ± 1 pups in controls to 5.8 ± 0.4 pups in anti-PD-L1–treated mice (Table 3). To track the maternal T cell responses to fetal expressed Bm12, they adoptively transferred anti-Bm12 (ABM) Tg T cells, which specifically recognize the paternally derived alloantigen (ie, Bm12) expressed on the fetus, into Bm12 mated Thy1.1 B6 female mice. Seven days after transfer, an increase in the percentage of activated ABM Tg T effector cells was noted in the spleens of PD-L1-treated mice when compared to controls (72% ± 1.8% versus 65% ± 1.7%, respectively). Together, these data provide additional evidence that inhibition of the PD-L1/PD-1 pathway during pregnancy results in an increase in fetal antigen specific T cells in the dam, which likely contributes to the T cell-mediated fetal rejection.
Effect of Anti-PD-L1 Treatment on Fetal Resorption and Survival in Allogeneic Pregnancy.a
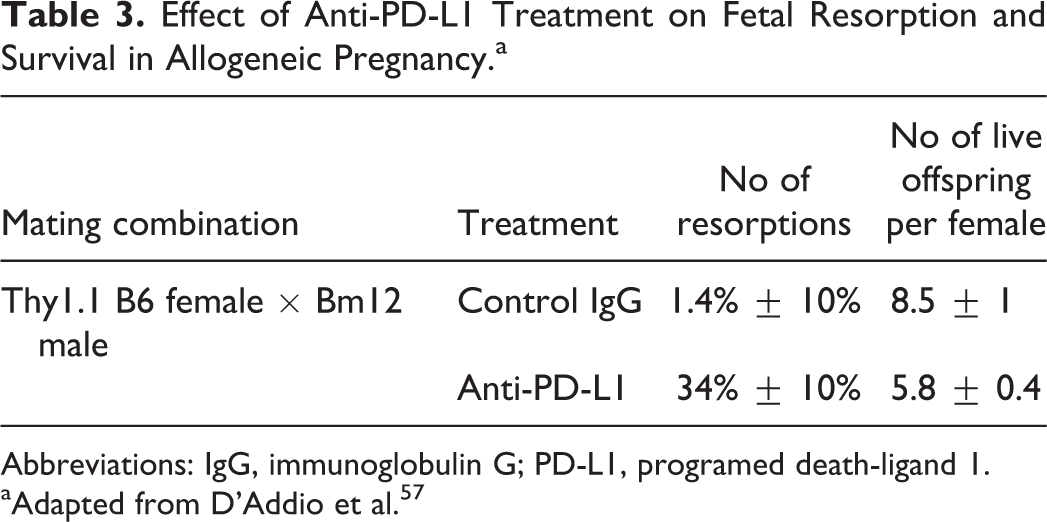
Abbreviation: IgG, immunoglobulin G; PD-L1, programed death-ligand 1.
aAdapted from D’Addio et al. 57
Inhibition of the PD-L1/PD-1 pathway has not been reported to result in teratogenic effects. Syngeneic homozygous knockout fetuses (PD-L1 or PD-1 knockouts) develop normally and have not shown skeletal or visceral defects. With regard to the potential risk on the developing offspring, several reports have demonstrated that PD-1-deficient mice and, to a lesser extent, PD-L1-deficient mice, develop strain dependent, spontaneous autoimmunity through loss of immune tolerance mechanisms. 43,44,50,64 In addition, polymorphisms in human PDCD1 (PD-1 gene) have been identified and are associated with several human autoimmune diseases including type 1 diabetes, systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, and Grave’s disease, suggesting that PD-1/PD-L interactions regulate and maintain peripheral tolerance. 64 Thus, the weight of evidence of the association between PD-L1/PD-1 inhibition and the increased risk of immune-mediated rejection of the developing fetus suggests there is a risk to the human fetus, including embryo lethality. Interestingly, administration of Opdivo (nivolumab), an Food and Drug Administration (FDA)-approved anti-PD-1 mAb, to cynomolgus monkeys from the onset of organogenesis through delivery resulted in increased abortion and premature infant death (nivolumab US FDA Summary Basis of Approval [SBA]). These data are consistent with the findings in the murine allogeneic pregnancy model and provide additional evidence that the allopregnancy model may be a relevant model to test for reproductive risks for CIT molecules. Alternatively, no nonclinical EFD or PPND studies were conducted for another FDA approved PD-1 inhibitor, Keytruda (pembrolizumab, US FDA SBA), presumably based on the evidence for the pathway reproductive liability, as summarized earlier.
Case Study 2: IDO Inhibitor
Indoleamine 2,3-dioxygenase is a rate-limiting enzyme in the catabolism of tryptophan and converts L-tryptophan into N-formylkynurenine. Indoleamine 2,3-dioxygenase 1 is expressed in a variety of cell types, especially antigen-presenting cells such as macrophages and dendritic cells in response to inflammatory stimuli. 65,66 Recently, there has been accumulating evidence from animal studies and early human clinical trials indicating that IDO is overexpressed in various tumors and dendritic cells in tumor draining lymph nodes and is critically involved in the evasion of antitumor immune responses. 67,68 Within the tumor microenvironment, IDO-mediated depletion of tryptophan and production of immunosuppressive tryptophan metabolites is thought to result in inhibition of tumor antigen-specific T cell responses and enhancement of regulatory T cell function. 69 Moreover, overexpression of IDO has been correlated with poor prognosis in patients with various carcinomas. 70 –72 As a result, currently there are a growing number of small molecule IDO-selective inhibitors in clinical development for oncology.
In addition to tumor immune tolerance, IDO has also been implicated in maternal–fetal tolerance. Since the conceptus genome is half paternal and half maternal, successful outcomes in allogeneic pregnancy depend upon regulation of maternal alloimmunity. In that capacity, IDO has been found to be highly expressed in the placenta—specifically within the synctiotropoblast cells of fetal origin—where it regulates maternal immunity against the conceptus. 1,61,73 –77 In humans, the placental activity of IDO is detectable during the first trimester and markedly increases during the course of gestation. 13,78,79 As with the tumor microenvironment, a combination of IDO-mediated tryptophan depletion and increased production tryptophan metabolites is thought to result in an immunosuppressive environment in the maternal–fetal interface thereby suppressing maternal effector T cell function.
In a series of experiments conducted by Munn and colleagues to elucidate the role of IDO at the maternal–fetal interface, pregnant mice carrying syngeneic (CBA × CBA) or allogeneic (CBA × B6) concepti were treated with 1-MT, an inhibitor of IDO, soon after implantation at 4.5 days postcoitus. 37 After approximately 4 days of treatment with 1-MT, the number of allogeneic concepti in treated females was significantly reduced, and extensive hemorrhaging was noted surrounding most of the remaining concepti. 37 After 5 to 9 days of treatment, no allogeneic concepti remained in any mice treated with 1-MT (Table 4). By contrast, neither the number nor the developmental status of syngeneic embryos was affected by exposure to 1-MT. Moreover, even a single paternally inherited MHC I alloantigen was sufficient to provoke potent maternal immunity upon IDO inhibition resulting in fetal rejection. 37 To investigate further the role of lymphocytes in fetal rejection, RAG-1−/− females carrying allogeneic concepti were exposed to 1-MT. Treatment of RAG-1−/− females with 1-MT resulted in healthy allogeneic concepti and delivery of normal litters. However, RAG-1−/− females lost all their allogeneic concepti when reconstituted with T cells specific to the paternally inherited MHC 1 alloantigen. Together, these data demonstrate that maternal T cells were essential for the rejection of allogeneic concepti upon inhibition of IDO.
Effect of 1-MT on Concepti Survival in Syngeneic and Allogeneic Pregnancy Models.a
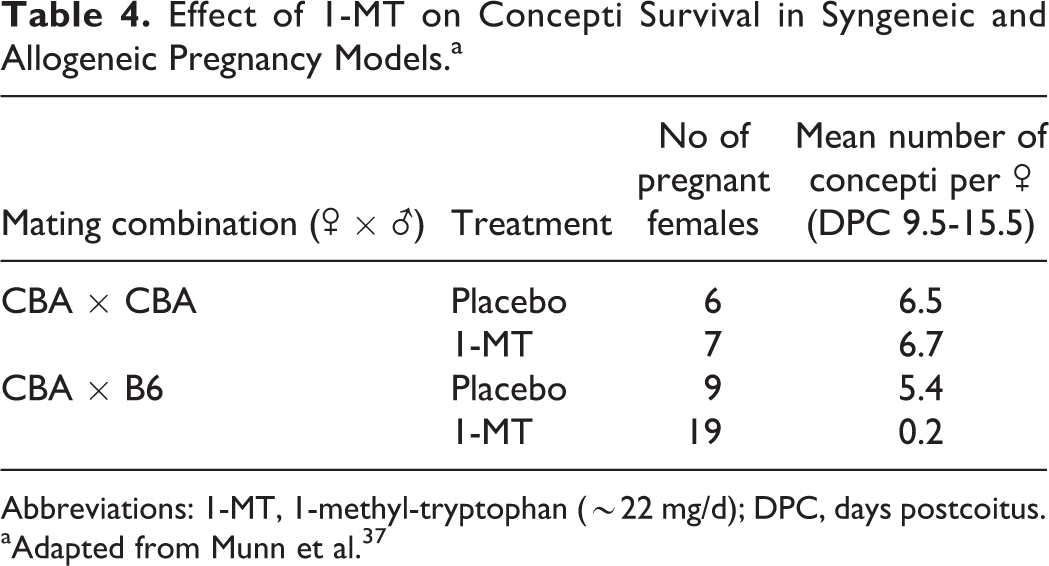
Abbreviations: 1-MT, 1-methyl-tryptophan (∼22 mg/d); DPC, days postcoitus.
aAdapted from Munn et al. 37
The same group further examined the mechanism of rejection of allogeneic concepti upon IDO inhibition. Crossing of various parental mouse strains in the presence of 1-MT revealed that (1) the degree of tissue incompatibility between parental strains determined the rate of pregnancy failure and (2) there was a clear dose–response of 1-MT with relation to concepti survival (Table 5). In addition, the mechanism of allogeneic fetal rejection upon exposure to 1-MT was accompanied by deposition of complement factor C3 and hemorrhagic necrosis at the maternal–fetal interface, and this inflammatory process was driven by T-cell recognition of fetal antigens. 42 The deposition of complement occurred despite expression of Crry, a complement inhibitory factor found at the maternal–fetal interface, suggesting that IDO inhibition during pregnancy can overcome normal inhibitory factors preventing fetal loss. Conversely, no inflammation, complement deposition, or T-cell infiltration was elicited in mice carrying syngeneic concepti exposed to 1-MT. This set of studies further established a role for IDO in the protection of the allogeneic concepti against maternal T cell-mediated local inflammatory responses and fetal rejection.
Effect of 1-MT Dose on Fetal Survival in Allogeneic Pregnancy.a
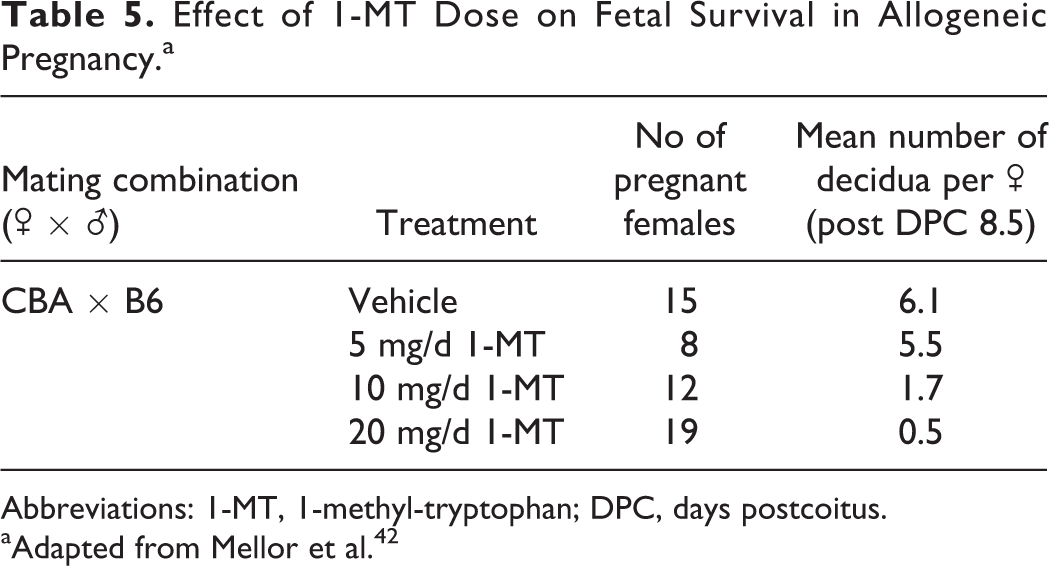
Abbreviations: 1-MT, 1-methyl-tryptophan; DPC, days postcoitus.
aAdapted from Mellor et al. 42
Given the clear function for IDO in maintenance of pregnancy in the above studies, it was quite surprising that genetic ablation of IDO did not affect pregnancy outcomes in mice. Using a variety of IDO−/− mouse crosses, Baban and colleagues demonstrated that neither allogeneic nor syngeneic pregnancy outcomes were affected by IDO deficiency, in either or both parents, relative to pregnancy rates in IDO-sufficient mice (Table 6). 74 Additionally, when IDO-deficient mice carrying allogeneic concepti were exposed to 1-MT, they also produced litters of comparable sizes and at similar rates as those exposed to placebo (Table 7). These results contrast with earlier studies showing that rejection of allogeneic concepti was dramatically increased in IDO-sufficient females exposed to 1-MT and imply that in IDO-deficient mice, other mechanisms compensate for the absence of IDO during pregnancy. It is likely that in the IDO-deficient mice, induction of immune tolerance at the maternal–fetal interface may be mediated by tryptophan dioxygenase (TDO), another enzyme involved in tryptophan catabolism. Indeed, the expression of TDO transcripts has previously been reported at the maternal–fetal interface in mouse and in human placentas. 80 –82
Pregnancy Outcomes in IDO-Sufficient and IDO-Deficient Syngeneic and Allogenic Matings.a
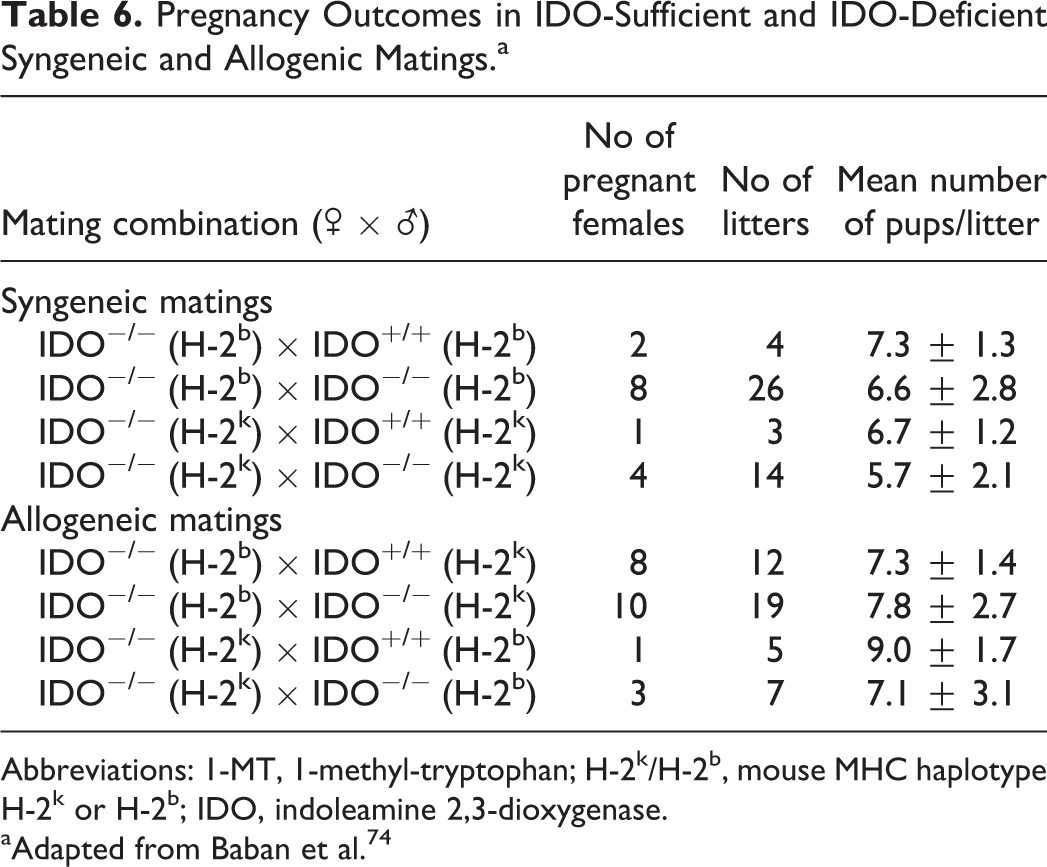
Abbreviations: 1-MT, 1-methyl-tryptophan; H-2k/H-2b, mouse MHC haplotype H-2k or H-2b; IDO, indoleamine 2,3-dioxygenase.
aAdapted from Baban et al. 74
Effect of 1-MT on Allogeneic Pregnancy Outcomes in IDO-Deficient Mice.

1-MT , 1-methyl-tryptophan; H-2k/H-2b, mouse MHC haplotype H-2 k or H-2b; IDO, indoleamine 2,3-dioxygenase.
a Adapted from Baban et al. 74
Taken together, these data indicate that in an IDO-sufficient condition, IDO function at the maternal–fetal interface is critical for protection of the concepti from maternal immune responses. Furthermore, pharmacological inhibition of IDO results in increased risk of allogeneic fetal loss via a mechanism involving activation of maternal T cells in mice. As with mice, IDO is expressed in human extravillous syncytiotrophoblast cells and is thought to have a similar role in the induction of immune tolerance at the maternal–fetal interface. 75,83 –87 Low IDO activity in human pregnancies has been linked to pre-eclampsia, a phenomenon also observed in IDO−/− mice, which may be related to induction of antifetal immune response. 88 –92
Discussion
Together, the 2 case studies described earlier provide clear evidence that a basic allopregnancy mouse model is capable of identifying reproductive hazards as a result of modulating the maternal immune system and interfering with fetomaternal tolerance mechanisms. It is also apparent that this model more closely resembles human allopregnancies, with clearly different parental phenotypes and genotypes, and can be used to interrogate both biotherapeutics and small molecule therapeutics that may be involved in maintaining maternal–fetal tolerance. Furthermore, this model appears to be very specific for defined immunomodulating pathways as demonstrated by the observation that inhibition of PD-L1/PD-1 pathway led to profound effects on fetal survival, whereas inhibition of PD-L2/PD-1 or CD28/B7.1 pathways had no effect. Unfortunately, a majority of the cancer immunotherapeutic targets have not been studied in an allopregnancy model and therefore lack published information on which to base a scientific rationale to use in lieu of additional NHP or standard rat and rabbit DART studies. We propose that conducting investigative studies in the murine allopregnancy model represents an appropriate and direct approach to identify on-target reproductive and developmental liabilities for cancer immunotherapeutic molecules. In the case of biotherapeutics, when the clinical candidate cross-reacts with the murine target, the clinical candidate can be used in this model to identify on-target liabilities. Alternatively, when the clinical candidate has restricted species reactivity, such as to humans and NHPs, a pharmacologically relevant surrogate molecule of the clinical candidate, such as the molecule used to demonstrate proof of activity in murine tumor models, could be considered to explore mechanistic relevance of the target to the pregnancy maintenance. The design of murine allopregnancy studies for CIT molecules should incorporate a subset of the standard rodent end points of embryo- and fetal-development studies, including maternal clinical signs, maternal body weights, and pathology assessments at cesarean section, potentially including number of corpora lutea, number of early and late resorptions, and number of live and dead fetuses. Other end points to assess maternal or fetal toxicity could also be considered on a “case-by-case” basis.
As highlighted above, there are potential developmental and reproductive hazards of CIT molecules secondary to the modulation of maternal–fetal tolerance; in contrast, direct effects on the fetus have not been demonstrated thus far. Therefore, standard rodent models, which have little to no maternal-versus-paternal genetic variability (ie, ranging from standard outbred laboratory stocks, such as CD1, to true syngeneic pregnancies), do not induce maternal immune activation and may not establish the maternal–fetal tolerance mechanisms due to the paucity of expression of distinct paternally derived alloantigens on the fetus. Therefore, the less genetically heterogeneous pregnancy models that are routinely used in pharmaceutical development to identify risks to the developing fetus may have limited utility for CIT molecules. Consistent with this concept, when CIT pathways are genetically manipulated, the reproductive liabilities are not highlighted as part of the knockout phenotype, as these mice are typically back-crossed to a syngeneic background prior to phenotyping. 45,64,74,93 Thus, the murine allopregnancy model represents a highly relevant, but nonstandard, rodent model in which to explore reproductive liabilities of both biotherapeutics and small molecule therapeutics targeting CIT pathways.
As demonstrated by Munn et al, mating of CBA mice with a single paternally inherited MHC I alloantigen was sufficient to provoke potent maternal immunity. 37 Therefore, mating any 2 strains of mice that are MHC I mismatched will induce maternal–fetal tolerance mechanisms following recognition of paternally derived alloantigens. Selection of specific strains of mice in which to mate could be influenced by additional factors such as the availability of genetically modified mice for the target of interest or strains that have been shown to be more responsive to therapeutic modulation of the target in proof of concept studies. Incorporation of genetically modified mice into the allopregnancy model could function as an additional approach to evaluate pathway liabilities for CIT molecules. In the 2 examples provided earlier, female C57Bl/6 mice were selected for the mating, likely due to the availability of PD-L1 knockout and IDO knockout animals on the C57BL/6 background. However, if genetically modified mice are available on different background strains of mice (ie, Balb/c), they could be tested in the allopregnancy model by mating with allo-mismatched male mice to characterize the pregnancy hazard.
Similar to the immunosuppressive tumor microenvironment, it is likely that multiple pathways are required for inducing and maintaining optimal maternal–fetal tolerance throughout the pregnancy. Individual pathways may play a more dominant role at various times during the pregnancy. This would be consistent with the observation that the number of Tregs peaks during the first trimester, suggesting a key role for Tregs in implantation or first exposure to fetal derived alloantigens, whereas PD-L1 expression peaks during the third trimester likely in response to chronic antigen stimulation. 51,94,95 However, the degree of similarity between pregnancies is likely higher than that of the tumor microenvironment since this represents a highly regulated and transient physiological process, while tumor microenvironments represent diverse dysregulated growth and differentiation that can differ between tumor types and locations. 6,9 Therefore, the degree of overlapping tolerance mechanisms between pregnancy and the tumor microenvironment has yet to be determined and the murine allopregnancy model could be a useful one in which to address this topic.
Summary/Conclusions
The advantage of the murine allopregnancy model is that it represents a relevant model that may allow for the identification of an expected, on-target pharmacological effect of CIT molecules in the drug development paradigm. If on-target immune-mediated reproductive hazards such as increased abortions and fetal losses are identified in this model, additional confirmatory studies in NHPs for biotherapeutics and rat and rabbits for small molecule therapeutics may not be warranted and additional confirmatory studies should be considered on a “case-by-case” basis.
Conversely, the absence of a liability in this model suggests that the molecular target for the CIT molecule is not uniquely critical for the maintenance of maternal–fetal tolerance. In this situation, conducting additional studies in more traditional DART models, as outlined in the ICH guidance documents [ICH S5(R2) and ICH S6(R1)], may be useful especially when there is concern for potential direct fetal effects other than immunologic pregnancy rejection. Moreover, this tiered approach is consistent with the principles of the National Centre for the Replacement, Refinement, and Reduction of Animals in Research, which was established to lead the discovery and application of new technologies and approaches that minimize the use of animals in research. 96 –98 While this approach does not eliminate animal use altogether, which still serve an important function in nonclinical safety testing, it does provide an opportunity to reduce the overall number of animals while still informing the drug label for reproductive hazards. In particular, a positive hazard identification in this model may obviate the need to conduct additional large studies, including those with pregnant NHPs.
Footnotes
Acknowledgments
The authors would like to thank Jorg Blumel, Donna Dambach, and Noel Dybdal for their critical review and insightful discussions. The opinions expressed herein are those of the authors and do not represent an official position of their employer or of the regulatory agencies.
Author Contribution
Prell, R., Halpern, W., and Rao, G. contributed to conception, contributed to interpretation, drafted the manuscript, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.