
Abstract
Nucleoside reverse transcriptase inhibitors (NRTIs)/nucleotide reverse transcriptase inhibitors are key components of combination antiretroviral therapy for HIV infection. First-generation NRTIs are associated with mitochondrial toxicity in patients, mainly due to inhibition of human DNA polymerase γ (hDNA polγ) that manifests as adverse events such as lipodystrophy, lactic acidosis, myopathy, cardiomyopathy, or nephropathy in patients. In chronic nonclinical studies in rodents and nonrodents, eukaryotic (host) mitochondrial toxicity manifests as some drug-specific toxicities similar to human toxicity. BMS-986001, a novel thymidine analog with minimal hDNA polγ inhibition, has demonstrated antiretroviral activity in early clinical studies. The primary toxicity of BMS-986001 in rats and monkeys is bone marrow dyserythropoiesis with associated decreases in red blood cell mass. Additionally, at high doses, severe platelet reductions accompanied by cutaneous petechiae began during weeks 8 and 11 in 3 of 60 monkeys in chronic toxicity studies. In a 6-month study, platelet reductions required euthanasia of the 2 affected monkeys (300 mg/kg/d) at week 14, but with dose reduction (200 mg/kg/d) remaining monkeys had no platelet changes. One affected monkey (200 mg/kg/d) in a 9-month study completed dosing and its platelet counts recovered during a 1-month recovery. Formation of platelet-bound immunoglobulin in the presence of BMS-986001, together with rapid and complete platelet recovery in the absence of BMS-986001, suggested that platelet decreases in monkeys may be immune mediated. No findings indicative of mitochondrial toxicity were observed in rats or monkeys given BMS-986001, suggesting an improved safety profile compared to marketed NRTI or tenofovir disoproxil fumarate.
Keywords
Introduction
Nucleoside reverse transcriptase inhibitors (NRTIs)/nucleotide reverse transcriptase inhibitors (NtRTIs), along with other antiretrovirals, are currently considered the backbone of combination antiretroviral therapy (cART) for the HIV infection. 1 –3 Despite their successful and widespread use in HIV treatment, mitochondrial toxicity in patients has been associated with the first-generation NRTIs such as stavudine (d4T), zidovudine (ZDV), didanosine (ddI), and zalcitabine (ddC). 4,5 Mechanisms that underlie NRTI eukaryotic mitochondrial toxicity include direct inhibition of the eukaryotic mitochondrial DNA replication enzyme (mtDNA polymerase γ [mtDNA pol γ]) without NRTI incorporation, 6 chain termination by incorporation of NRTI triphosphate (NRTI TP) into eukaryotic mitochondrial DNA by mtDNA polγ, persistence of the incorporated analogs in the mitochondrial DNA because of inefficient excision and resultant defective template mtDNA, or a combination of the above-mentioned mechanisms. 7 Inhibition of human DNA polymerase γ (hDNA polγ) or direct mitochondrial damage is clinically linked to lipodystrophy/lipohypertrophy (d4T, ddC, and ddI), 8 –10 muscle toxicity (d4T [skeletal], ZDV [skeletal and cardiac]), 11 –13 and metabolic acidosis (ddI, ddC, d4T, and ZDV) 14 –16 The molecular mechanism of peripheral neuropathy seen with NRTIs (ddI, ddC, and d4T) 17,18 is unclear. Due to toxicity, many first-generation NRTIs are no longer the first choice as HIV therapy. Tenofovir disoproxil fumarate (TDF), a first-line, second-generation NtRTI, has a higher selectivity toward hDNA polγ and greatly improved mitochondrial toxicity profile compared to the first-generation NRTI. 19 Nevertheless, mitochondrial toxicity seen in kidney proximal tubule (PT) cells in patients treated with TDF is associated with acute and chronic renal toxicity and, in some cases, irreversible renal failure. 20 –24 Therefore, there is a need for improved NRTIs/NtRTIs that exhibit potent anti-HIV activity, a high barrier to HIV drug resistance and an improved toxicity profile.
In nonclinical studies, first-generation NRTIs have shown notable mitochondrial toxicity, including reduction in mitochondrial DNA content and increased lactate production in multiple cell types such as hepatocytes, preadipocytes, muscle, and neuronal cells in vitro, 25 with in vivo nonclinical toxicities including (1) d4T-induced hepatic changes characterized by increased liver weight, centrilobular hypertrophy, and increased foci of cytologic alterations 26 ; (2) ZDV-induced striated muscle toxicity (including cardiotoxicity) in both rats and monkeys 27 ; (3) ddC- and ddI-induced gastrointestinal (GI) and hepatic toxicity in rats and dogs 28,29 ; and (4) ddC-induced microscopic changes in various organs and tissues (including lungs) and hemorrhage in multiple organs. 28 Hepatic, GI, and muscle toxicities are attributed to inhibition of mtDNA polγ enzyme, as well as mitochondrial DNA synthesis, due to chain termination by incorporation of NRTI-TP into mitochondrial DNA by the enzyme. Similar to the clinical renal findings with TDF (Fanconi syndrome or acute kidney injury), mitochondrial toxicity in PT epithelial cells is a frequent presentation in nonclinical studies with TDF in vitro (reduction in mitochondrial DNA content and transmission electron microscopic [EM] changes in renal PT epithelial cells [RPTECs]) 30 and in vivo (increased oxidative stress and mitochondrial damage confirmed by EM, renal tubular necrosis, and increased excretion of low-molecular weight proteins in the urine). 31,32
BMS-986001, a thymidine analog, is a next-generation NRTI, with potent antiviral activity against several HIV-1 subtypes in vitro, reduced inhibition of hDNA polγ, and an improved genetic resistance profile when compared with the first-generation NRTI. 33,34 Since DNA polγ is the only DNA polymerase in eukaryotic mitochondria, selectivity toward this polymerase is crucial for the maintenance and integrity of eukaryotic mitochondrial DNA and improvement in the safety profile of potential therapeutic agents in patients. Compared to d4T (inhibition constant [Ki] 1 μmol/L), BMS-986001 is 100× less potent in inhibiting eukaryotic, specifically hDNA polγ (Ki 100 μmol/L). Consistent with this improved selectivity profile toward hDNA polγ, BMS-986001 showed the least mitochondrial toxicity (reduction in DNA content and increased adenosine triphosphate synthesis and lactate production) in HepG2 cells and in primary cultures of human muscle, preadipocytes, and RPTECs). 35 Although TDF is also selective for hDNA polγ (K i 59.5 μmol/L) compared to the earlier NRTI, TDF is transported by organic anion transporter 1 and actively accumulates in RPTECs, leading to concentrations that result in depletion of eukaryotic mitochondrial DNA and kidney toxicity (mitochondrial damage, increased kidney weight, and microscopic tubular epithelial changes). A similar mechanism of host mitochondrial toxicity is also seen with TDF in nonclinical rodent and nonrodent studies. 31,36
To support clinical development, the toxicity and toxicokinetic profile of BMS-986001 was characterized following oral administration in Sprague Dawley (SD) rats for 6 months and in cynomolgus monkeys for 6 and 9 months. The choice of the rodent and nonrodent species for the pivotal studies was based on their use in previous toxicity studies of shorter duration (≤3 months) and comparable metabolism and disposition of BMS-986001 to humans. BMS-986001 does not undergo any discernible oxidative or conjugative metabolism in rats, monkeys, or humans, and there are no unique human metabolites. Similar to other NRTIs, BMS-986001 is phosphorylated to its active (antiviral activity) TP (BMS-986001 TP) form by intracellular kinases. Therefore, exposure to BMS-986001 and BMS-986001 TP was characterized in rats and monkeys. BMS-986001 presented a generally favorable nonclinical safety profile compared to first-generation NRTIs and TDF in chronic studies.
Methods and Materials
Compound and Formulation
BMS-986001 was formulated for dosing in a vehicle and carrier of aqueous 0.5% (wt/vol) methylcellulose (4000 cPs).
Animals and Duration of Studies
Rats, Crl: CD(SD), 8 weeks of age and weighing 120 to 210 g (males) or 100 to 190 g (females) at the start of dosing were obtained from Charles River (St Constant, Quebec, Canada). Cynomolgus monkeys (Vietnamese origin) were 2 to 3.6 years of age, weighing ∼1.5 to 3.0 kg at the start of dosing, and were obtained from Covance Research Products Inc, (Alice, Texas). Rats were used in a 6-month study (1-month recovery) and monkeys were used in 6- and 9-month (1-month recovery) studies. All studies were conducted in compliance with the US Food and Drug Administration Good Laboratory Practice regulations (21 CFR Part 58), the Animal Welfare Act on Good Laboratory Practice (ENV/MC/CHEM [98]17), and in accordance with applicable standard operating procedures. Protocols were reviewed by the Institutional Animal Care and Use Committees of the relevant facilities.
Randomization and Dosing
Male and female rats were separately assigned by body weight randomization to 4 treatment groups of 25 rats/sex/group. Following randomization, rats were given a unique identification number using an implanted chip (Bio Medic Data Systems, Inc, Plexx, the Netherlands). In all, 20 rats/sex/group were assigned for end of dose (EOD; week 26) evaluations, and 5 rats/sex/group were assigned to a 1-month recovery (EOR; week 30) evaluation. Following a minimum acclimation period of 14 days, rats were administered BMS-986001 by oral gavage once daily at doses of 0 (vehicle control), 50, 100, or 300 mg/kg/d for 6 months. Area under the concentration time curve (AUC) margins at the low, intermediate for consistency with other portions of the manuscript and high doses were approximately 2×, 4×, and 21×, respectively, the projected exposure at the recommended human dose (RHD; 400 mg).
In the 6-month study, monkeys were randomized to 4 treatment groups of 4 monkeys/sex/group, stratified by body weight as explained earlier. All monkeys were assigned to EOD evaluations (week 26). Following a 6-week acclimation period, monkeys received BMS-986001 by oral gavage once daily at doses of 0 (vehicle control), 50, 100, or 300 mg/kg/d for 6 months. The 300 mg/kg/d dose was reduced to 200 mg/kg/d starting at week 15 of the dosing period, following euthanasia of 2 females (week 14) due to severe platelet reductions. In the 9-month study, monkeys were randomized to 4 treatment groups of 6 monkeys/sex/group, stratified by body weight. Four monkeys/sex/group were assigned to EOD evaluations (week 39) and 2 monkeys/sex/group were assigned to a 1-month EOR (week 43) evaluation. Following a 6-week acclimation period, monkeys received BMS-986001 by oral gavage once daily at doses of 0 (vehicle control), 50, 100, or 200 mg/kg/d for 9 months. The AUC margins at the low, mid, and high doses in the 9-month study were approximately 2.5×, 7×, and 20×, respectively, the projected exposure at the RHD (400 mg).
Toxicokinetic Evaluation
Blood samples were collected in EDTA tubes from 3 rats/sex/dose/time point on day 1 and during weeks 13 and 26, and from all monkeys on day 1 and during weeks 26 (6-month study), and 39 (9-month study) at 0.5, 1.0, 2.0, 4.0, 8.0, and 24 hours postdose. Blood samples were centrifuged within 1 hour of collection at 4°C and approximately 1200g. Following centrifugation, plasma was collected and stored at −20°C until analysis. For the determination of BMS-986001 concentration, plasma samples were extracted using a 50-μL aliquot volume and a solid-phase extraction (SPE) procedure (Waters Oasis HLB 96-well plates, Waters Corporation, Milford, Massachusetts). The SPE plate was conditioned with 450 µL of methanol, followed by 450 µL of a 10 mmol/L ammonium acetate buffer solution. Samples and the internal standard (d4T) were added at a concentration of 500 ng/mL and were diluted with 400 µL of 10 mmol/L ammonium acetate before applying to the plate. The plate was subsequently washed with 450 µL of water. The analyte and the internal standard were eluted with 500 µL of methanol. All solutions were passed through the plate by applying vacuum using a Tecan Genesis Liquid Handler (Tecan US, Durham, North Carolina). The samples were evaporated to dryness and reconstituted in 200 µL of methanol–water (10:90, v/v) prior to analysis by liquid chromatography-tandem mass spectrometry (LC/MS/MS). Chromatographic separation was achieved at room temperature using an Atlantis dC18, 3 µ, 50 × 2.1 mm analytical column (Waters Corporation). Mobile phase A consisted of 10 mmol/L ammonium formate and 0.1% formic acid in water. Mobile phase B consisted of methanol. BMS-986001 and the internal standard were eluted from the column using a flow rate of 0.4 mL/min with a gradient of 10% to 50% mobile phase B over 1.9 minutes. The total cycle time was 3 minutes. An API 4000 (AB Sciex, Framingham, Massachusetts) was operated in the Selected Reaction Monitoring mode under optimized conditions for detection of BMS-986001 and internal standard positive ions formed by electrospray ionization. BMS-986001 concentrations were calculated with a 1/x 2 linear regression using a concentration range of 5 to 5000 ng/mL.
Toxicokinetic parameters of BMS-986001 were calculated from the plasma concentration–time profiles using the eToolbox/Kinetica pharmacokinetic software (Thermo Fisher Scientific, Philadelphia, Pennsylvania). Maximum plasma concentration (Cmax), time to Cmax (Tmax), T-half and the AUC over a 24-hour period (AUC [0-24 hours]) were reported.
Assessment of BMS-986001 Metabolite
Steady state intracellular levels of BMS-986001 TP were assessed in peripheral blood mononuclear cells (PBMCs) isolated from monkeys in the 9-month study. 37 The PBMCs were isolated from blood samples collected during week 34 at 0, 4, 12, and 24 hours postdose using a standard Ficoll Paque gradient density method, and cell pellets were resuspended in 70% methanol. Extraction was performed using organic precipitation. Specifically, 25-µL aliquot of standards, quality control, and study samples were pipetted into a 96-well plate; 50 µL of internal standard working solution was added to each sample followed by an addition of 50 µL of methanol. After mixing and centrifugation, the supernatant was transferred to a new plate and dried under nitrogen. The final extracts were reconstituted in 100 µL of water containing 2% dimethyhexylamine and 1% acetic acid in water and stored at 4°C in the autosampler before injection. BMS-986001 TP levels were analyzed by LC/MS/MS and data were reported as TP (ng) per million (106) cells as described in detail previously. 37 To be consistent with the clinical protocol, no phosphatase inhibitors (to inhibit endogenous phosphatase activity) were used in the determination of BMS-986001 TP in monkey PBMCs; TP levels are therefore variable and may represent underestimated values.
Assessment of Toxicity
Parameters evaluated were survival, clinical observations, body weight, food consumption, physical and ophthalmological examinations, clinical pathology, organ weights, and gross and microscopic pathology. Clinical examinations were carried out daily. Body weight and food consumption were measured weekly. A thorough physical examination was carried out prior to initiation of dosing and at EOD and EOR. Ophthalmology examinations included funduscopic (indirect ophthalmoscopy in rats and direct and indirect ophthalmoscopy in monkeys) and biomicroscopic (slit lamp) examinations once prior to dosing and at EOD. Electrocardiography (ECG) tracings were conducted in monkeys, once prior to the first day of dosing and again at 2 hours (±1 hour) after a daily dose during weeks 25 (6-month study) and 38 (9-month study). The ECG recordings were measured on monkeys restrained in a sling using limb leads I, II, and III, and the augmented limb leads (right [aVR], left [aVL], and foot [aVF]). Tracings were evaluated (qualitatively and quantitatively) by a consultant board-certified cardiologist. All waveforms were qualitatively evaluated to detect rhythm or conduction disturbances, including verifying normal PR and QRS intervals. Quantitative evaluation consisted of the manual measurement of the heart rate (HR), PR, QRS, and QT intervals and the calculation of Fridericia HR-corrected QT interval. Arterial oxygen saturation was measured in monkeys by pulse oximetry once prior to dosing and once after a daily dose during weeks 1, 11/12, 25/26 (6-month study), and 37 to 38 (9-month study). Hematology, coagulation, clinical chemistry, and urinalysis evaluations were conducted for rats at the end of weeks 4/5, 12/13, EOD, and at EOR, and for monkeys prior to dosing and during weeks 4, 11/12, and EOD (both studies), and EOR (9-month study).
Clinical pathology
Blood was collected from the jugular vein for rats and the femoral vein for monkeys. Blood coagulation evaluations were conducted in conjunction with scheduled necropsies for both rats and monkeys; blood samples for coagulation evaluations were collected from the abdominal aorta following isoflurane anesthesia in rats. Food, but not water, was removed overnight prior to scheduled blood and urine collection. Urine samples were collected overnight (approximately 18 hours on ice) and stored in a refrigerator at 4°C (when analyzed on the same day) or in a freezer at 20°C for future analysis. Hematology parameters measured using an ADVIA 120 analyzer (Seimens; Canada) included erythrocyte count, hemoglobin (Hb), hematocrit (Ht), red cell distribution width (RDW), mean corpuscular volume (MCV), mean corpuscular Hb (MCH), MCH concentration, platelet count (light scatter), mean platelet volume, absolute reticulocyte count (RETIC), and absolute total and differential leukocyte counts. Cell morphology was evaluated by microscopic examination and scores were assigned: negative (not observed), 1+ (trace), 2+ (few), 3+ (moderate), or 4+ (severe). Coagulation parameters (turbidometric assay) measured using an ACL advance analyzer (Beckman Coulter; Canada) included prothrombin time, activated partial thromboplastin time, and plasma fibrinogen. Serum chemistry parameters measured using a Modular Analytics analyzer (Roche, Canada) included aspartate aminotransferase, alanine aminotransferase, γ-glutamyl transferase, blood urea nitrogen (BUN), alkaline phosphatase, creatinine, total bilirubin, conjugated and unconjugated bilirubin, total protein, albumin (A), globulins (G), A/G ratio, total cholesterol, triglyceride, glucose, calcium, sodium, phosphorus, potassium, chloride, and bicarbonate. Urinalysis was conducted by automated reflectance using a Clinitek ATLAS instrument (Siemens, Canada). Urinalysis end points included volume, color, appearance, specific gravity, glucose, bilirubin, specific gravity, blood, pH, ketone, urobilinogen, protein, total protein output, and centrifuged deposits (microscopy). Serum thrombopoietin (TPO) levels were determined in the 6-month (weeks 18 and 26) and 9-month (weeks 29 and 38) monkey studies using a monkey cross-reactive human enzyme-linked immunosorbent assay (ELISA) assay (Rules Based Medicine, Texas). The lower limits of detection for the assay was 1.13 ng/mL. Urinary biomarkers included β2 microglobulin, calbindin D28, clusterin, neutrophil gelatinase-associated lipocalin (NGAL), and creatinine; evaluations were carried out using rat- and human- (cynomolgus monkey cross reactive) specific multiplex ELISA assays (Rules Based Medicine).
Euthanasia and anatomic pathology (organ weights, gross, and microscopic examination)
Rats and monkeys were fasted overnight before scheduled necropsy and terminal body weights were recorded. Rats under isoflurane anesthesia were euthanized by exsanguination, and blood was collected from abdominal aorta. A similar proportion of rats from each group and sex were euthanized on any one day and a complete gross pathology examination of the carcass was conducted immediately. Ketamine HCl was administered by intramuscular injection to monkeys before transport to the necropsy area. Next monkeys were anesthetized under isoflurane and euthanized by intravenous injection of sodium pentobarbital, followed by exsanguination by incision of the axillary or femoral arteries. Monkeys were subjected to a complete necropsy examination including evaluation of the carcass and musculoskeletal system; all external surfaces and orifices and cranial cavity and external surfaces of the brain, thoracic, abdominal, and pelvic cavities along with the associated organs and tissues. Pathology evaluations included assessments of organ weights (at EOD and EOR only) and gross examinations. A standard list of protocol-specified organs and tissues were collected and evaluated. Representative samples of each organ and tissue (except eyes and testes) and all gross lesions were fixed in 10% neutral-buffered formalin. Eyes and testes were fixed in Davidson fluid and modified Davidson fluid, respectively. Tissues and gross lesions were processed and embedded in paraffin. For rats, all tissues from the control and high-dose groups, potential target organs from low- and intermediate-dose groups and gross lesions from all groups; and for monkeys, all tissues from all dose groups were sectioned, stained with hematoxylin and eosin, and examined by light microscopy. Histopathology data were subjected to an additional peer review to confirm findings.
Direct platelet-associated immunoglobulin
In the 6-month study, 2 female monkeys given 300 mg/kg/d were euthanized because of severe platelet reductions during weeks 13 and 14. To address the possible cause of platelet reduction, serum antiplatelet antibody was evaluated (ELISA assay). Two high-binding, 96-well plates were coated with a pool of platelets isolated from naive monkey donor blood and immobilized at densities of ∼1 × 106 platelets/mL and ∼5 × 106 platelets/mL. Following overnight incubation, the platelet-coated plates were blocked with binding buffer (10% milk, 0.1% bovine serum albumin, and 0.05% Tween) to reduce nonspecific binding. Serum samples collected from the affected monkeys were added to the plates to capture any free antiplatelet antibodies. Unbound antibody was washed away before the secondary detection antibody (antihuman immunoglobulin [Ig] G conjugated with horseradish peroxidase [HRP]) was added. Detection reagent used for color development was the chromogenic substrate 3, 3′, 5, 5′-tetramethylbenzidine and HRP. To confirm that the absence of positive signal was not due to the absence of platelets on the surface of the plates, control wells were analyzed with mouse antihuman CD61 and antimouse IgG HRP antibodies.
Platelet-bound platelet-associated immunoglobulin
In the 9-month study, additional investigational analysis of platelet-associated immunoglobulin (PAIg) was completed to determine the potential cause of the platelet reduction observed in 1 female monkey between weeks 11 and 38. During weeks 16, 24, and 29, blood samples were collected from all groups for total platelet-bound PAIg assessment by flow cytometry (fluorescence-activated cell sorting [FACS]) using a FACS Calibur instrument (Becton Dickenson, California). During weeks 24 and 29, additional isotype analyses (IgM, IgG, and IgA) were conducted using platelets from the affected monkey. The PAIg assessment was conducted per THROMBOCYTEST immune kit instructions (Glycotope Biotechnology, Germany). Briefly, on assay day 1, platelet-rich plasma was prepared from whole blood. The isolated platelets were washed 4 times with wash buffer and left overnight at 4°C in the same buffer. On assay day 2, platelet counts were adjusted to ≤ 20 000 platelets/μL (suspended in wash buffer) and incubated with goat serum to prevent false-positive signal due to Fc binding. For detection of PAIg, the Fc-blocked platelet suspension was sequentially incubated with an antihuman Ig phycoerythrin (PE) antibody, washed, and then incubated with an anti-CD42a FITC antibody (CD42a[GP1b/IX]: platelet surface glycoprotein marker). The negative control tube received antirabbit Ig PE alone. In addition to evaluation of platelet-bound PAIg, circulating antiplatelet antibody was also measured in the serum. Data collection for platelet-bound Ig assessments was performed using FACS and CellQuest Pro software, version 4.0.2. (Becton Dickenson).
Statistical Analysis
For comparison between groups where N ≥ 3, mean and standard deviations were calculated. Group means for individual parameters, separated by sex, were compared between groups treated with BMS-986001 or vehicle by Dunnett t-test, using a 1-way analysis of variance model. For each pairwise comparison, statistically significant differences of P ≤ 0.05 and P ≤ 0.01 were reported.
Results
Toxicokinetic Summary of BMS-986001
In rats, mean BMS-986001 AUC (0-24 hours) values on day 1 and in week 26 increased dose proportionally from 50 to 100 mg/kg/d and greater than dose proportionally from 100 to 300 mg/kg/d (Table 1). In monkeys, mean BMS-986001 AUC (0-24 hours) values at weeks 26 (6-month study) and 39 (9-month study) increased in a greater than dose proportional manner across all doses from 50 to 300/200 mg/kg/d in the 6-month study (Table 2) and from 50 to 200 mg/kg/d in the 9-month study (Table 3). After repeated dosing for both species, AUC values of BMS-986001 were similar to day 1, indicating no accumulation or loss of exposure to BMS-986001.
Toxicokinetic Summary of BMS-986001 in a 6-Month Study Conducted in Rats.
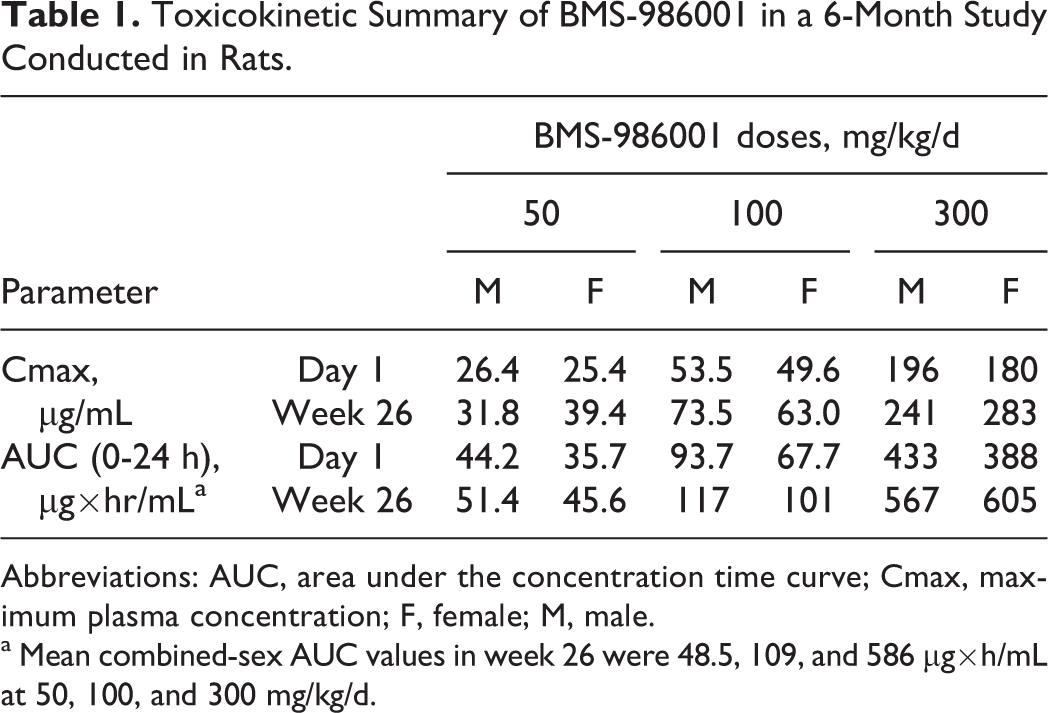
Abbreviations: AUC, area under the concentration time curve; Cmax, maximum plasma concentration; F, female; M, male.
a Mean combined-sex AUC values in week 26 were 48.5, 109, and 586 μg×h/mL at 50, 100, and 300 mg/kg/d.
Toxicokinetic Summary of BMS-986001 in a 6-Month Study Conducted in Cynomolgus Monkeys.a
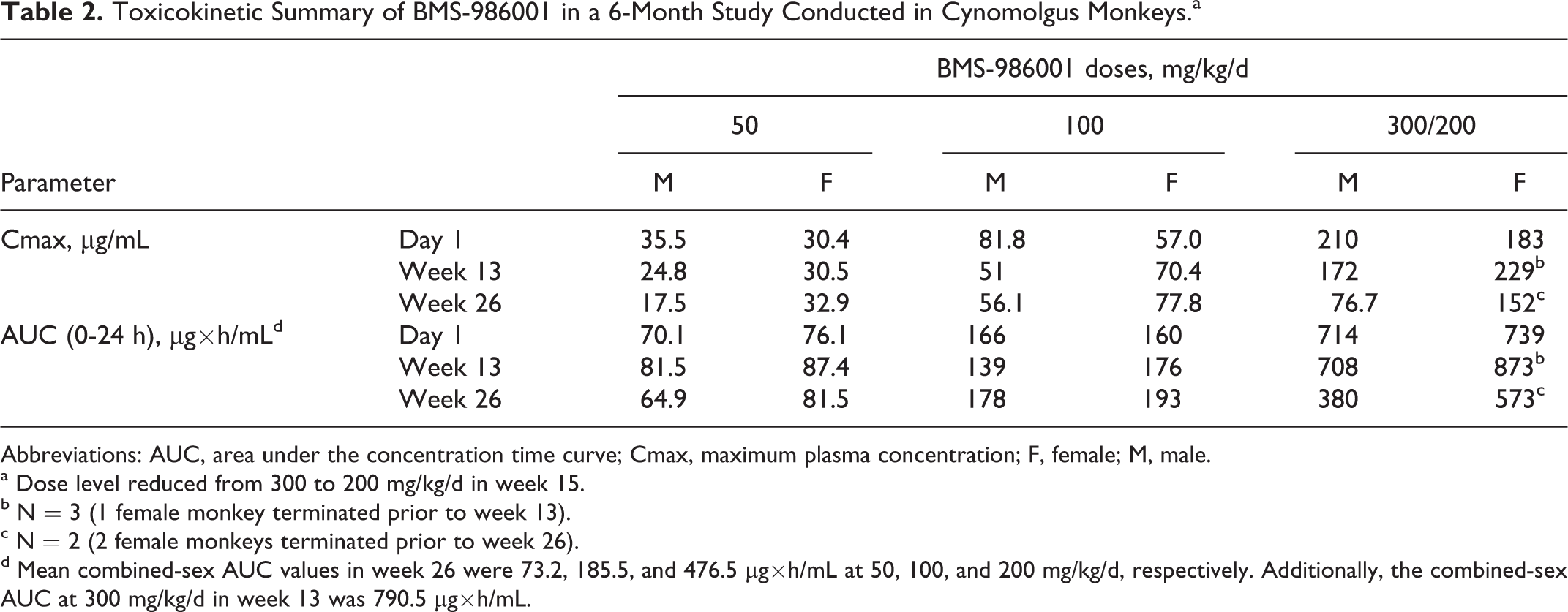
Abbreviations: AUC, area under the concentration time curve; Cmax, maximum plasma concentration; F, female; M, male.
a Dose level reduced from 300 to 200 mg/kg/d in week 15.
b N = 3 (1 female monkey terminated prior to week 13).
c N = 2 (2 female monkeys terminated prior to week 26).
d Mean combined-sex AUC values in week 26 were 73.2, 185.5, and 476.5 μg×h/mL at 50, 100, and 200 mg/kg/d, respectively. Additionally, the combined-sex AUC at 300 mg/kg/d in week 13 was 790.5 μg×h/mL.
Toxicokinetic Summary of BMS-986001 in a 9-Month Study Conducted in Cynomolgus Monkeys.
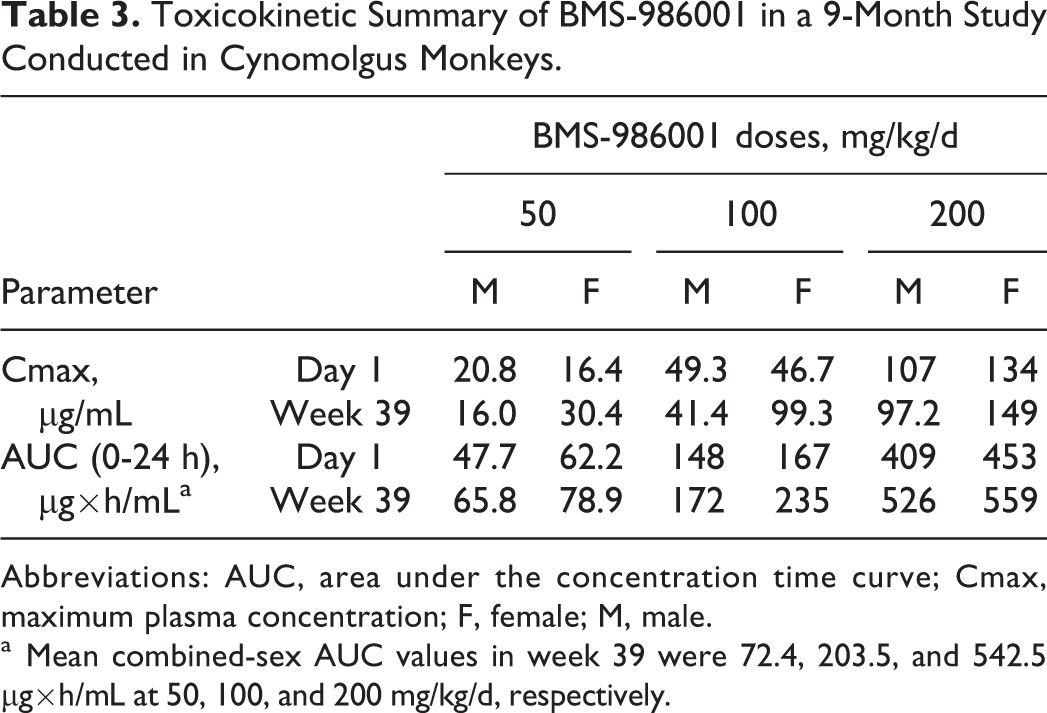
Abbreviations: AUC, area under the concentration time curve; Cmax, maximum plasma concentration; F, female; M, male.
a Mean combined-sex AUC values in week 39 were 72.4, 203.5, and 542.5 µg×h/mL at 50, 100, and 200 mg/kg/d, respectively.
BMS-986001-TP Metabolite Analyses in Monkey PBMCs
BMS-986001-TP was demonstrated in rat PBMCs following administration of a single dose of 50 mg/kg[14C] BMS-986001 in a previous metabolism study (data not shown). BMS-986001-TP was measured in monkey PBMCs during week 34 in the 9-month study (Table 4). The concentrations of BMS-986001 TP in PBMCs of monkeys receiving 200 mg/kg/d ranged from 30 to 120 pg/106 cells. The BMS-986001-TP values at the 4, 12, and 24 hours postdose were variable, most probably due to the absence of a phosphatase inhibitor (stabilizing agent for BMS-986001-TP moiety) during the PBMCs collection. At ≤ 100 mg/kg/d, BMS-986001-TP concentration in PBMCs was below the lower limit of quantification (LLOQ). The presence of BMS-986001-TP in the PBMCs of both rats and monkeys confirmed that these were appropriate nonclinical toxicology species, as BMS-986001-TP is also formed in humans.
Summary of BMS-986001 (Plasma) and BMS-986001-TP (PBMC) Levels in Cynomolgus Monkeys at 200 mg/kg/d during Week 34 in the 9-Month Study.a

Abbreviations: F, female; M, male; PBMCs, peripheral blood mononuclear cells.
a Values represent the mean concentrations; N = 6 for M and 5 for F.
b Time points represent plasma and PBMC collections at 4, 12, and 24 hours postdose during week 34.
Toxicity Profile of BMS-986001 in SD Rats
BMS-986001 was clinically well tolerated at ≤300 mg/kg/d (AUC ≤ 586 μg/h/mL) for ≤6 months, and there were no drug-related effects on survival, body weight, ocular or physical evaluations, coagulation, clinical chemistry and urinalysis parameters, organ weights, or gross pathology findings.
BMS-986001-related clinical findings were limited to mild (∼8%) reductions in food consumption and body weight gain at 300 mg/kg/d. The main hematology findings considered adverse at 300 mg/kg/d (compared to the vehicle control) included mild to moderate increase in dyserythropoiesis in the bone marrow (BM; characterized by the presence of erythroid precursors containing a mature cytoplasm and immature and/or misshapen/fragmented nuclei, Figure 1), resulting in minimal decreases in the BM myeloid to erythroid (M:E) ratio and a reciprocal decrease in red blood cell (RBC) mass (Table 5). The decreased M:E ratios reflected a predominance of erythroid precursors, which without any evidence of concurrent increases in RETICs indicated ineffective erythropoiesis. The RBC changes (300 mg/kg/d) at week 26 (Table 5; Figure 2A and B) included decreases (22%) in RBC count, Hb, and Ht and compensatory increases in MCV, MCH, and RDW. Minimal dyserythropoiesis, decreases in M:E ratios, and decreases in RBC mass (10%) were also seen at 100 mg/kg/d (Table 5). The hematologic changes at 300 mg/kg/d were considered adverse because the changes in RBC mass was >10% and the severity of the BM changes was generally mild to moderate; whereas, decreases in RBC mass at 100 mg/kg/d were not adverse because they are <10%. The BM and hematologic changes were reversible after the 1-month recovery period with the exception of continued, minimal decreases in RBC (≤ 7%) and increases in MCV (≤ 5%) at 300 mg/kg/d. There were no drug-related gross pathologic findings or organ weight changes in the 6-month rat study. Microscopic changes were limited to increased extramedullary hematopoiesis in the spleen (minimal to moderate) and liver (minimal) at all doses that were not considered adverse but rather an adaptive response to the BM alterations and the associated hematologic changes. All other pathologic changes were low in incidence and severity and commonly reported as background findings, randomly distributed between the groups (including vehicle group) and therefore not considered to be attributed to BMS-986001. The no observed adverse effect level (NOAEL) dosage for ≤6 months in rats was 100 mg/kg/d (AUC ≤ 109 μg×h/mL; AUC margin ∼4× to the exposure at the RHD).
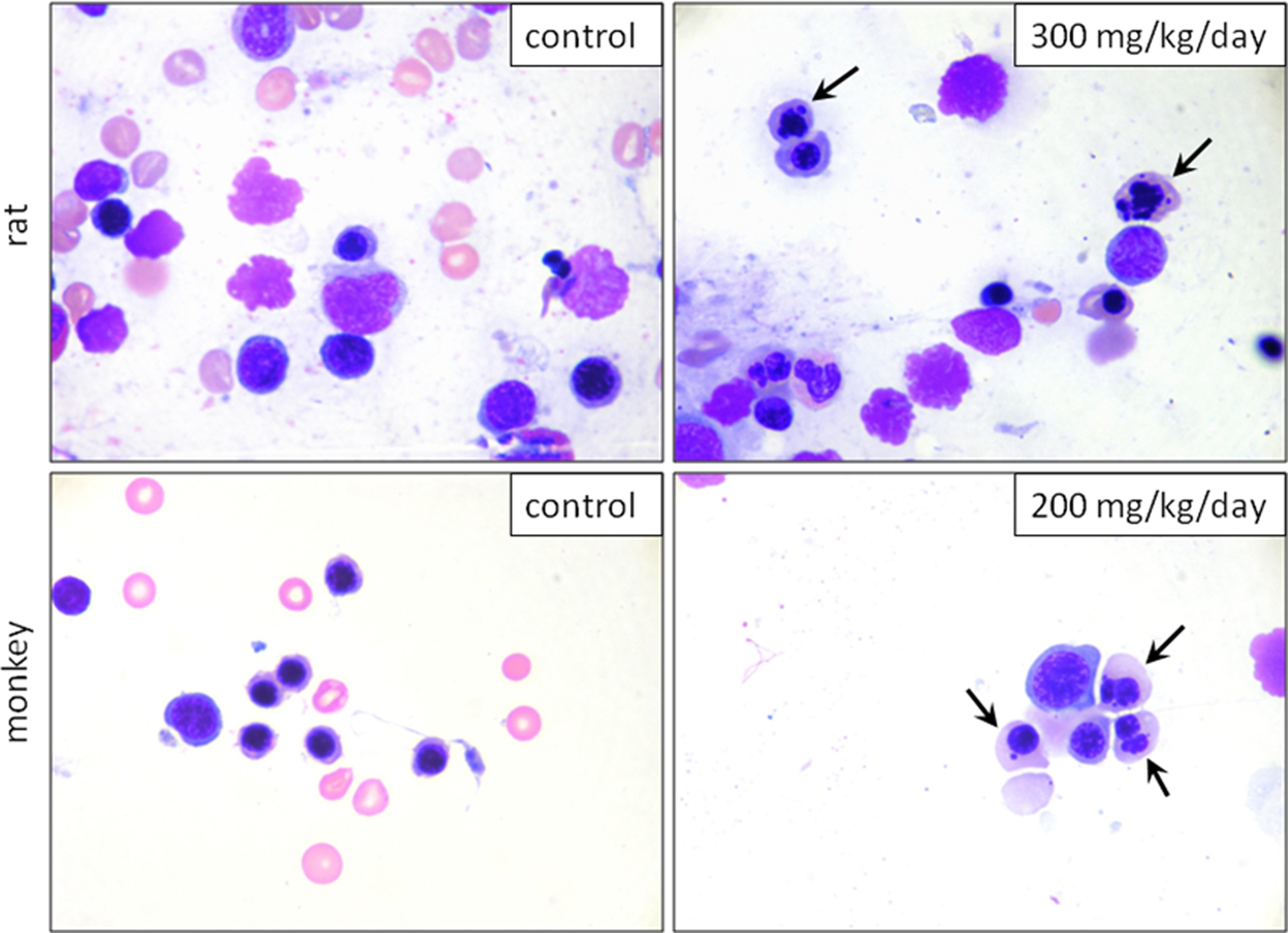
Representative photomicrographs of femoral bone marrow smears at the end of dose (EOD) from rats (week 26) and monkeys (week 39). Arrows indicate erythrocyte precursors with mature cytoplasm but misshapen, lobulated, or fragmented nuclei characteristic of BMS-986001-related dyserythropoiesis. Wright-Giemsa stain, 1000× magnification.
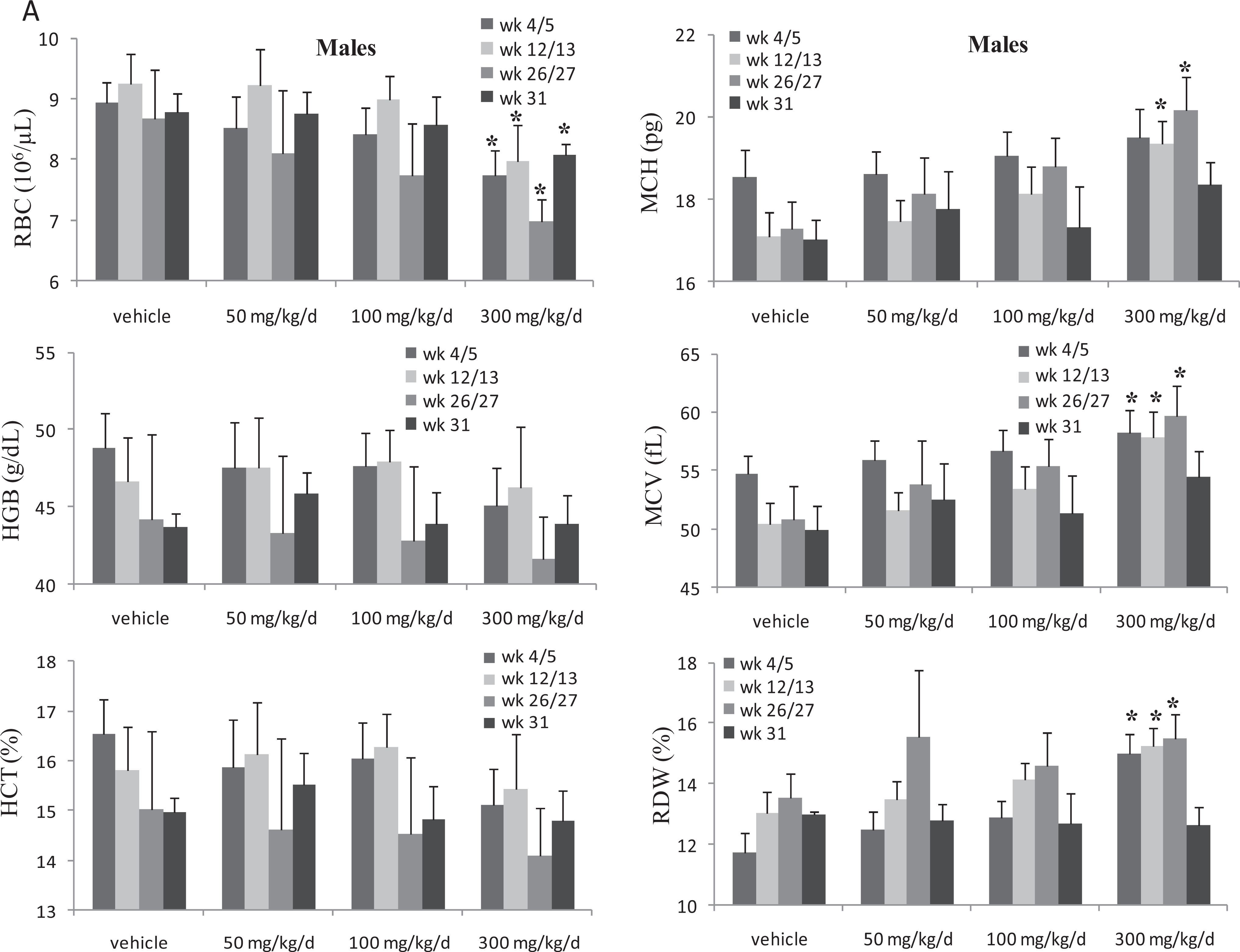
Summary of red cell changes in male Sprague Dawley (SD) rats administered BMS-986001 for 6 months. Changes in red cell parameters in male rats. Dosing phase was for 6 months (week 26) and drug-free recovery phase was for 1 month; week 26: end of dose (EOD); week 31: end of recovery (EOR); N = 24 rats/dose at weeks 4/5 and 12/13 and N = 19 rats/dose at EOD; 5 rats/dose at EOR, significantly different from vehicle group value:*P ≤ 0.05 (Dunnett).
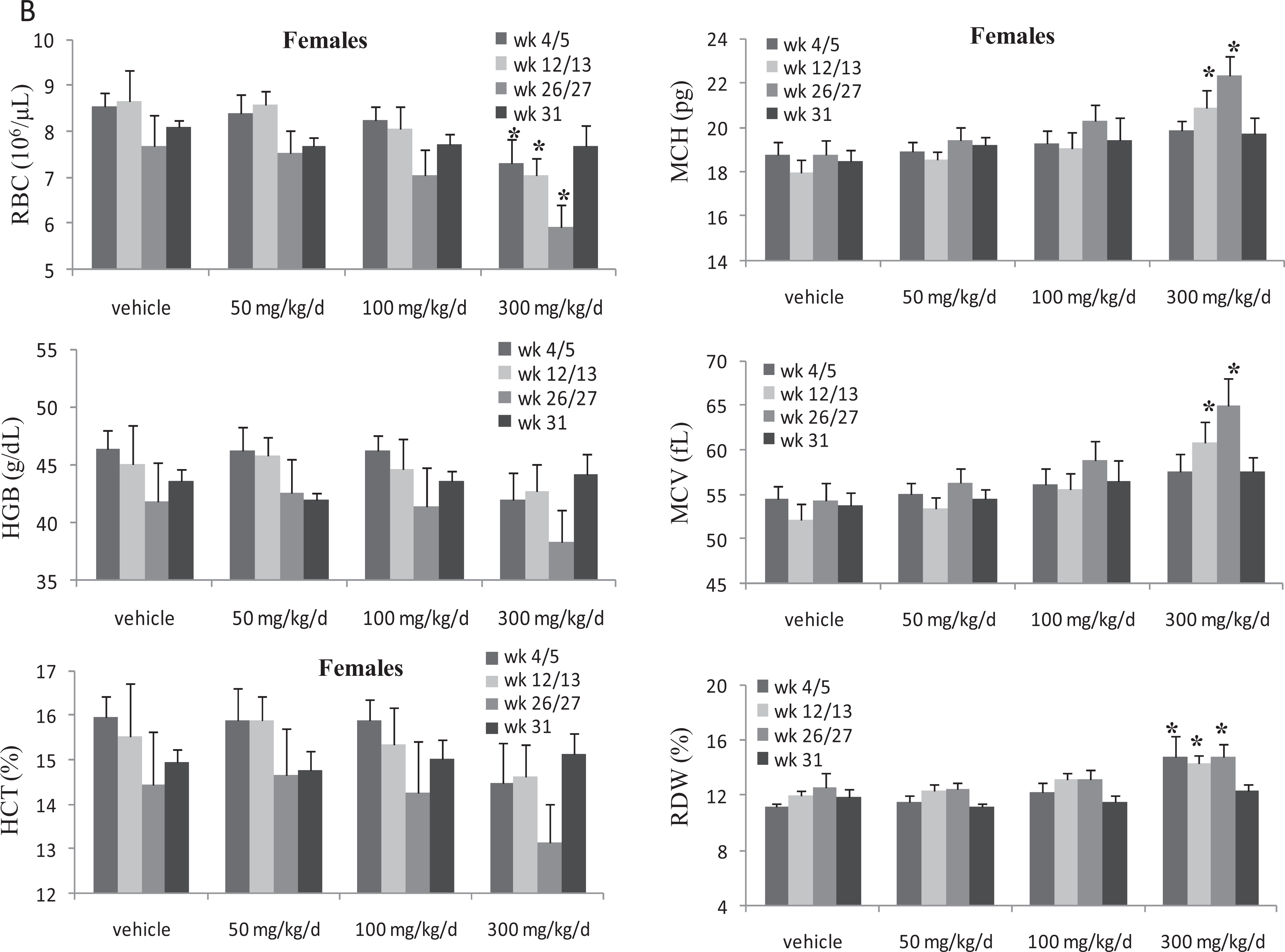
Summary of red cell changes in female SD rats administered BMS-986001 for 6 months. Changes in red cell parameters in female rats. Dosing phase was for 6 months (week 27) and drug-free recovery phase was for 1 month; week 27: EOD; week 31: EOR; N = 24 rats/ dose at weeks 4/5 and 12/13 and N = 20 rats/dose at EOD; 5 rats/dose at EOR; significantly different from vehicle group value: *P ≤ 0.05 (Dunnett).
Summary of Hematology and Bone Marrow Changes in a 6-Month Study Conducted in SD Rats.a

Abbreviations: EOD, end of dose; EOR, end of recovery; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; M:E, myeloid to erythroid ratio; RBC, red blood cell; RDW, red cell distribution width; SD, Sprague Dawley.
a N = 20 rats/sex/group for EOD; 5 rats/sex/group for EOR.
b Percentage decrease or increase in group mean (males and females combined) for each parameter compared to vehicle control group at week 26 (EOD).
c Dyserythropoiesis is characterized by abundance of erythroid precursors with mature cytoplasm and immature and/or misshapen nuclei.
d Predominance of erythroid precursors.
e 1 to 2 misshapen metarubricytes/polychromatic rubricytes/300 nucleated cells.
f Findings were partially reversible after 1-month recovery period.
g 12 to 13 misshapen metarubricytes/polychromatic rubricytes/300 nucleated cells.
Toxicity Profile of BMS 986001 in Cynomolgus Monkeys (6- and 9-Month Studies)
BMS-986001 was clinically well tolerated at ≤ 200 mg/kg/d in monkeys for ≤9 months (AUC ≤ 543 μg/h/mL) and there were no effects on survival, body weight, food consumption, ocular, physical or ECG evaluations, arterial oxygen saturation, clinical chemistry, coagulation and urinalysis parameters, organ weights, or gross pathology findings.
Except for the new findings of decreased platelet counts (described subsequently), the primary BMS-986001-related findings in the 6- and 9-month studies (Tables 6 and 7) were consistent with those identified in studies of shorter duration in monkeys and rats. The main hematology findings in both monkey studies, considered adverse at ≥200 mg/kg/d, included mild to moderate increases in BM dyserythropoiesis (Figure 1), resulting in minimal to mild decreases in M:E ratios and correlative decreases in the RBCs (Tables 6 and 7). As with rats, there was ineffective erythropoiesis characterized by the decreased M:E ratios that reflected a predominance of erythroid precursors without increases in RETICs. Relative to predose and control values, the hematologic changes at 300/200 mg/kg/d (6-month study; no recovery period; Table 6) and at 200 mg/kg/d (9-month study; 1-month recovery; Table 7; Figure 3A and B) consisted of decreases in RBC counts (14 21%), Hb, and Ht and increases in MCV, MCH, and RDW. Most of the changes in RBC parameters were seen as early as 3 months in both monkey studies, with minimal progression through the EOD. Minimal dyserythropoiesis, decreases in M:E ratios, and decreases in RBC mass (9%-10%) were also seen at ≤100 mg/kg/d dose groups (Tables 6 and 7). The hematology changes at ≥200 mg/kg/d were considered adverse because the changes in RBC mass are >10%; whereas, changes at 100 mg/kg/d are not adverse because the decreases in the RBC mass are <10%. Although BM dyserythropoiesis was present at 100 mg/kg/d, the corresponding hematology changes were generally minimal and considered not to be adverse. There was partial to complete recovery of all hematology and BM findings in monkeys following the 1-month recovery period in the 9-month study. The NOAEL for ≤9 months in monkeys was 100 mg/kg/d (≤204 μg×h/mL; AUC margin ∼7× to the exposure at the RHD).
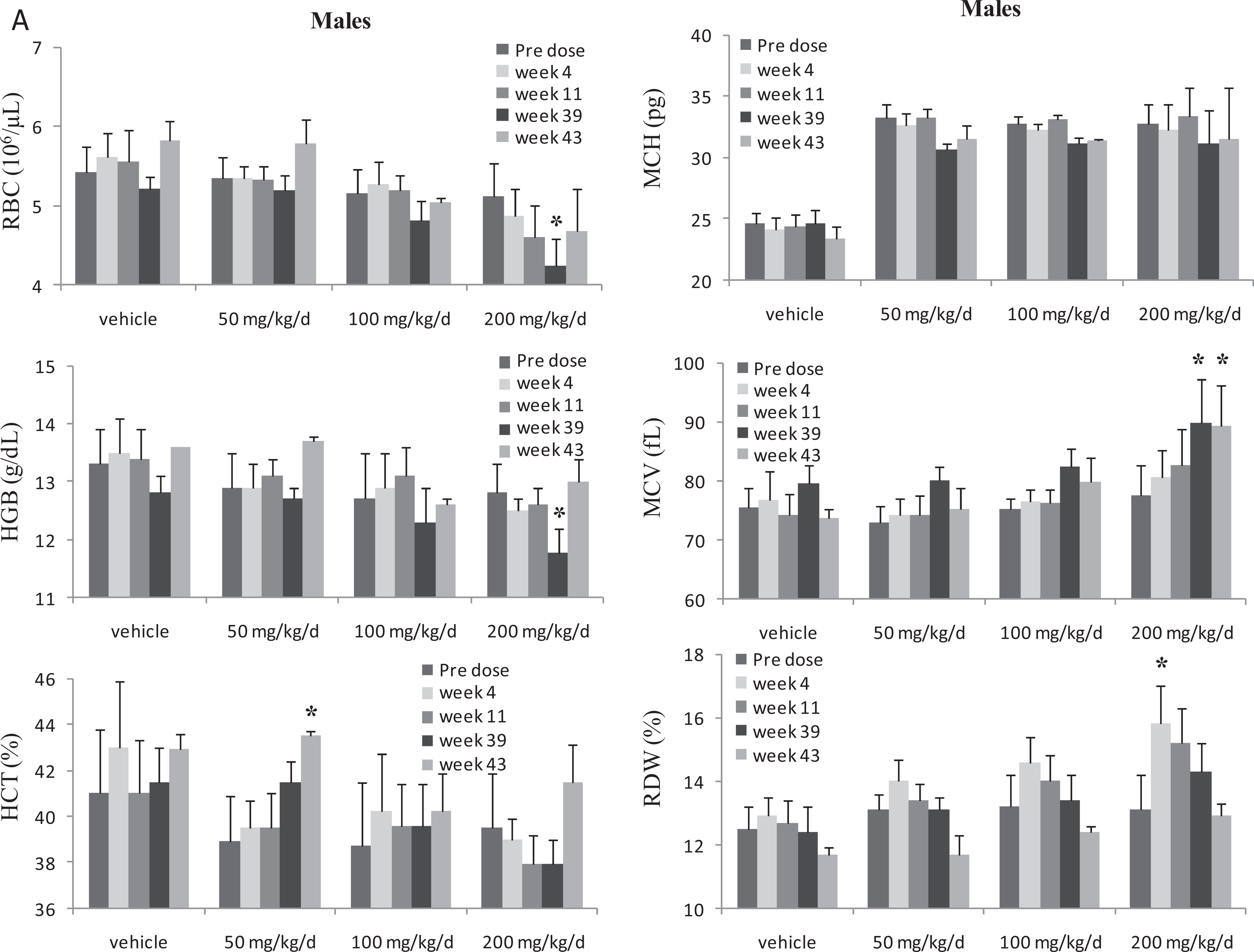
Summary of red cell changes in male cynomolgus monkeys administered BMS-986001 for 9 months. Changes in red cell parameters in male monkeys. Dosing phase was for 9 months (week 39) and drug-free recovery phase was for 1 month; week 39: end of dose (EOD); week 43: end of recovery (EOR); N = 6 monkeys per dose at predose, weeks 4, and 13 and N = 4 monkeys/dose at EOD; 2 monkeys/dose at EOR; significantly different from vehicle group value: *P ≤ 0.05 (Dunnett).
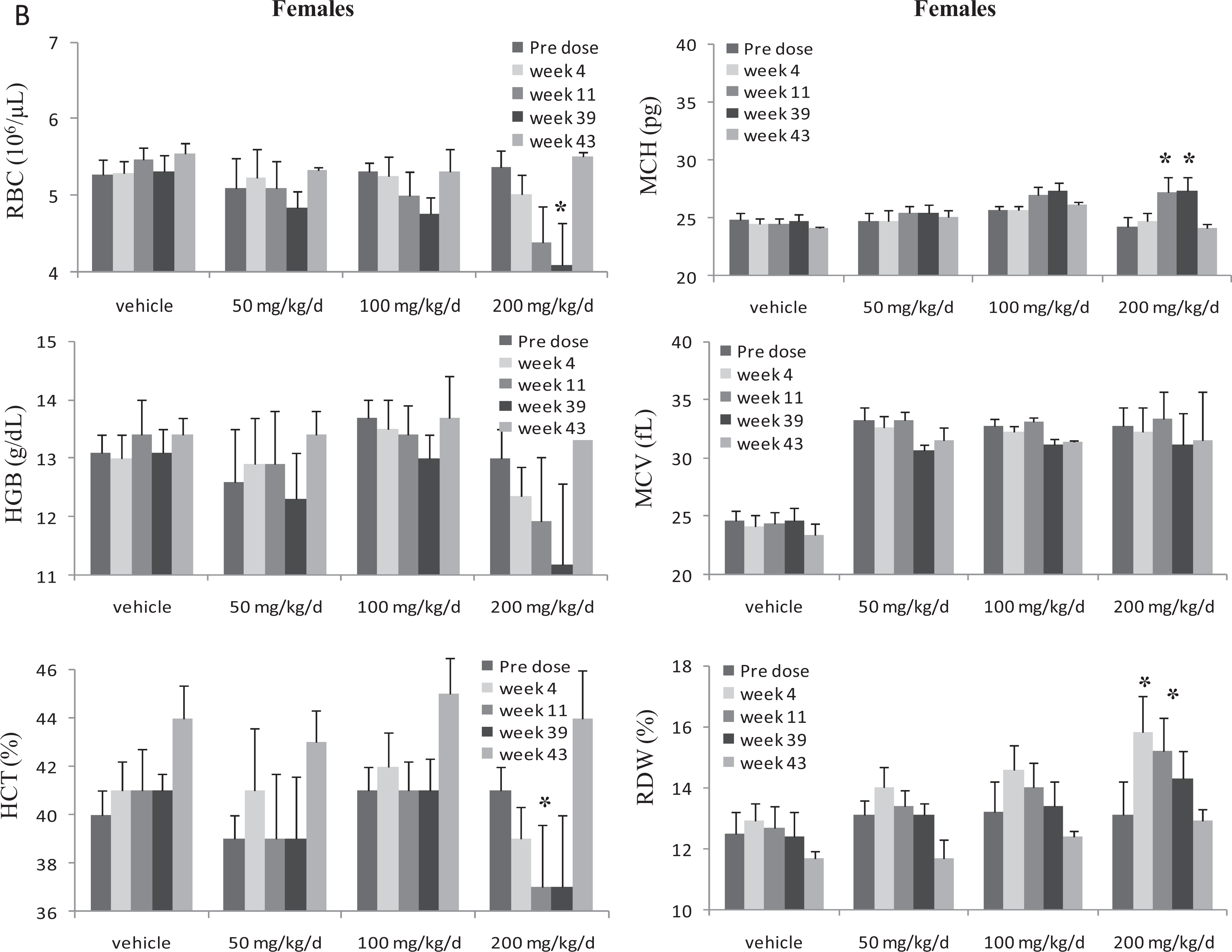
Summary of red cell changes in female cynomolgus monkeys administered BMS-986001 for 9 months. Changes in red cell parameters in female monkeys. Dosing phase was for 9 months (week 39) and drug-free recovery phase was for 1 month; week 39: EOD; week 43: EOR; N = 6 monkeys per dose at predose, weeks 4, and 13 and N = 4 monkeys/dose at EOD; 2 monkeys/dose at EOR; significantly different from vehicle group value: *P ≤ 0.05 (Dunnett).
Summary of Hematology and Bone Marrow Changes in a 6-Month Study Conducted in Cynomolgus Monkeys.a

Abbreviations: EOD, end of dose; EOR, end of recovery; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; M:E, myeloid to erythroid ratio; RBC, red blood cell; RDW, red cell distribution width.
a N = 4 monkeys/sex/group in all groups except N = 2 females at 300/200 mg/kg/d for EOD.
b Percentage decrease or increase in group mean (males and females combined) for each parameter compared to vehicle control group at week 26 (EOD).
c Dyserythropoiesis is characterized by abundance of erythroid precursors with mature cytoplasm and immature and/or misshapen nuclei.
d Predominance of erythroid precursors.
e 1 to 2 misshapen metarubricytes/polychromatic rubricytes/300 nucleated cells
f Dose reduced to 200 mg/kg/d during week 15; 2 females euthanized (weeks 11-14) due to low platelet counts.
g 12 to 13 misshapen metarubricytes/polychromatic rubricytes/300 nucleated cells.
Summary of Hematology and Bone Marrow Changes in a 9-Month Study Conducted in Cynomolgus Monkeys.a

Abbreviations: EOD, end of dose; EOR, end of recovery; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; M:E, myeloid to erythroid ratio; RBC, red blood cell; RDW, red cell distribution width.
a N = 6 monkeys/sex/group in all groups except; N = 4 (EOD); N = 2 (EOR).
b Percentage decrease or increase in group mean (males and females combined) for each parameter compared to vehicle control group at week 39 (EOD).
c Dyserythropoiesis is characterized by abundance of erythroid precursors with mature cytoplasm and immature and/or misshapen nuclei.
d Predominance of erythroid precursors.
e 1 to 2 misshapen metarubricytes/polychromatic rubricytes/300 nucleated cells.
f Findings were partially reversible after 1-month recovery period.
g 12 to 13 misshapen metarubricytes/polychromatic rubricytes/300 nucleated cells.
New findings in the 6-month monkey study were moderate to marked decreases (0.3%-45% of predose and control values, respectively) in platelets clinically manifesting as petechiae in 2 female monkeys (onset at weeks 10 and 11, respectively) at 300 mg/kg/d, and these monkeys were euthanized during weeks 13 to 14 and the high dose was reduced to 200 mg/kg/d during week 15, with the remaining monkeys completing the study without additional incidences of platelet reduction. Decreased platelets were also observed in 1 female monkey at 200 mg/kg/d in the 9-month study (onset at week 11; Figure 4A). Platelets counts in this single monkey recovered to predose level by the end of the 1-month recovery period (weeks 39-43). The AUC values in monkeys with low platelets were similar to those in unaffected female monkeys in both the 6- and 9-month studies (data not shown), thereby not providing an exposure based rationale for the platelet decreases.
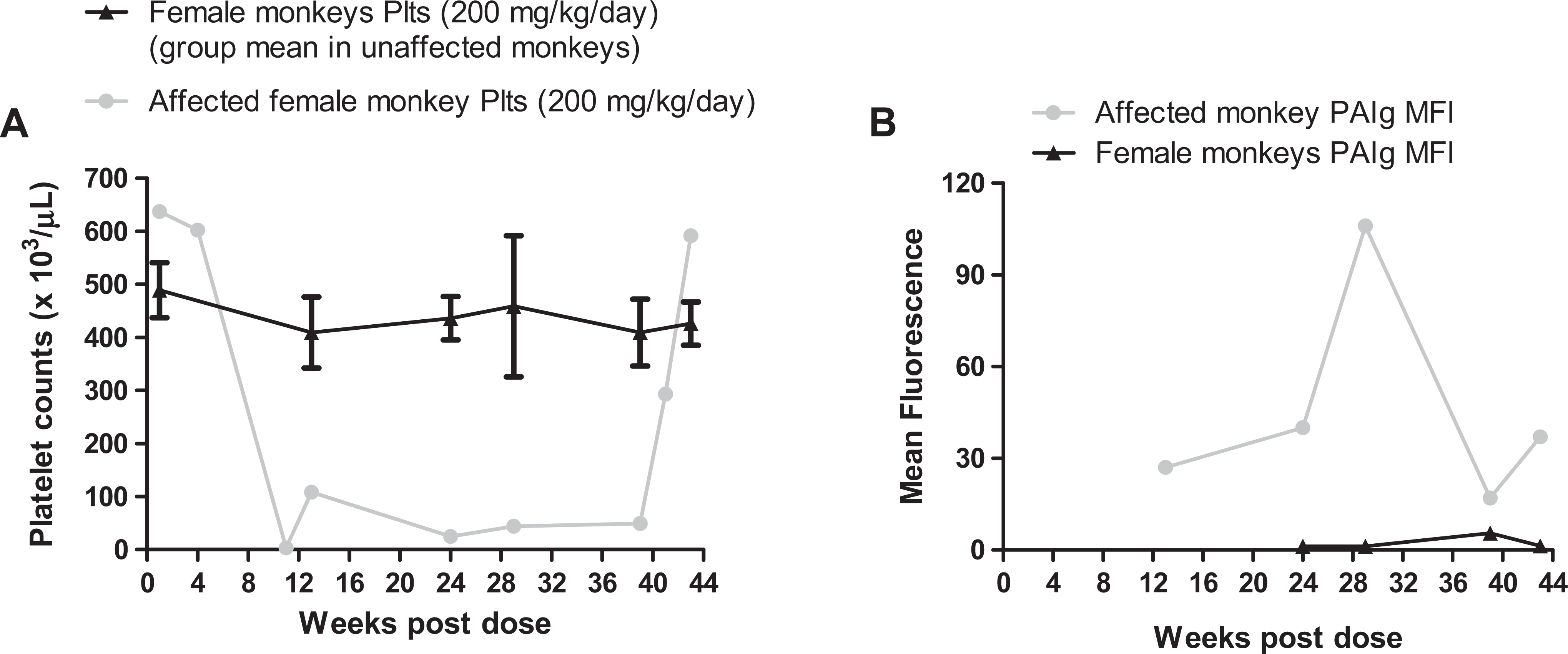
A, Platelet reduction in a single female monkey administered 200 mg/kg/d BMS 986001 in the 9-month study. Y-axis represents platelet changes in 1 affected female monkey (gray line) versus mean platelet changes in the unaffected female monkeys in the 200 mg/kg/d dose group (black line; N = 5), for the dosing period (weeks 1-39) and during the recovery period (weeks 40-43). B, Platelet-bound platelet-associated immunoglobulin (PAIg) in a single female monkey administered 200 mg/kg/d BMS 986001 in the 9-month study. Y-axis represents mean fluorescence intensity (MFI) of platelet-bound PAIg in 1 affected female monkey (gray line) versus MFI of platelet-bound PAIg in unaffected female monkeys in the 200 mg/kg/d dose group (black line; N = 5), for the dosing period (weeks 1-39) and during the recovery period (weeks 40-43).
Investigation into the mechanism of platelet reduction did not detect circulating antiplatelet antibody in the serum of the affected monkeys. However, additional investigations using a more sensitive direct assay revealed the presence of platelet-bound PAIg (Figure 4B). The higher platelet-bound PAIg mean fluorescence intensity correlated with the severity of the platelet reductions in the affected monkey (Figure 4B). In addition, the platelet counts returned to predose levels in the absence of BMS 986001 at the end of the 1-month recovery period (Figure 4A), despite the presence of low levels of platelet bound PAIg (Figure 4B). Additionally, there was no BMS-986001-related effect on megakaryocyte numbers or morphology in the BM or changes in serum thrombopoietin values in the affected monkeys from the 6- or 9-month studies (data not shown). Therefore, the low incidence of BMS-986001-induced platelet reduction, reversible upon BMS-986001 withdrawal, appears to be immune-mediated destruction of platelets in the presence of platelet-bound PAIg and BMS-986001. In the 6-month monkey study, the noteworthy BMS-986001-related macroscopic and microscopic findings in the 2 female monkeys at 300 mg/kg/d with severely reduced platelets consisted of hemorrhages in the skin, stomach, heart, and thymus, which correlated with macroscopic observation of dark areas/foci. Hemorrhage in the skin and/or heart was seen in both the affected monkeys (6-month study), while minimal erythroid hypercellularity in the BM and moderate centrilobular hepatocellular vacuolation were observed only in 1 of the 2 euthanized monkeys. No hemorrhage was seen in the affected monkey in the 9-month study that underwent scheduled necropsy at the EOR. All other macroscopic and microscopic changes in the tissues examined in the 6- and 9-month studies at the EOD or at EOR (9-month study) were low in incidence, incidental, or spontaneous in nature and considered to be within the expected biological variation in the test species and therefore, not considered to be drug related.
Although BMS-986001 is primarily (>90%) eliminated unchanged in the urine, there were no signs of renal toxicity as determined by kidney histopathology, clinical chemistry (serum creatinine, cystatin C, and BUN), urinalysis (total protein, calcium or phosphorus, and urine volume), or novel urinary biomarkers in rats or monkeys. Specifically, there was no change in levels of urinary β2 microglobulin, a low-molecular weight protein marker of PT damage, in rats (Supplemental Table 4A) or monkeys (Supplemental Table 4B) at the EOD or at the EOR. Additional biomarkers of kidney toxicity such as NGAL, calbindin D28, and clusterin remained unaffected (data not shown) in BMS-986001-treated animals of both species in the chronic studies.
Therefore, the nonclinical toxicity profile and the associated exposure values of BMS-986001 were generally consistent across species, sex, and study durations. The primary effect in both species was BM dyserythropoiesis, resulting in lower M:E ratios in the BM and reversible decrease in RBC mass. The mechanism for high-dose, BMS-986001-induced BM changes has not been established. However, the improved selectivity for hDNA polγ (100× less potent inhibition) compared to its analog d4T and lack of organ toxicity indicative of mitochondrial damage (seen with marketed NRTI and TDF) in rats and monkeys support a favorable and differentiated toxicity profile from the first-generation NRTI and from TDF for BMS-986001.
Discussion
With concerns over the long-term toxicity of the NRTI class of antiretroviral drugs and TDF (backbone of cART therapy) and evidence suggesting links between these drugs and mitochondrial toxicity, 5,7,8 there is need for a new NRTI/NtRTI with potent anti-HIV activity and an improved safety profile. We have demonstrated that chronic oral administration of BMS-986001 in rats and monkeys shows a differentiated and generally favorable nonclinical toxicity profile compared to the first-generation NRTIs and TDF.
The major findings with BMS-986001 in rats and monkeys were minimal to moderate dyserythropoiesis (characterized by the presence of erythroid precursors with mature cytoplasm and immature and/or misshapen/fragmented nuclei) in the BM resulting in minimal to mild decreases in the M:E ratio in the BM and associated decreases in RBC mass, without increase in RETICs, indicating ineffective erythropoiesis. The maximum changes in some red cell parameters occurred as early as 3 months following initiation of dosing (Figures 2 and 3), with minimal progression following chronic dosing. Although the exact mechanism for the dyserythropoiesis seen with BMS-986001 is unclear, it does not appear to be due to mitochondrial toxicity, given the complete absence of any organ toxicity typically seen with drugs that target the mitochondria in the chronic studies. Organ toxicities typically seen with drugs (including NRTI) that target the mitochondria include hepatic, skeletal and cardiac muscle, kidney, and/or GI toxicity. Additional support for the conclusion that the BM dyserythropoiesis seen with BMS-986001 is not related to mitochondrial toxicity is also based on the hematologic changes seen with other thymidine analogs such as ZDV and d4T, where the rank order of mitochondrial toxicity profile (based on inhibition [K i] of DNA polγ) is d4T > ZDV > BMS-986001. Hematologic toxicity is seen only with ZDV in chronic (6-month duration) nonclinical studies (macrocytic anemia) and clinically in patients (anemia and neutropenia) but is not a common finding in patients with d4T or BMS-986001 (to date). In vitro studies using BM progenitor cells demonstrated that ZDV-induced toxicity directly correlated with higher ZDV monophosphate levels and not due to effects on the mitochondrial DNA. Finally, mechanistic studies have correlated the ZDV-induced RBC decreases with effects on heme expression, suggesting that the BM and hematologic toxicity seen with ZDV probably occur through mechanisms other than mitochondrial toxicity. 5 Therefore, the dyserythropoiesis seen with BMS-986001 is probably due to mechanisms other than mitochondrial toxicity.
An additional finding in monkeys treated with BMS-986001, not observed in other species or in previous studies in monkeys of shorter duration (≤3 months), was severe platelet reductions in 3 of 60 total monkeys administered BMS-986001 in the 6- and 9-month studies. The cause of the platelet reductions appeared to be a reversible immune-mediated event due to the presence of platelet-bound PAIg (mainly IgG isotype) that was confirmed in the single affected monkey in the 9-month study. Despite the presence of platelet-bound PAIg at the end of the 1-month, drug-free recovery period, platelet counts returned to baseline (predose) values in the affected monkey in the 9-month study, suggesting a role of both BMS-986001 and the platelet-bound PAIg in inducing platelet destruction. Similar platelet destruction at a high dose, although at a much higher incidence (3 of 8 monkeys), was seen with LY127210 (a peripheral arterial vasodilator) in a 1-year rhesus monkey study. 38 In the absence of drug-specific antibody in the sera, a “neoantigen hypothesis,” which suggested that platelets coated with drug express an unstable antigen formed by a drug-induced conformational changes in the platelet membrane was proposed for the mechanism of platelet destruction by LY127210. This initial event may generate drug-specific, platelet-bound PAIg, which can stabilize the platelet-bound PAIg drug complex and trigger platelet destruction. 39 Based on the absence of circulating antiplatelet antibodies, a similar mechanism can be hypothesized for BMS-986001-induced platelet reduction in the monkeys. An alternate explanation for the return of platelets to predose levels following recovery could also be a threshold effect due to the amount of platelet-bound PAIg. However, it is important to note that there were adequate number of megakaryocytes in the BM of the affected monkeys indicating that BMS-986001 did not reduce production of the platelet progenitors. In the face of severe reduction in platelets, an increase in serum TPO was expected but levels were unchanged in affected monkeys compared to unaffected monkeys in the same dose group or in the vehicle control group. Since TPO is released in short bursts to affect platelet maturation, it is possible that the exact window of TPO release was not captured in our measurements due to suboptimal sampling. The complete reversibility of platelet counts in the absence of the drug (Figure 4A) indicates that BMS-986001-induced thrombocytopenia is a reversible effect.
No biochemical, gross, or microscopic changes that were predictive of liver, muscle (skeletal or cardiac), or kidney toxicity occurred with BMS-986001 (4-ethylene d4 T) in the chronic toxicology studies in rats (≤300 mg/kg/d) or in monkeys (200 mg/kg/d). In contrast, the chronic toxicity studies with d4T in rats (≤6 months) and monkeys (≤ 1 year) demonstrated liver changes characterized by mild centrilobular hypertrophy, proliferation of smooth endoplasmic reticulum, and increased liver weights, and, in only rats, foci of cytologic alterations and multifocal hepatocellular cytoplasmic vacuolation in the absence of any transaminase changes. Studies in rats and monkeys with other NRTIs such as ddC and ddI have demonstrated multiorgan toxicities that were attributed to mitochondrial toxicity. The ddC-induced changes in rats, dogs, and monkeys (≥3-month studies) that included mortalities (due to secondary infections); GI and gait disturbances; pulmonary edema; severe hematologic toxicity (decreased erythrocytes, leukocytes, and platelets); lymphoid depletion in spleen; thymus, cecum, and lymph nodes; BM hypoplasia (erythroid and myeloid); and microscopic changes in various organs and tissues including multisystemic hemorrhage and inflammation. The ddI-induced changes in chronic toxicity studies (up to 1 year) in rats and dogs included GI and hematologic toxicities similar to ddC, serum transaminase changes, and histopathologic alterations in various organs and tissues (liver, heart, and skeletal muscles). The nonclinical data with BMS-986001 are therefore consistent with the improved in vitro mitochondrial toxicity profile and selectivity (100× less potent compared to d4T) toward the eukaryotic DNA polymerases. 33
There was no evidence of kidney toxicity by biomarker or microscopic evaluation following chronic administration of BMS-986001 in either species. This is in contrast to the data from chronic studies with TDF, where renal toxicity characterized at least in part by proximal tubulopathy (inhibition of mitochondrial replication in PT) was seen in rats and monkeys. 31 In a chronic toxicity study in rats with TDF (≤ 43 weeks), pale coloration of kidneys, slight to mild renal tubular karyomegaly, and increased renal tubular pigment accumulation were noted at ≥30 mg/kg/d with early onset of findings at higher doses. 31 Recent data in rats also demonstrated increases in low-molecular weight proteins in the urine that were not identified but confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis following 5 weeks of TDF administration. 36 The mitochondrial damage in the PT was seen in the same study at the EOD (6 weeks) and was mechanistically linked to oxido nitrosative stress (increased lipid peroxidation and protein oxidation as well as depletion of the antioxidant system) in the kidney. By EM analyses, TDF-induced ultrastructural changes included reduced numbers and damaged mitochondria, the later characterized by swollen (giant) mitochondria of varying shape, disrupted cristae, and accumulation of amorphous deposits in the mitochondrial matrix. 36 Renal toxicity was also seen in rhesus monkeys administered TDF for 56 days. Changes in kidneys included pale coloration, increased weights (relative to body weight), and associated histological changes in tubular epithelial cells (karyomegaly, generalized swelling, and fine cytoplasmic granulation) at all doses (≥30 mg/kg/d). Additional findings in the rhesus monkeys at the intermediate and high doses included increased serum BUN and creatinine (indicative of more extensive kidney damage) and individual cell necrosis in the PT. 31
In a phase IIa monotherapy trial in treatment-experienced HIV-1-infected patients, BMS-986001 was generally safe, well tolerated, and efficacy was comparable to previous studies with TDF. 40 Therefore, given the nonclinical toxicity data and the decreased hDNA polγ inhibition profile (ddC > ddI > d4T > ZDV > BMS 986001), BMS-986001 presents a differentiated and generally improved safety profile compared to the first-generation NRTI and TDF, supporting further clinical development.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.