
Abstract
[14C]-Labeled arruva [sodium/potassium (2R,4R)-2-amino-4-carboxy-4-hydroxy-5-(3-indolyl) pentanoate] was administered as a single gavage dose (10 mg/kg bw) to male and female Beagle dogs and 1 bile duct-cannulated male. The mean peak arruva plasma concentration equivalent of 1.2 µg/g occurred at first sampling time point of 1 hour postdosing. The mean area under the concentration versus time curve from 0 hour postdosing to the last time point was approximately 20 µg·h/g and the mean terminal plasma elimination half-life ranged from 15 hours in females to 21 hours in males. Over 168 hours postdosing, 35% to 50% of the administered arruva was eliminated in the urine with 44% to 53% eliminated in feces; 1.3% of the administered dose was recovered in bile. Arruva and its derivatives were identified using tandem mass spectrometry, and the relative percentage of each substance was quantified via radio high-performance liquid chromatography. Over a 168-hour collection period, combined urine and feces extract data from the 6 noncannulated dogs showed that approximately 91% of the dose was excreted as unchanged parent arruva (41% in urine and 50% in feces). In the cannulated male, 95.3% was excreted as unchanged parent arruva; 50.2% in urine, 43.9% in feces, and 1.3% in bile. Lactone and lactam derivatives of arruva and 1 unidentified substance were detected in urine only during the first 24 hours postdosing with the greatest amounts detected during the first 6 hours of collection; up to 1% of lactone or lactam derivatives were detected in bile samples. Plasma pharmacokinetics data indicated rapid absorption of arruva with the majority of radioactivity located in the feces collected in the first 48 hours.
Introduction
Arruva is a salt of the α-amino acid, 2-hydroxy-2-(indol-3-ylmethyl)-4-aminoglutaric acid, which was determined to be responsible for the sweetness of the root bark of Sclerochiton ilicifolius, a spiny-leaved hardwood shrub native to South Africa. 1,2 Four possible stereoisomers (R,R-, S,S-, R,S-, and S,R-) of this amino acid exist and are collectively referred to as “monatin.” Among the 4 isomers, the R,R-monatin salt, that is, arruva (Figure 1), is considered the sweetest with a potency of approximately 3000 times that of 5% aqueous sucrose. 3 Under acidic aqueous conditions, arruva is known to undergo cyclization to either its lactam form (2-amino-4-hydroxy-4-carboxy-5-(3-indoyl)-pentanoic acid lactam) or its lactone form (2-amino-2-carboxy-4-hydroxy-5-(3-indolyl) pentanoic acid lactone), Figure 1; increasing temperature favors lactam formation over lactone formation.
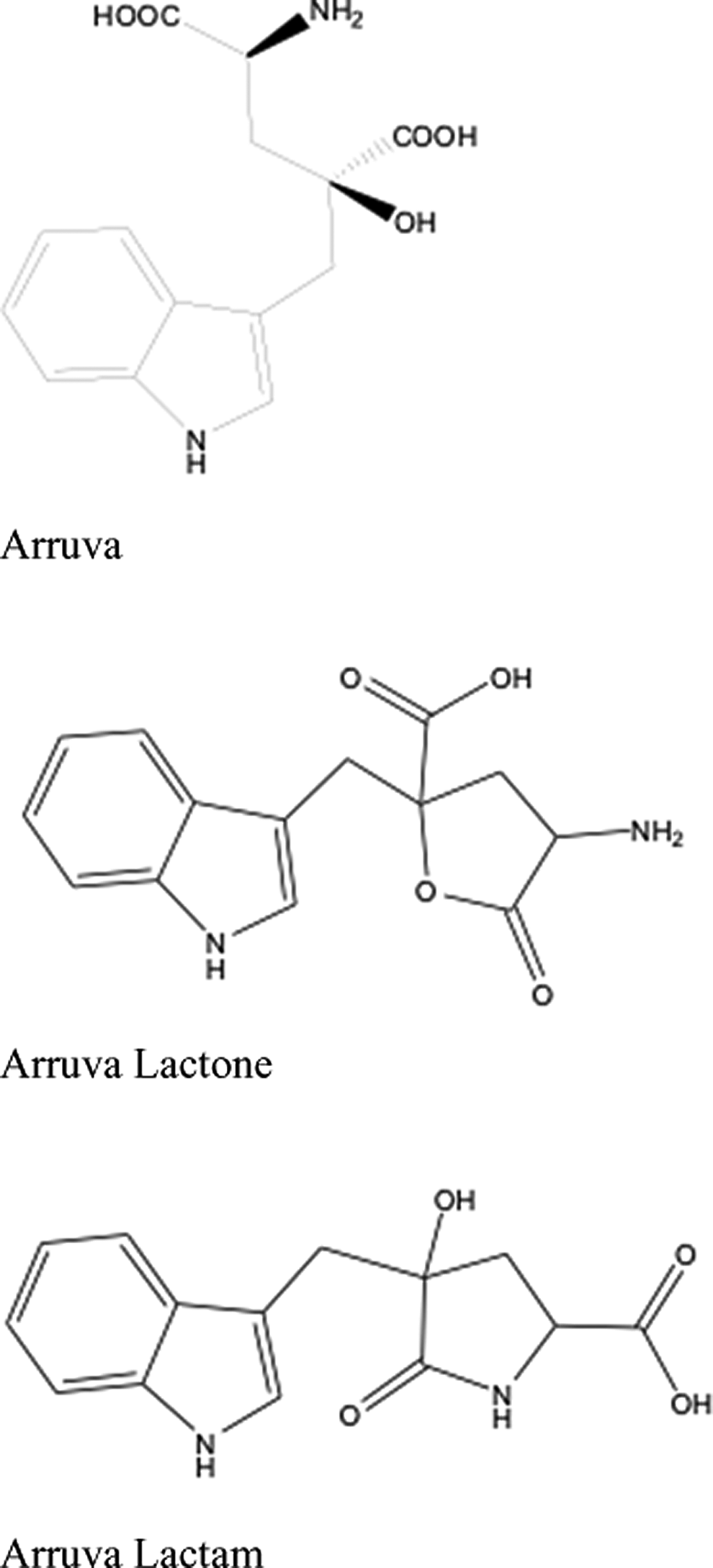
Structure of arruva, arruva lactone, and arruva lactam.
The safety of arruva has been demonstrated in previous 90-day dietary rat and mouse studies. 4,5 In the mouse study, no observed adverse effect levels (NOAELs) of 5764 and 8013 mg/kg bw/d were achieved in male and females, respectively; both NOAELs were equivalent to dietary concentrations of 35 000 ppm. In the rat study, a NOAEL of 2368 mg/kg bw/d occurred in males (equivalent to a 35 000 ppm dietary concentration), whereas the female rat NOAEL was 1544 mg/kg bw/d (equivalent to 20 000 ppm) due to significantly reduced body weight at the highest dietary arruva concentration. Arruva was also evaluated in a rat embryo/fetal toxicity study in which groups of pregnant animals consumed dietary arruva at 0, 15 000, 30 000, and 50 000 ppm during gestation days 6 to 21. 6 Both maternal toxicity, characterized by lower mean body weights, body weight gain, and feed efficiency, and fetal toxicity, characterized by decreased mean fetal body weights, were noted at the highest dietary concentration, thereby resulting in a NOAEL of 30 000 ppm (equivalent to 2564 mg/kg bw/d).
Included in the development of a body of evidence, human safety database for arruva as a potential food ingredient was an investigation of the plasma pharmacokinetics and routes of excretion associated with oral administration of radiolabeled arruva to Beagle dogs. A concurrent investigation of the absorption, distribution, metabolism, and excretion (ADME) pathways in rats was also performed and will be reported separately. In addition to parent compound identification, formation of arruva’s lactam and/or lactone derivatives and its potential metabolites was evaluated in this study.
Materials and Methods
This study was conducted in compliance with the US Food and Drug Administration (FDA) Good Laboratory Practice (GLP) Regulations 7 and the Organization for Economic Cooperation and Development (OECD) Principles of GLP. 8 This study was conducted in general accordance with the FDA Redbook II Guidelines 9 and the OECD Guidelines. 10
Test Substance
The nonradiolabeled test substance used in this study was a powder of R,R-monatin’s sodium/potassium salt, hereinafter termed arruva, which was synthesized and provided by Cargill, Inc (Wayzata, Minnesota) at 98.1% purity (corrected for diastereomeric purity). The radiolabeled test substance used for this study, [14C]-monatin sodium salt (hereinafter termed [14C]arruva), was provided by Moravek Biochemicals, Inc (Brea, California) on behalf of Cargill, Inc. The radiochemical purity ranged from 98.0% to 99.1%.
To prepare the dose formulation, [14C]arruva was isotopically diluted with nonradiolabeled arruva in deionized (DI) water. The radioconcentration and radiopurity of the dosing formulation were assessed prior to and after dosing by liquid scintillation counting (LSC) and high-performance liquid chromatography (HPLC), respectively. The formulation was uniform with respect to radioactivity as indicated by a low coefficient of variation, that is, <1.7%, between triplicate analyses performed pre- and postdosing and was 109% of the target radioconcentration. The measured mean radiochemical purity of the dosing formulation prior to and after dosing was 100%, which was similar to the reported radiopurity of 98.0% to 99.1%, indicating stability of the radiolabeled compound over the time course of dosing.
Animals, Housing, and Environmental Conditions
In all, 5 male and 3 female beagle dogs were obtained from Ridglan Farms, Inc (Mt Horeb, Wisconsin). Prior to arrival, all dogs were immunized and stool samples tested negative for parasites. Two males were surgically implanted with bile duct cannulae, and from those 2, the 1 producing the most bile was selected for use in the study. Animals were maintained in washout conditions for 10 to 14 days prior to dosing. They were housed individually in stainless steel cages located in an environmentally controlled room maintained at 20°C ± 3°C and 50% ± 20% relative humidity with daily 12-hour light–12-hour dark photoperiods and at least 10 fresh air changes per hour. Prior to dosing, the cages were equipped with a 1/4-in mesh screen to keep the feces elevated and to minimize potential urine contamination. After dosing, the dogs were observed twice daily for changes in appearance or behavior and remained in their cages until the final urine or feces collection. Studies were conducted under an institutionally approved protocol and complied with all applicable sections of the Final Rules of the Animal Welfare Act regulations (9 CFR), the Guide for Care and Use of Laboratory Animals, and the Public Health Service Policy on Humane Care and Use of Laboratory Animals. In addition, the laboratory’s policy and procedures are compliant with the American College of Toxicology’s Policy Statement on the Use of Animals in Toxicology.
The dogs were fed approximately 400 g of PMI Nutrition International, LLC-Certified Canine LabDiet 5007 PMI Nutrition International, LLC every day with reverse osmosis-treated water being available ad libitum. The number of animals utilized in the study was considered the minimum needed to yield scientifically valid data.
Dose Administration
At the start of dosing, the animals were approximately 10 to 15 months old with body weight ranges of 11.9 to 13.5 kg for males and 8.1 to 10.3 kg for females. Dogs without bile duct cannulation (3/sex) and the 1 bile duct-cannulated male were administered the test substance in a single 10 mg/kg bw gavage dose at a dose volume of 5 mL/kg bw. The target radioactivity was 250 μCi/animal. The dosage level of 10 mg/kg was selected as this dose was not expected to result in adverse effects in the animals or in the potential saturation or alteration in the test-article ADME pathways.
Sample Collection
Blood samples
Jugular vein blood samples of approximately 4 mL were collected from 1 animal/sex prior to dosing and from each animal at approximately 1, 2, 4, 8, 12, 24, 48, 72, 120, and 168 hours postdosing. All blood samples were immediately placed on wet ice following collection. Plasma was isolated by centrifugation under refrigerated conditions within 30 minutes of sample collection and then stored frozen at approximately −70°C.
Urine samples
Urine was collected on ice from 1 animal/sex overnight prior to dosing and from each animal at postdosing intervals of 0 to 6, 6 to 12, and 12 to 24 hours. Thereafter, collection continued at approximately 24-hour intervals through 168 hours postdosing. After each urine collection, cage bottoms were rinsed with an appropriate amount of DI water with the rinse retained as a separate sample. Each urine sample was weighed and then stored frozen at approximately 70°C. Cage rinses were weighed and then stored frozen at approximately −20°C.
Fecal samples
Feces were collected from 1 animal/sex overnight prior to dosing and from each animal at the postdosing intervals of 0 to 12 and 12 to 24 hours and at approximately 24-hour intervals thereafter through 168 hours. Fecal homogenates were prepared at the time of feces collection and stored frozen approximately −70°C. The processing of those homogenates is described under the section Analytical Procedures.
Bile samples
Bile was collected from the cannulated animal for approximately 2 hours on the day prior to dosing and over the postdose intervals of 0 to 1, 1 to 2, 2 to 4, 4 to 6, 6 to 8, 8 to 12, 12 to 24, 24 to 48, and 48 to 72 hours. Bile replacement salt solution was infused continuously throughout the postdosing bile collection period. The collected bile was stored frozen at approximately −70°C.
Cage wash
Following the final excreta collection, each cage was washed with DI water with the wash waters retained as separate samples that were weighed and stored frozen at approximately −20°C. Cages were also wiped with absorbent paper or Exodontia sponges, Henry Schein, Inc which were then stored frozen at approximately −20°C.
Analytical Procedures
Sample processing
Plasma, urine, bile, cage rinse, and cage wash samples were analyzed without processing. Fecal samples were homogenized with DI water at an approximate feces-to-water ratio of 2:1 (w/w) using a food processor or stainless steel spatula. Cage wipes were subjected to an overnight digestion in a methanol (MeOH)–50% sulfuric acid (H2SO4) solution that was prepared at a ratio of 2:25 (v/v) followed by dilution with DI water prior to analysis via LSC.
Liquid scintillation counting analyses
Radioactivity was determined using a Beckman Model LS 6000TA or LS 6500 liquid scintillation spectrophotometer (Beckman Instruments, Inc, Fullerton, California). Background radioactivity was determined for each day of analysis using the same scintillation media used for the preparation of the samples. Background radioactivity was automatically subtracted, and count data were automatically corrected for chemiluminescence and chemical quench. Samples were prepared for analysis of total [14C] radioactivity in duplicate, where possible, with aliquots from each plasma, bile, urine, cage rinse, cage wipe digest, and cage wash sample being mixed with 10 mL of Ultima Gold (PerkinElmer, Boston, Massachusetts) liquid scintillation cocktail for direct analysis by LSC.
Solid samples (eg, feces) analyzed by combustion were weighed into combustion boats and combusted in a Harvey Biological Materials Oxidizer (Model OX500 or OX501; RJ Harvey Instrument Co, Hillsdale, New Jersey). The liberated [14C]O2 was trapped in Permafluor E+–Carbosorb E (2:1, v/v) combined with liquid scintillation cocktail and CO2 absorber (PerkinElmer).
Sample processing for parent compound derivative and/or metabolite identification
Urine was pooled by combining equivalent volumes of urine (by sex and time point) and then centrifuged with the supernatant fraction collected for analysis; bile samples were analyzed without processing. Fecal samples were pooled by combining an equivalent mass taken from each sample by sex and time point and DI water. Resulting pooled samples were mixed while the sample containers were immersed in an ice water bath; the samples kept in the ice water bath for 1 minute after mixing. The mix–rest cycle was repeated 2 additional times. The homogenate was then centrifuged, and the supernatant fraction was transferred to a clean tube and placed on ice. The pellet was then extracted once more with water and then twice with an acetonitrile (ACN)–MeOH–water mixture (3:2:1, v/v/v) using the same process. A 100-µL aliquot of the initial fecal homogenate and each supernatant fraction was collected and analyzed for total radioactivity in order to determine the extraction efficiency. The remaining solids were oxidized and counted by LSC to determine the amount of radioactivity that was not extracted.
Among all sample pools, an approximate mean of 76% of the radioactivity was collected in the supernatant fractions from the first and second water extractions. A maximum of 2% of the radioactivity was extracted in the 2 ACN–MeOH–water extractions for each sample pool. The remaining radioactivity was retained in the sample pellet; after summing the radioactivity from all extractions and the oxidized pellet, >94% of the radioactivity in the initial pools was recovered. A 1-mL aliquot of the supernatant fractions from the first water extraction was centrifuged again prior to analysis by radio-HPLC and tandem mass spectrometry (MS/MS).
Protein in plasma samples was precipitated with ACN. An equivalent volume of ACN was added to plasma, vortex-mixed, and centrifuged. The supernatant fraction was transferred to a clean vial for radio-HPLC analysis. There was insufficient radioactivity in the plasma samples, and no radioactive peaks were apparent in the radiochromatograms; therefore, further analysis and metabolite identification were not conducted.
Radio-HPLC analysis of dose formulation
Radio-HPLC analyses of the dosing formulation were performed using an Agilent 1200 series HPLC system (Agilent Technologies, Inc, Santa Clara, California) equipped with an Agilent Quaternary Pump (G1311A), Agilent Degasser (G1322A), Agilent Variable Wavelength Detector (G1314B) set at 280 nm, and an Agilent autosampler (G1329A). Radioactivity in the effluent was monitored using an IN/US ß-Ram model 4B Radio-HPLC detector (IN/US Systems, Inc, Tampa, Florida). Radiochromatograms were processed using the Laura Lite software package (LabLogic Systems Ltd, Sheffield, England). The mean radiochemical purity for the formulation prior to and postdosing was approximately 100%. The validity of the radio-HPLC procedure was assessed by measuring the recovery of radioactivity applied to the HPLC system using a dilution of the dosing formulation. Recovery was approximately 99.8%.
Parent compound derivative and/or metabolite identification
The HPLC analyses were performed using a Thermo Electron Surveyor series HPLC system (Thermo Fisher Scientific, Waltham, Massachusetts) equipped with a Thermo Electron Surveyor Auto sampler, a Thermo Electron Surveyor LC Pump, and a Thermo Electron Surveyor PDA Plus Detector set at 280 nm. Radioactivity in the effluent was monitored using an IN/US ß-Ram Model 4B Radio-HPLC detector (IN/US Systems, Inc).
For parent compound derivative and/or metabolite identification, the postcolumn flow was split between an ion trap mass spectrometer and the radiometric detector in an approximately 1:4 ratio. The mass spectrometer, LCQ Deca XP Max (Thermo Fisher Scientific), was equipped with an electrospray ionization probe operated in the positive mode with Xcalibur version 2.0 software (Thermo ScientificTM).
Calculations
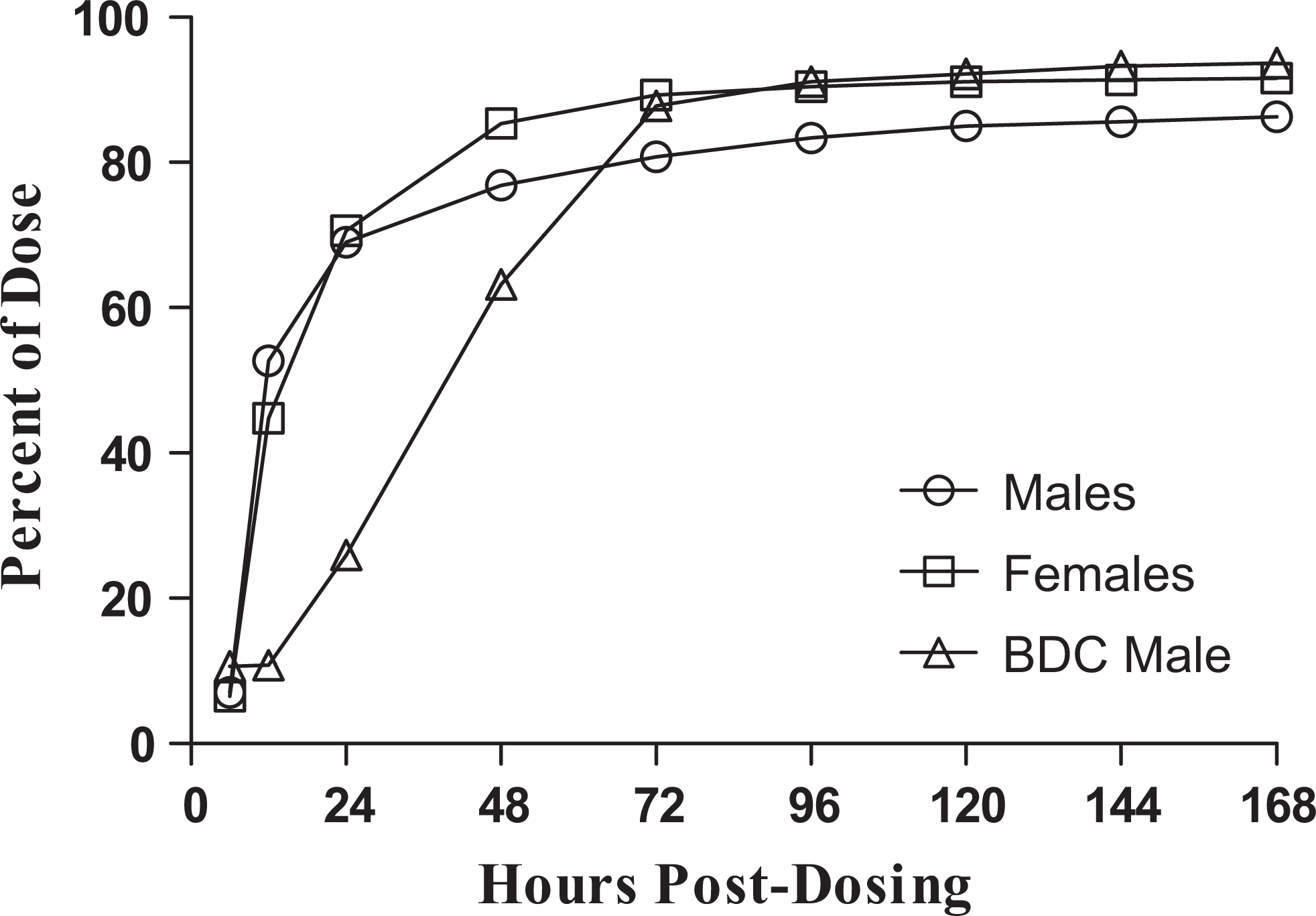
All calculations were performed with Microsoft Excel spreadsheets using full floating decimal point calculations and WinNonlin 5.2 (Pharsight Corporation, Mountain View, California). Maximum mean concentration of the test substance (Cmax), time at which Cmax occurred (Tmax), sampling time of the last plasma analyte concentration above the lower limit of quantitation (Tlast), area under the concentration versus time curve from 0 hours postdosing to the last time point (AUClast), estimate of the area under the plasma concentration versus time curve from the time of dosing to infinity (AUC0-∞), terminal elimination rate constant for the compound (Kel), and half-life for the compound (T1/2) were calculated as follows:
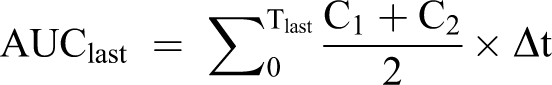
where C1 and C2 are successive plasma or blood analyte concentrations and Δt is the sampling interval, in hours, between C1 and C2.
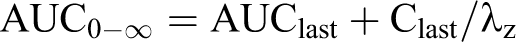
where AUClast was defined previously, Clast is the last measurable plasma or blood concentration, and λz is defined subsequently.
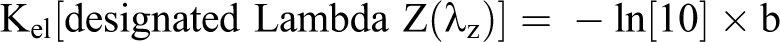
where b is the slope of the least-squares linear regression line of the log plasma or blood concentrations through at least 3 time points after Tmax.
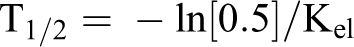
where Kel was defined previously.
Results
Actual Dosage Administered
The mean actual dose of the [14C]arruva formulation administered to male and female dogs was 10.8 ± 0.12 mg/kg bw or approximately 108% of the target dose, and the mean radioactivity administered to each animal was 274 ± 43 µCi.
Plasma Pharmacokinetics
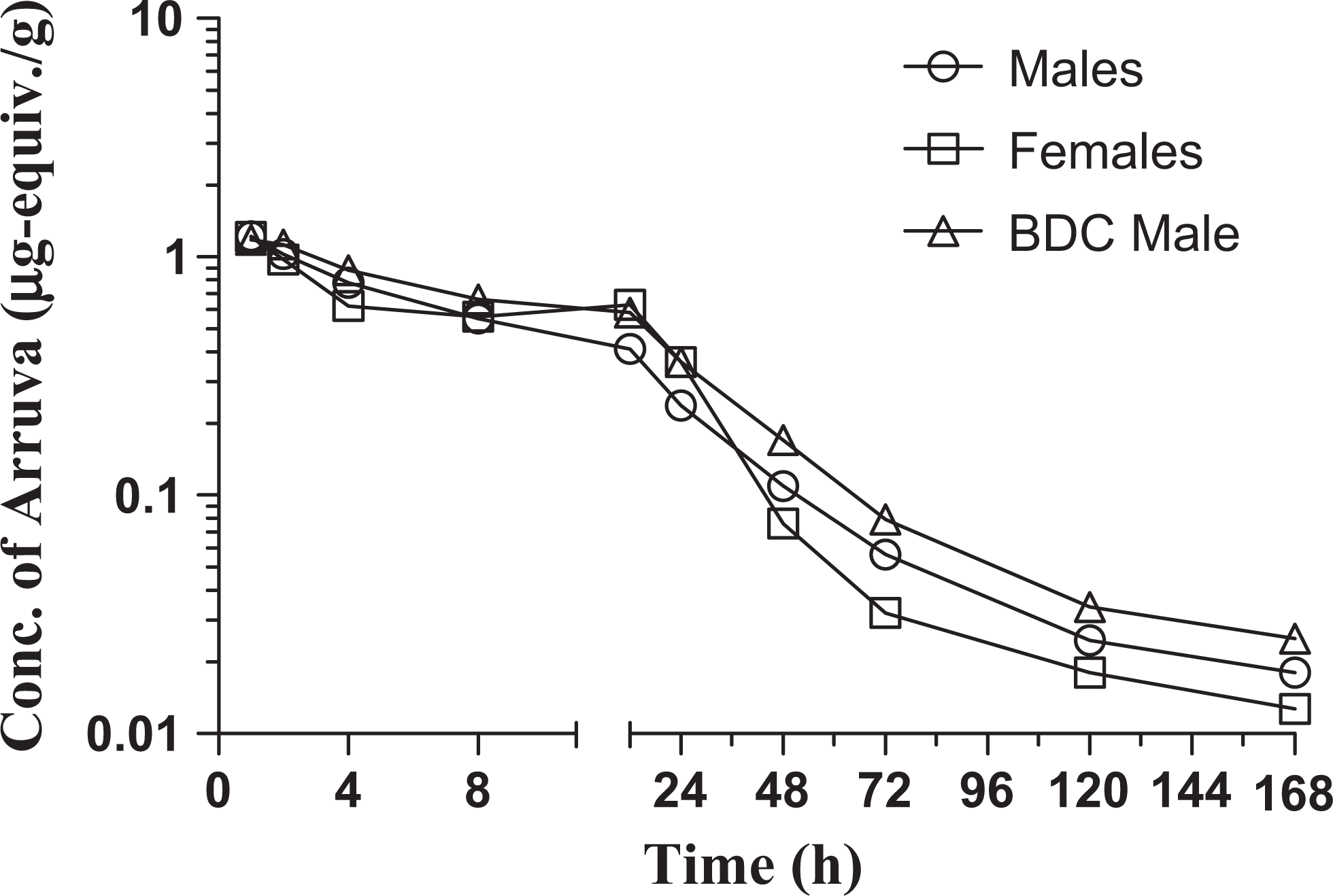
The concentration of arruva equivalents in plasma and the calculated pharmacokinetic parameters are shown in Figure 2 and Table 1, respectively. Following oral administration of approximately 10 mg arruva/kg bw, the mean measured Cmax of arruva equivalents in plasma from male and female dogs was approximately 1.2 µg/g and occurred at 1 hour postdosing (ie, the first sampling time point). The mean plasma AUClast for arruva equivalents was 18 µg·h/g in normal males, 21 µg·h/g in females, and 24 µg·h/g in the bile duct-cannulated male. The elimination kinetics of arruva equivalents appeared to be multiphasic over the 168-hour time course. The elimination rate constant (Kel) was calculated from 12 to 72 hours postdosing; the mean terminal elimination phase half-life (T1/2) was approximately 21 hours in males and 15 hours in females.
Concentration of [14C]arruva-derived plasma radioactivity following single gavage administration to Beagle dogs.
Concentration (± Standard Deviation) and Kinetics of Arruva Equivalents in Plasma of Dogs Following Single Gavage Administration.
Abbreviations: BDC, bile duct cannulated; Cmax, maximum mean concentration of the test substance; Tmax, time at which Cmax occurred; AUClast, area under the concentration versus time curve from 0 hours postdosing to the last time point; AUC0-∞, estimate of the area under the plasma concentration versus time curve from the time of dosing to infinity; T1/2, half-life for the compound; conc, concentration.
Elimination in Urine, Feces, and Bile
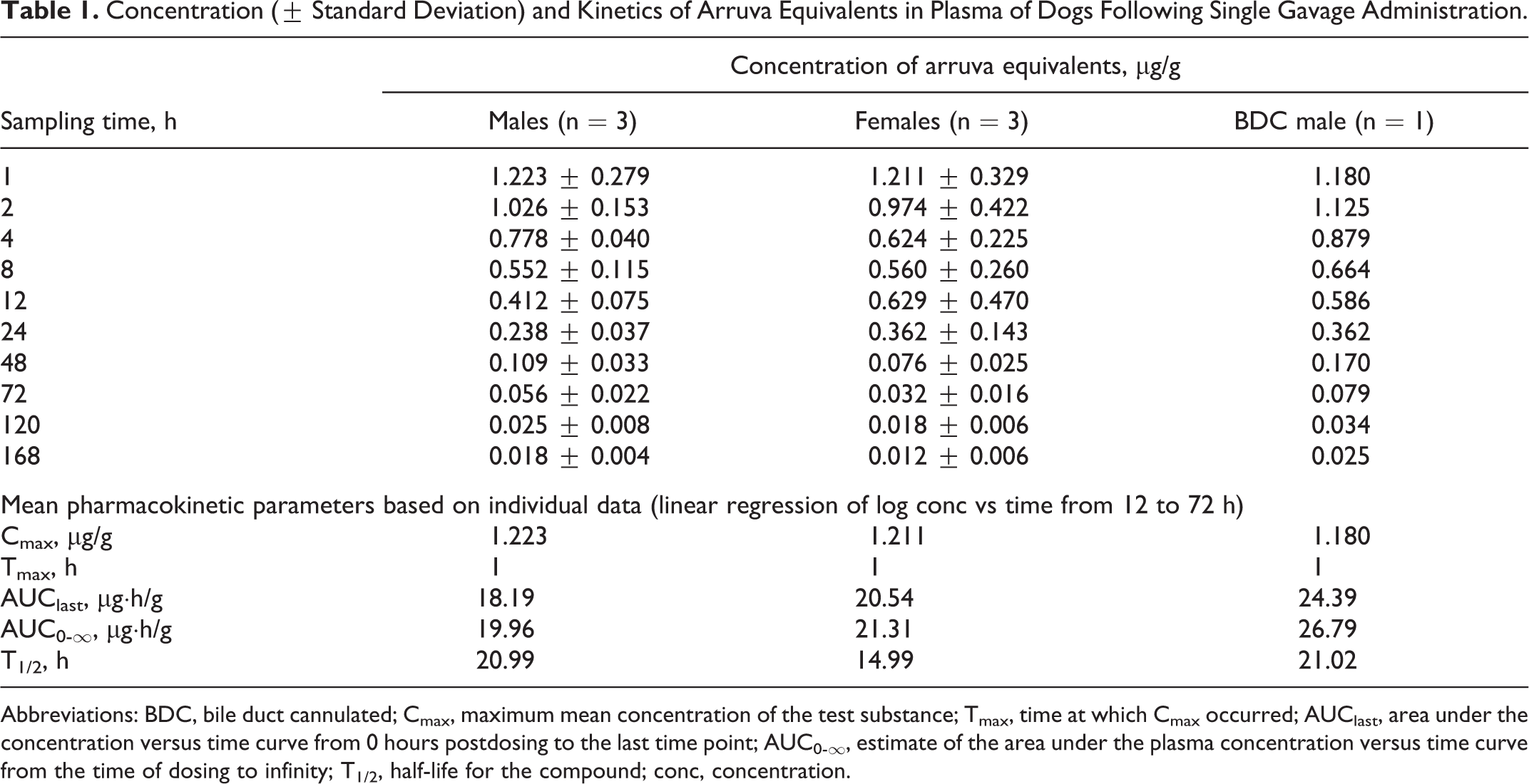
The percentage of the [14C]arruva dose eliminated in urine, feces, and bile is summarized in Table 2 and cumulative excretion is shown in Figure 3. Approximately 35% to 50% of the administered radioactivity was eliminated in the urine and approximately 44% to 53% was eliminated in feces during 168 hours postdosing, for a total of 91% of the dose recovered among all animals (based on mean values for males and females). A small percentage (1.3%) of the dose was recovered in the bile. The majority of the radioactivity in the urine and feces was recovered over the first 48 hours postdosing.
Cumulative excretion of [14C]arruva-derived radioactivity in urine and feces following single gavage administration to Beagle dogs.
Percent (Mean ± Standard Deviation) of [14C]arruva Dose in Urine, Feces, and Bile of Dogs Following Single Gavage Administration.
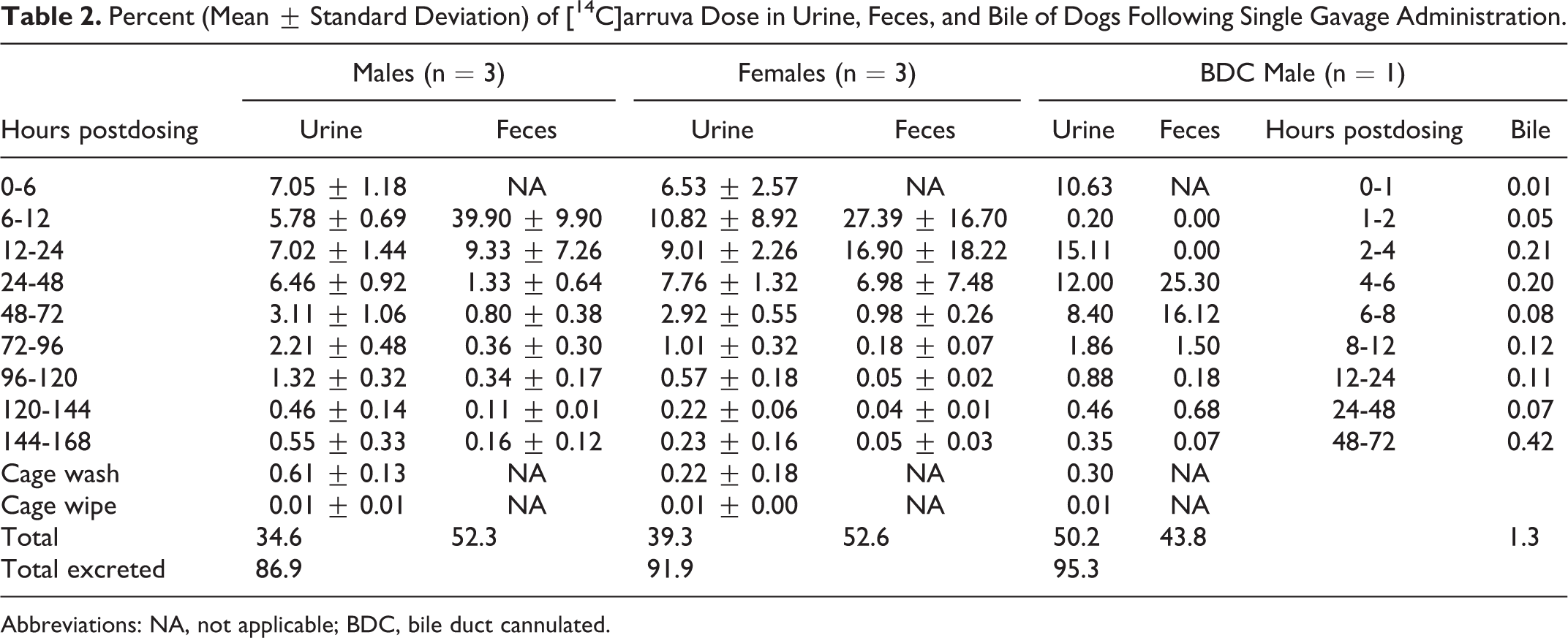
Abbreviations: NA, not applicable; BDC, bile duct cannulated.
Parent Compound Derivative and/or Metabolite Identification Using HPLC/MS/MS and Quantitation Using HPLC–Radiochromatography
The reference MS/MS spectra were obtained from a sample of arruva, which had been incubated in water and MeOH at 20°C for 3 days prior to analysis. Four primary components eluted at approximately 6.5, 7.0, 7.9, and 8.4 minutes. The mass spectrometry (MS) and MS/MS spectra of these components were used for their identifications. The HPLC/MS chromatograms and the MS and MS/MS spectra of the 6.5-, 7.0-, 7.9-, and 8.4-minute components showed them to be consistent with arruva lactone, arruva, an arruva methyl ester, and arruva lactam, respectively. Regarding the 7.9-minute elution component, although its MS spectra was close to where the methyl ester of monatin would elute, unlike the other 3 MS spectra, that 7.9-minute spectra only suggested—but did not verify—the structure as being an arruva methyl ester. A large background component eluted at that particular point thereby obscuring the metabolite’s identity, which remained unknown at the end of the study.
Arruva and its lactone and lactam forms identified using the HPLC/MS/MS techniques described previously were quantified based on their radiochromatograms. The results from the analysis of urine, fecal extract, and bile samples are presented in Table 3. In all samples, the major component identified was arruva. In addition to arruva, urine collected from 0 to 6 hours postdosing had a relative contribution of approximately 4% lactone, 1% lactam, and 9% from the unidentified peak at the 7.9-minute retention time. Although identification of the peak at the 7.9-minute retention time was attempted, coeluting components of the urine sample precluded the laboratory’s ability to conclusively identify the mass. In the remaining urine collections, 97% to 100% of the radioactivity present was attributed to arruva.
Relative Percentagesa of Arruva Parent Compound, Derivatives, and Unidentified Substance in Urine, Bile, and Fecal Extracts of Dogs Following Single Gavage Administration.
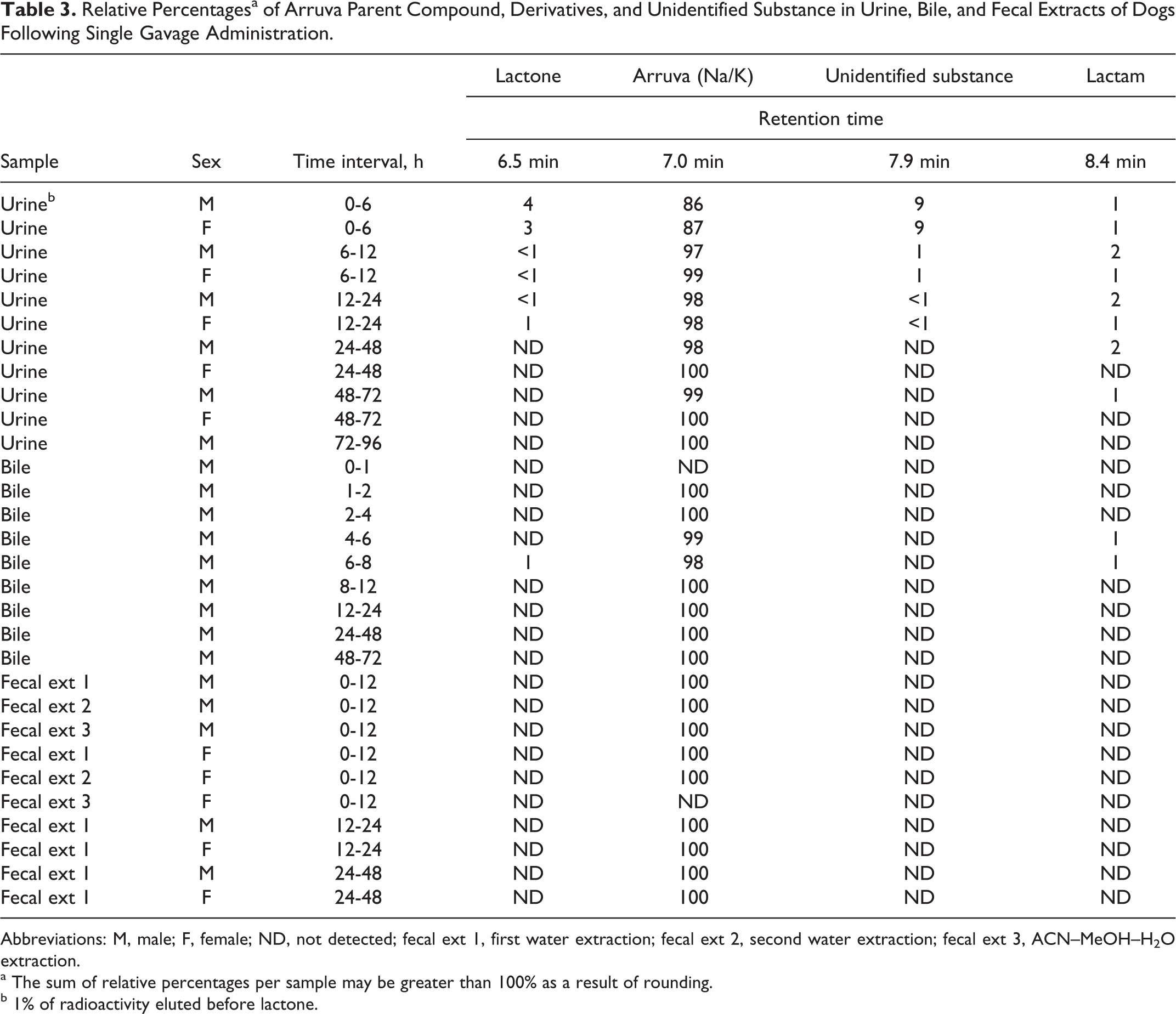
Abbreviations: M, male; F, female; ND, not detected; fecal ext 1, first water extraction; fecal ext 2, second water extraction; fecal ext 3, ACN–MeOH–H2O extraction.
a The sum of relative percentages per sample may be greater than 100% as a result of rounding.
b 1% of radioactivity eluted before lactone.
In bile samples, 98% to 100% of the radioactivity was attributed to arruva. A small percentage of the lactone and/or lactam forms (1%) were present in 2 bile samples collected from 4 to 6 or 6 to 8 hours postdosing.
In the supernatant fraction collected from the first and second water extraction of the 0- to 12-hour fecal pools, arruva was the only analyte present. Very little radioactivity (≤2.0%) could be extracted in the presence of organic solvent using the ACN–MeOH–water mixture and, while arruva could be detected using MS, no derivative forms or metabolites were apparent in the radiochromatogram. For the remaining fecal pools (12-24 and 24-48 hours), only the first water extraction was analyzed, and only arruva was present.
Discussion and Conclusions
[14C]arruva administered by gavage to male and female Beagle dogs was excreted predominantly in the urine and feces as unchanged parent arruva. [14C]arruva equivalents were excreted in both the urine and the feces (35%-50% and 44%-53% of the dose, respectively), with a mean total of approximately 91% of the administered dose recovered over 168 hours postdosing. There appeared to be a slight increase in the fraction of the dose eliminated in the urine of the bile duct-cannulated dog versus the noncannulated dogs; however, <2% of the administered dose was recovered in the bile collected from the cannulated dog over 168 hours.
Plasma pharmacokinetics indicated rapid absorption of arruva with the majority of radioactivity eliminated within 48 hours. The mean measured Cmax of arruva equivalents in plasma was approximately 1.2 µg/g, and the mean AUClast ranged from 18 to 24 µg·h/g. The terminal elimination rate constant (Kel) was calculated from 12 to 72 hours postdosing, and the apparent mean terminal T1/2 was approximately 21 hours in male dogs and 15 hours in female dogs. Radioprofiling and identification of derivative forms and/or metabolites indicated that the entire radioactivity in feces was associated with unchanged arruva. In some bile samples, small percentages of arruva’s lactone and lactam derivatives were measured but ≥98% of the radioactivity was associated with unchanged arruva. The first urine collection (0-6 hours postdosing) contained up to approximately 4% arruva lactone, 1% arruva lactam, 9% of an unidentified substance with a retention time of 7.9 minutes, and the remainder (86%-87%) was unchanged arruva. For all urine collections >6 hours postdosing, 97% to 100% of the radioactivity in urine was associated with unchanged arruva. Based on the study parameters, there were no apparent differences in excretion parameters between sexes and no indication of enterohepatic metabolism through biliary excretion.
Footnotes
Acknowledgments
The authors thank Ms Elke Kennepohl of Write-Tox Consulting/Equinox Scientific Services for her assistance in the initial preparation of this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported 100% by Cargill, Inc, Wayzata, Minnesota.