
Abstract
Aflatoxins (AFs) are highly hazardous fungal biometabolites usually present in feeds and foods. Aflatoxin B1 (AFB1) is the most toxic and a known carcinogen. Toll-like receptors (TLRs), highly expressed by myeloid dendritic cells (DC), are key innate immune-surveillance molecules. Toll-like receptors not only sense pathogen-associated molecular patterns but also contribute to infections and cancer. To assess AFB1–TLR interactions on human myeloid DC, pure CD11c+ DC were generated from monocytes isolated from healthy individuals and then exposed to relevant level of AFB1 for 2 hours. Both quantitative polymerase chain reaction and flow cytometric assays were used to quantify, respectively, expression of TLR2 and TLR4 at the messenger RNA (mRNA) and protein levels in these DC. Levels of interleukin (IL) 1β, IL-6, and IL-10 were also analyzed in AFB1- and mock-treated DC. Compared to nontreated CD11c+ DC, expression levels of both TLR2 and TLR4 mRNA and proteins were significantly upregulated in AFB1-treated cells. Further, although IL-10 levels in AFB1-treated DC were similar to those in the mock-treated DC, the AFB1-exposed DC secreted higher amounts of IL-1β and IL-6. Dendritic cells are sensitive to environmentally relevant level of AFB1, and TLR2 and TLR4 are involved in sensing AFB1. Considering the broad roles of TLR2, TLR4, and DC in immunity and infections, our novel findings open a new door to understanding the molecular mechanisms and functional consequences of AFB1 in inducing immunodysregulation, immunotoxicity, and thus (non)infectious diseases in humans.
Introduction
Aflatoxins (AFs) are one of the most hazardous mycotoxins produced by Aspergillus flavus and Aspergillus parasiticus. 1 These mycotoxins have intensively been studied, and their potent toxic and carcinogenic effects in humans and animals have classified AF as among notifiable biological/environmental toxins. 1 –4 Aflatoxin B1 (AFB1) is frequently found in many feeds and foods, and its unanticipated consumption poses alarming risks to public health worldwide. 4,5 The broad and nonselective toxic effects of AFB1 in exposed humans and animals are well documented 1,6,7 ; the International Agency for Research on Cancer has classified AFB1 as a group 1 carcinogen. 8
The immunotoxic effect of increasingly relevant doses of AFB1 on 1 class of pivotal immune surveillance molecules, pattern recognition receptors (PRRs), is still unclear. The vital role of PRR in inflammation to clear microbial infection 9 –11 is well documented. Dendritic cells (DC) are key antigen-presenting cells (APCs) and professionally link innate and acquired immunity. 10,12 Expression of PRR allows sensing of conserved structures designated pathogen/damage-associated molecular patterns (P/DAMPs) of exogenous as well as endogenous danger ligands. 10 Toll-like receptors (TLRs) are well-characterized PRRs; they are involved in cross talk with many other molecules and cell signaling, resulting in inflammatory responses and subsequent cellular activation. 13 –15 Recognition of P/DAMPs by TLR on DC triggers an intracellular signaling pathway; this induces production of proinflammatory cytokines, chemokines, and type I interferons and thus activates APC and other immune cells to link the innate and adaptive parts of the immune system to eventually protect humans from invading pathogens. 10,11,13,14,16 The TLR2, which recognizes bacterial lipopeptides, 16 and TLR4, which in the presence of an adaptor protein, MD2 and coreceptor CD14, recognizes lipopolysaccharide (LPS) from many different gram-negative bacteria, are 2 important well-studied PRR in humans and animals. 7,13,14,16 The TLR2 and TLR4 are involved in inflammation, cancer, and many degenerative diseases, and their unnecessary activations, for example, via AFB1, lead to the production of many inflammatory molecules and free radicals, thus damaging neighboring cells and tissues. 7,17 –19
Due to extremely tough measures, the occurrence of AFB1-contaminated feeds and foods in developed countries (but not in developing countries) has decreased substantially. 1,20 –22 However, experimental, analytical, and epidemiological data reveal that, even in Europe, food-producing animals and humans are routinely exposed to low levels of AFB1. 23 –26 Aflatoxin-contaminated food/feed is a silently current and future's public health issue, worldwide 4,11 ; because AFB1 is primarily a hazard in animal feeds and pose a risk to humans through unanticipated consumption. We therefore provide information on how environmentally relevant level of AFB1 modifies TLR expression, leading to a functional disruption in DC. Generally, AFB1 affects DNA in any cells in vivo and thus promotes the risk of cancer. Aflatoxin B1 also impairs the redox status of immune cells. 6 Further, pathogen recognition is weakened, 7 and key cytochrome P450 (CYP) enzymes with TLR4 are activated 26 in human lymphocytes and monocytes (MN) by AF treatment, deriving immune cells to pro-oxidant/inflammation status, 6,27,28 thereby enhancing immune dysregulation 29 and risk of infections and cancer. 11
Research has never been conducted on the influence of AFB1 on human PRR, especially the TLR on DC. Two pivotal TLRs, TLR2 and TLR4, are highly expressed by DC. Prolonged activation of TLR2 and TLR4 via, for example, AFB1 might lead to unnecessary production of many cytokines, 5 –7,10,11,15,27,29 potentiating leukocytes disarmament and thus improper immunity. The issue of proinflammatory concepts of TLR2 and TLR4 lead us to conduct the key pro- and anti-inflammatory cytokine analyses to ascertain further functional consequences of the early hours of AFB1 exposure. Whether and how intensely TLR2 and TLR4 are coexpressed at messenger RNA (mRNA) and protein levels in AFB1-exposed human DC remain unclear, and elucidating the impact of naturally occurring level of AFB1 on TLR2 and TLR4 in human DC could provide better insight into understanding the AFB1-associated immunotoxicity in chronically exposed people.
To verify our hypothesis on the interaction of relevant level of AFB1 with the human immune system, we therefore determined mRNA and protein expression of TLR2 and TLR4 in AFB1-exposed DC.
Materials and Methods
Reagents and Media
Aflatoxin B1 was obtained from Sigma-Aldrich Chemie (Taufkirchen, Germany); for this study, AFB1 was first dissolved in 96% ethanol (0.1 mg/mL), and further dilutions were made with Dulbecco phosphate-buffered saline (DPBS; Sigma-Aldrich, Deisenhofen, Germany). 6 Recombinant cytokines (granulocyte macrophage colony-stimulating factor [GM-CSF] and interleukin [IL] 4) and anti-CD11c antibody were purchased from R&D Systems (R&D, Minneapolis, Minnesota); and both fluorescein isothiocyanate (FITC)-conjugated antihuman CD282 (TLR2) and phycoerythrin (PE)-conjugated antihuman CD284 (TLR4) PE were bought from eBioscience (San Diego, California).
All human peripheral blood mononuclear cells (PBMC) and DC were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (Biochrom, Berlin, Germany) supplemented with 2% human (AB) serum (BioWhittaker, Walkersville, Maryland), 2 mmol/L
In Vitro Experimental Design, Isolation of PBMC, and Production of MN-Derived CD11c+ DC
To isolate PBMC, blood samples were aseptically collected into heparinized vacutainer tubes from 20 healthy male individuals (age 23 ± 2 years). The procedures of blood sampling from the volunteers for the in vitro cell culture assays and the experiments were in accordance with the local human/animal welfare regulations and were approved by the ethical committee of Ferdowsi University of Mashhad. Total numbers of circulating white blood cells were determined using a Coulter Counter (MEK-6450K; Nihon Kohden, Tokyo, Japan). Differentiation of all nucleated blood cells was performed microscopically on blood smears. 6,30 To isolate the PMBC, each blood sample was diluted 1:4 in DPBS (without Mg2+ and Ca2+ ions) and then layered atop 15 mL Ficoll-Paque plus (Lympholyte; Zierikzee, the Netherlands). After centrifugation (1100g, 20°C, 40 minutes), the layer of PBMC was collected and the purified PBMC washed once (450g, 4°C, 10 minutes) with a solution of 98% DPBS, 1% penicillin/streptomycin (P/S), and 1% inactivated FCS. The pellets were resuspended and washed (180g, 4°C, 5 minutes) twice with RPMI 1640 containing 10% inactivated FCS, 1% P/S, 1% kanamycine, and 1% gentamycine 29,31 ; this procedure yielded >98% viable PBMC. The isolated PBMC from each sample were used both for the production of DC and assessment of the effects of AFB1. The time between blood collection and start of the DC production from pure MN was always ∼3 hours.
Isolated PBMC were dispensed in 3 mL (107 PBMC/mL RPMI-FBS) volumes into culture plates (3 cm diameter) and incubated for 2 hours in a 37°C chamber containing 95% humidity and 5% CO2. Nonadherent cells were removed by washing with warm Mg2+ and Ca2+-free DPBS. To obtain DC, the adherent cell fractions were then incubated (37°C, 5% CO2, 95% humidity) in DC medium containing RPMI 1640 supplemented with 10% FCS, 2 mmol/L
Harvested DC were washed in DPBS before examination by flow cytometry using a FACSCalibur System (Beckton Dickinson, Franklin Lakes, New Jersey). This washing process was specifically employed to exclude any remaining AFB1 in the media, thus avoiding the probability of any potential florescent effects from AFB1 in fluorescence activated cell sorting (FACS) assays. Briefly, flow cytometric analysis was performed as follows: a suspension of DC (200 µL, at 106 DC/mL) was incubated with 10 μL of 1:10 diluted mouse FITC-conjugated antihuman CD11c antibody for 30 minutes at 4°C in the dark. The cells were then washed twice (180g, 4°C, 10 minutes), resuspended in 1 mL DPBS, and then immediately analyzed in the flow cytometer using forward scatter–side scatter (FSC-SSC) gating, which excluded any dead cells, clump, or debris. The DC gate was also checked using Leukogate reagent (Becton Dickinson, Immunocytometry Systems, San José, CA, USA). The FITC-conjugated nonspecific antibody was also used for each batch of DC samples to account for any nonspecific staining. A minimum of 10 000 events was acquired for each sample. All data files were analyzed with Cell Quest software (BD Biosciences, Palo Alto, California). Data were presented as percentage of total DC population expressing the CD11c marker.
In Vitro Exposure of CD11c+ DC to AFB1, RNA Extraction, and Complementary DNA Synthesis
The CD11c+ DC were washed with culture media without phenol red, seeded at 3 × 106 cells/mL in culture plates, and treated with 0 or 10 ng AFB1/mL for 2 hours (37°C, 5% CO2, 95% humidity); phenol red-free media were used to provide as much closer physiological microenvironment conditions as possible, thus nullifying any possible AFB1–phenol red interaction during the 2 hours of DC-AFB1 exposure and any potential autofluorescence from phenol red during flow cytometric assays. The AFB1 concentration was chosen based on previous study using bovine granulocytes, 6 as well as human PBMC 7,27 and porcine DC. 29 The CD11c+ DC were also treated with 10 ng LPS/mL (TLR4 agonist and positive control) for 2 hours. The suspensions of CD11c+ DC were then centrifuged (350g, 5 minutes, 4°C), and the cell pellets were stored at −80°C for molecular analysis. The generated supernatants were saved at −80°C until used for cytokine analyses.
Total RNA from AFB1-treated and -nontreated DC was extracted with TriPure isolation reagent (Roche Diagnostics, Mannheim, Germany) according to the manufacturer instructions. The extracted total RNA (∼1 μg) was treated with DNase I (Roche); the resultant RNA dissolved in nuclease-free water (Promega, Madison, Wisconsin) and immediately stored at −80°C for later polymerase chain reaction (PCR) analyses. Prior to freezing, the concentration and quality of the RNA were determined using a NANODROP 2000 spectrophotometer (Thermo Scientific, Langenselbold, Germany) and gel electrophoresis, respectively.
First-strand complementary DNA (cDNA) was synthesized from 0.5-1 µg RNA in a 20-μL final volume using a cDNA synthesis kit (RevetAid™ First Strand cDNA synthesis kit; Fermentas, Vilnius, Lithuania). All samples were reverse transcribed under the same conditions (65°C for 5 minutes, 42°C for 1 hour, and 70°C for 5 minutes). The concentrations of all cDNA samples were then quantified with the nanodrop spectrophotometer.
Polymerase Chain Reaction Assays for Relative Quantification of TLR2 and TLR4 mRNA
Before quantitative PCR (qPCR) assays, conventional PCR was performed with the designed primers for TLR2, TLR4, and glyceraldehyde phosphate dehydrogenase (GAPDH) genes (Table 1), using an AccuPower™ PCR PreMix (Korea, Seoul, Pat No. 162770). The primers were designed using a CLC program with specific sequences, annealing temperature, and amplicon size (Table 1). These primers were also used in qPCR assays. Briefly, cDNA (400-500 ng) was mixed with 250 mmol/L deoxyribonucleotide triphosphates, 1× reaction buffer (Fermentas), forward and reverse primers (10 pmol/L), and 0.4 U Taq polymerase in a 25-μL reaction volume. The PCR conditions were 1 cycle of 94°C for 4 minutes followed by 36 cycles of 1 minutes at 94°C, 53°C (depending on primers) for 1 minute, and 72°C for 1 minute followed by 1 cycle at 72°C for 10 minutes. Reactions without cDNA (RT-minus samples) and nontemplate control (NTC) were conducted in parallel. An aliquot (10 μL) of each PCR product was then electrophoresed over a 1% agarose gel containing ethidium bromide and visualized with a ultraviolet light.
Designed Primers Used in PCR Assays in This Study.
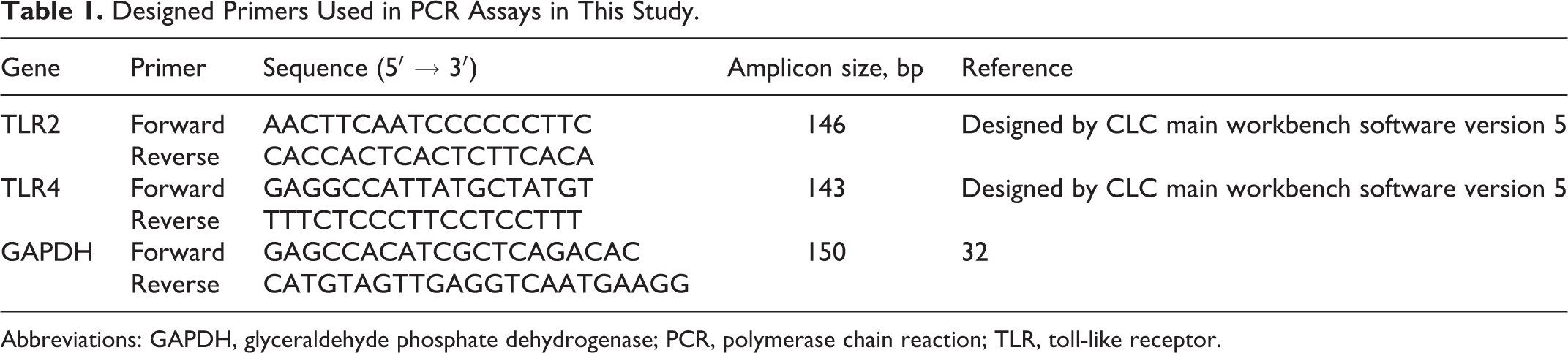
Abbreviations: GAPDH, glyceraldehyde phosphate dehydrogenase; PCR, polymerase chain reaction; TLR, toll-like receptor.
To optimize the qPCR assays, different concentrations (0.5, 0.05, 0.005, and 0.0005 μg/μL) of cDNA from experimental DC were analyzed using the same specific primers for the reference and target genes as mentioned earlier. Eventually, 0.5 µL aliquots of the test cDNA (at 0.5 μg/μL) were used as templates for the TLR2-, TLR4-, and GAPDH-specific assays with SYBR green (Fermentas) using a Swift Spectrum™ 48 Real-Time Thermal Cycler PCR (Esco Micro Pte Ltd, Singapore). The final concentrations of forward and reverse primers were each 10 pmol/L. The qPCR conditions were 94°C for 5 minutes followed by 35 cycles at 94°C for 50 seconds (denaturation) and 53°C for 50 seconds (annealing and elongation). Identification of the single PCR products amplified by the TLR2-, TLR4-, and GAPDH-specific primer pairs was again confirmed by gel electrophoresis; each primer set yielded a single-specific qPCR product.
For the qPCR assays on the cDNA from DC, experimental samples were run in duplicate with the same concentration of cDNA per reaction. To check the amplicon contamination, each run contained an NTC. For each set of qPCR assays, RT-minus samples and an NTC were also employed. Complementary DNA of LPS-treated DC was used as positive control in each AFB1 treatment in qPCR assays. Cycle threshold (Ct) values were recorded. Confirmation of single qPCR products was applied using dissociation curves and agarose gel electrophoresis. Data were normalized using GAPDH and transformed using the comparative Ct method. 32,33 The relative quantification of gene expression changes (2−ΔΔCt and/or folding) was calculated (after normalizing GAPDH gene expression) using the formula: ΔΔCt = Σ[(CtGOI − CtHKG)control − (CtGOI − CtHKG)]AFB1-treated, where GOI is gene of interest, HKG is the housekeeping gene, and 2−ΔΔCt is the fold change of the AFB1-treated GOI expression relative to that in nontreated cells. After normalizing and calculating with that formula, the fold change of the TLR2 and TLR4 mRNA in AFB1-exposed DC was finally converted to the “logarithmic (log 10) scale” for better readability of the results.
Flow Cytometry Analysis of Expression of TLR2 and TLR4 Proteins
Two-color flow cytometry (FACS) was performed with the isolated CD11c+ DC as outlined previously to analyze the protein expression of TLR2 and TLR4. The selected FSC/SSC-gated cells, expressed CD11c, were used for the analyses and calculation of the corrected geometric mean fluorescence intensity (cMFI). In the assay, CD11c+ DC were labeled with saturating quantities of FITC-antihuman CD282 (TLR2) and PE-antihuman CD284 (TLR4). To account for any nonspecific antibody binding, staining of parallel samples of CD11c+ DC was performed using mouse immunoglobulin (Ig) G2a (κ isotype; control, FITC) and IgG2a (κ isotype; control, PE). In CD11c+ DC gate, the exact position of quadrants was specified from isotype control samples and then applied for batch of samples. A minimum of 10 000 events was acquired for each sample.
To be sure and get rid of possible dead cells and debris, we further excluded the low FSC/SSC region (doublet exclusion) in the flow cytometric analyses of TLR2/TLR4. The CD11c+ DC were treated with AFB1 and stained with α-TLR2 and α-TLR4; they were also compared to isotype-stained DC that were treated with/without AFB1 when calculating the cMFI. For compensations of the double-stained DC with FITC and PE, the percentage of compensation was always adjusted to yield an identical MFI for FITC-positive and PE-positive DC. To do this, we first calculated the compensation by correcting the spillover of FITC in the PE channel (ie, FITC-positive DC was prepared, run, and then compared with the MFI of the FITC-negative DC and the FITC-positive DC in the PE channel). Similar analyses and adjustments were performed with PE spillover in the FITC channel. Protein expression of the TLR2 and TLR4 was eventually expressed in terms of cMFI, that is, cMFI = MFIreference Ab/MFIisotype Ab. Using PE-antihuman CD284, TLR4 expression at the membrane level on LPS-treated DC was also assessed as a positive control for each AFB1 treatment.
Analysis of IL-1β, IL-6, and IL-10 Secretion by DC
Dendritic cells were generated and stimulated with AFB1 as mentioned earlier. After 2 hours, DC culture supernatants were collected and their IL-1β, IL-6, and IL-10 cytokine concentrations determined using commercially available ELISA kits (eBioscience), according to manufacturer instructions. Samples and standards were analyzed in duplicate. All cytokine concentrations (in pg/mL) were calculated using curve-fitting algorithms applied against standard curves generated in parallel. The sensitivities of the IL-1β, IL-6, and IL-10 kits were <1 pg/mL.
Statistical Methods
All data were analyzed using SAS ® statistical software (Version 9.0; NC STATE University, USA). For the relative gene and protein expression values, differences between the AFB1-treated and nontreated samples were analyzed using a Student t test and further compared using a 1-way analysis of variance. A P value <0.05 was considered significant.
Results
Aflatoxin B1 Upregulates Expression of TLR2 and TLR4 mRNA in Human CD11c+ DC
In these experiments, DC were derived from the blood of healthy individuals; CD11c+ DC production yielded >92% CD11c+ DC with a >93% viability (Figure 1A-D). To optimize the PCR assays, not only conventional PCR (Figure 1E) but also qPCR was used to quantify the specific single PCR products for TLR2, TLR4 and GAPDH (Figure 1F and G). Representative results of the qPCR analyses using different concentrations of cDNA for TLR2, TLR4, and GAPDH genes are shown in Figure 1G. The analyses of the qPCR data revealed that expression levels of both TLR2 and TLR4 mRNA in 2-hour, AFB1-treated CD11c+ DC were significantly higher than in nontreated cells (P < 0.05); in other words, the increased expression of TLR2 and TLR4 mRNA was ∼11-fold and ∼69-fold higher than their control counterparts, respectively (Figure 2). The TLR4 mRNA levels in LPS-treated CD11c+ DC were ∼111-fold higher than their control counterparts (Figure 2B; P < 0.05).
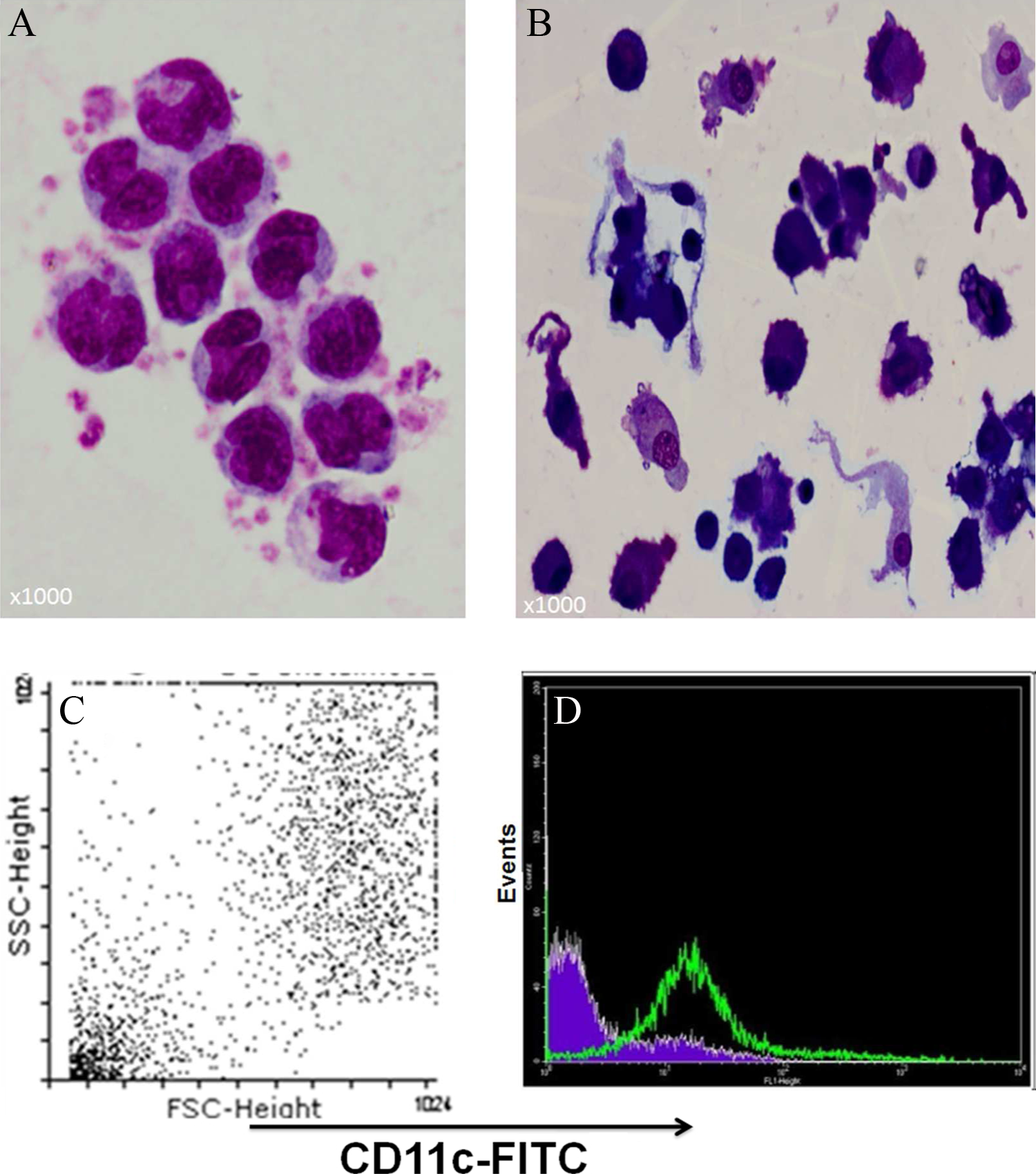
Production of CD11c+ dendritic cells (DC) from pure adhered monocytes (MN) and subsequent molecular analyses. Light microscopic image (×1000) of (A) MN and (B) MN-derived DC after 6 days of incubation of adherent MN with interleukin 4 (IL-4) and granulocyte macrophage colony-stimulating factor (GM-CSF). C, Confirmation of CD11c+ DC in gated dot plot by flow cytometry; >92% of cells were CD11c+ DC (with >93% viability). D, The DC were stained with anti-CD11c+ antibody showing shift to right in forward scatter/side scatter dot plot cytogram. E, Conventional PCR and (F) qPCR results for TLR2, TLR4, and GAPDH genes in CD11c+ DC; the band intensity for the AFB1-treated DC is more intense than the control DC, and there is no variability in the GAPDH bands. Both reactions without cDNA (RT-minus samples and nontemplate control (NTC) were used to confirm PCR procedure accuracy. G, Representative results of qPCR analyses using different concentrations of complementary DNA (cDNA) for TLR2, TLR4, and GAPDH to optimize qPCR for quantification of relative abundance of TLR2 and TLR4 messenger RNA (mRNA) in the AFB1-treated and -untreated CD11c+ DC. AFB1 indicates aflatoxin B1; GAPDH, glyceraldehyde phosphate dehydrogenase; qPCR, quantitative polymerase chain reaction; TLR, toll-like receptor.
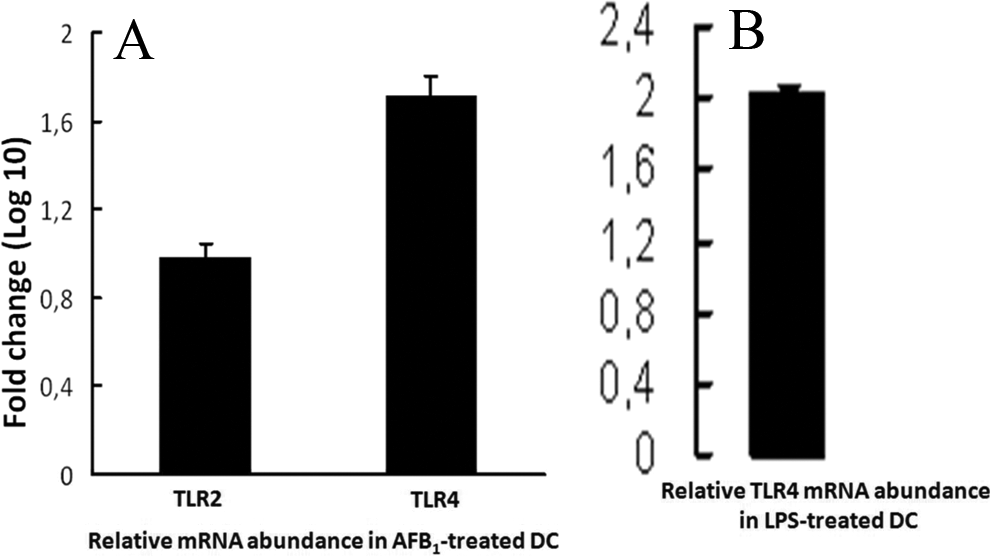
Overexpression of TLR2 and TLR4 messenger RNA (mRNA) in CD11c+ DC treated 2 hours with 0 or 10 ng/mL of AFB1. Compared to untreated controls, relative gene expression (fold change, “logarithmic, log 10, scale”) of both TLR2 (∼11-fold) and TLR4 (∼69-fold) in AFB1-treated DC was significantly higher than their control counterparts (P < 0.05). The CD11c+ DC were also treated with 10 ng LPS /mL for 2 hours as positive control of TLR4 (B). The log 10 of ∼2.2-fold change in LPS-treated CD11c+ DC (ie, ∼111-fold higher than their untreated counterparts) in the quantitative polymerase chain reaction (qPCR) assay (P < 0.05). Values are mean (±SEM) from samples isolated from 20 individuals. AFB1 indicates aflatoxin B1; DC, dendritic cells; LPS, lipopolysaccharide; SEM, standard error of the mean; TLR, toll-like receptor.
Aflatoxin B1 Modulates Expression of TLR2 and TLR4 Proteins in Human CD11c+ DC
Two-color flow cytometry using FITC-conjugated α-TLR2 and PE-conjugated α-TLR4 clearly revealed overexpression of TLR2 and TLR4 proteins in 2-hour, AFB1-treated CD11c+ DC (Figure 3A and B). The shift up (TLR4) and to the right (TLR2), that is, double-positive region (upper right) in the quadrants, was more pronounced in AFB1-treated CD11c+ DC compared to among untreated counterpart cells (Figure 3A1 and A2), clearly confirming there was an augmentation in the levels of TLR4/TLR2 double-positive cells. An overall comparison of the results of flow cytometric assessments in 2-hour, AFB1-exposed and -unexposed CD11c+ DC showed that the protein expression levels were significantly (P < 0.05) upregulated both for TLR2 (Figure 3B1) and TLR4 (Figure 3B2). Expression of TLR4 at the membrane level in the 2-hour, LPS-treated CD11c+ DC was remarkably higher than in their control counterpart DC (Figure 3C; P < 0.05).
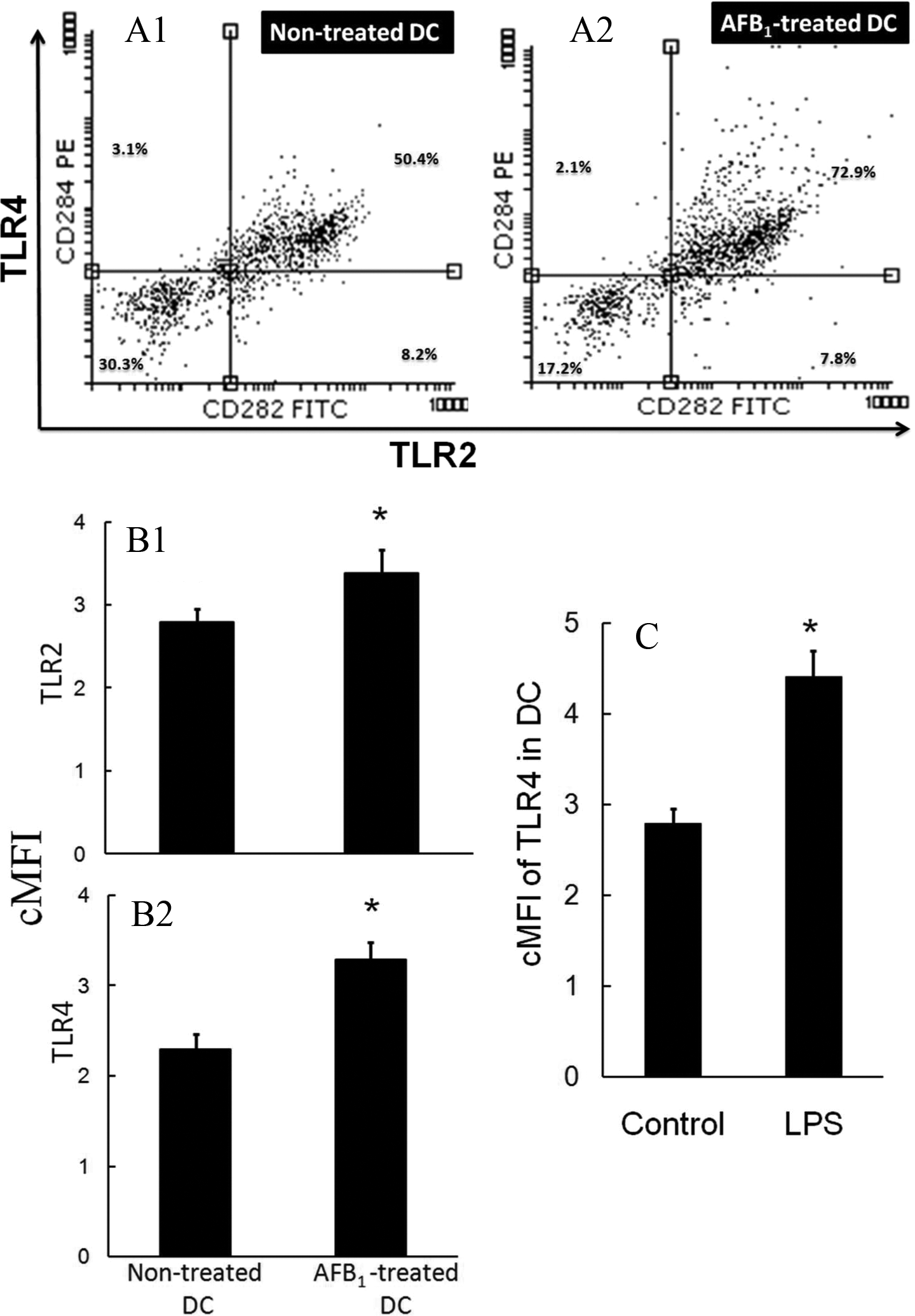
Flow cytometric analyses of DC with dual-color method with fluorescein isothiocyanate (FITC)-conjugated antihuman CD282 for TLR2 and phycoerythrin (PE)-conjugated antihuman CD284 for TLR4. Results indicate overexpression of TLR2 and TLR4 proteins in 2-hour, AFB1-treated CD11c+ DC. Representative DC population displayed in the scatter plot clearly show the augmentation of TLR4/TLR2 double-positive cells; the percentage of positive cells for single positive (upper left plus lower right), double positive (upper right), and negative (lower left) is shown in the representative quadrants. In AFB1-treated CD11c+ DC (A2), the shift up (TLR4) and right (TLR2) are more pronounced compared to untreated counterpart cells (A1). Overall comparison of TLR2 (B1) and TLR4 (B2) protein expression in CD11c+ DC compared with untreated control DC, TLR2, and TLR4 expression was significantly (*P < 0.05) increased in AFB1-treated CD11c+ DC. Also, TLR4 protein expression level on the cell membrane of lipopolysaccharide (LPS)-treated CD11c+ DC was higher (*P < 0.05) than that in control counterpart DC (C). Values are mean (±SEM) of cells isolated from 6 individuals. Corrected geometric mean fluorescence intensity (cMFI) is corrected mean fluorescence intensity. AFB1 indicates aflatoxin B1; DC, dendritic cells; SEM, standard error of the mean; TLR, toll-like receptor.
Effects of AFB1 on IL-1β, IL-6, and IL-10 Secretion by DC
As AFB1 tends to create a proinflammatory milieu in situ/in vivo/in vitro, this study sought to determine the effects of AFB1 on the secretion of selected key pro- and anti-inflammatory cytokines by DC. As illustrated in Figure 4, there was a substantive increase (P < 0.05) in IL-1β and IL-6 secretion by 2-hour, AFB1-exposed DC, although IL-10 secretion appeared unaffected by the toxin treatment of DC.
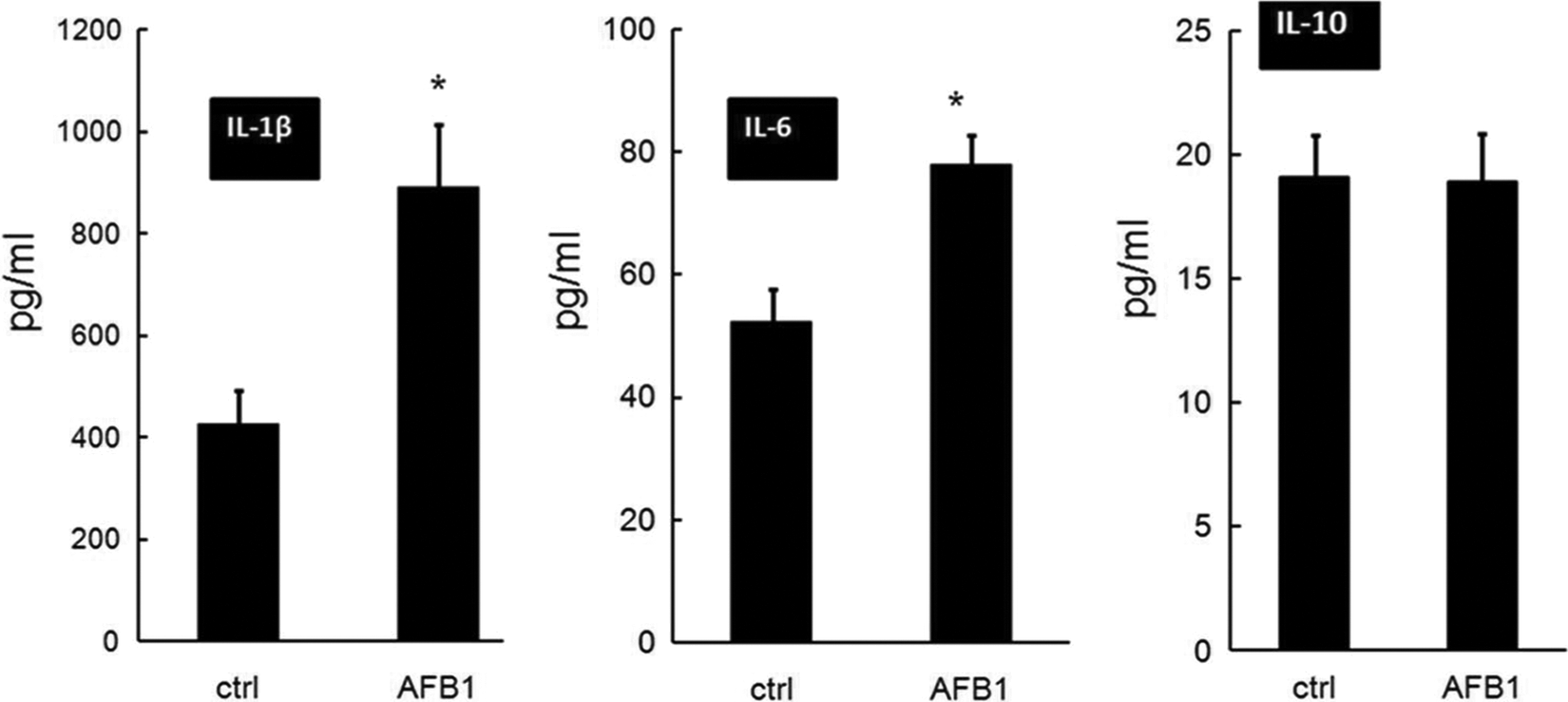
Secretion of IL-1β, IL-6, and IL-10 by CD11c+ DC treated for 2 hours with 0 or 10 ng AFB1/mL. Expression levels were assessed in culture supernatants after 2 hours using ELISA assays. Although IL-10 secretion was unaffected, AFB1 induced a greater (*P < 0.05) secretion of both IL-1β and IL-6 by the treated DC. Values are mean (±SEM) of cells isolated from 6 individuals. AFB1 indicates aflatoxin B1; DC, dendritic cells; IL, interleukin; SEM, standard error of the mean.
Discussion
This study focused the in vitro effects of a naturally occurring level of AFB1 on some key TLR in human DC. The roles of TLR in immune regulation, immune- and inflammation-mediated diseases, sepsis, autoimmune disorders, allergy, cancer, and many other infectious/noninfectious diseases remain the focus. 9,10,11,20,22,34 –36 Although continuous efforts have been made to consume AFB1-free feeds and foods, 20,37 unfortunately, humans and especially farm animals are still inevitably exposed 21 ; this is more frequent in developing countries. 1,38
Although it is believed that the exposure of food-producing animals and humans to AFB1 is relatively low in Europe, 23 –25 but occupational exposure to AFB1 is alarming, especially for indoor farmers/workers, whose exposure is by both the inhalation and the oral routes as well as healthy skin; as such, the selected level of AFB1 is increasingly reasonable, even in Europeans, 26 unquestionably making the dose rationale in our study relevant. Globalization, harsh climate changes like gas emission, flood, drought, earthquake, and typhoon (in)directly ease the issue of AFB1 in feeds/foods and thus immunotoxicity, worldwide. Surely, the relevant dose in developing countries is much higher than this selected dose. In light of the toxicokinetics/toxicodynamics properties of AFB1, gastrointestinal tract in monogastric animals including humans, mice, rats, and nonhuman primates differs from, for example, ruminants. 39 The latter possess more metabolic biological pathways to detoxify AFB1 before entering the blood stream; nevertheless, in ruminants, a fast spread of AFB1 via healthy skin, mouth, esophagus, gastrointestinal tract, and vagina to the blood stream is very likely. 23,40 Mainly due to ethical issues, little toxicokinetics/toxicodynamics studies have been conducted in humans; therefore, examining the toxicokinetics of AFB1 in human model supports researchers to strongly defend any selected AFB1 doses for their future in vitro/in situ tests. In contrast, in a variety of nonruminant mammals, those ethically difficult studies have been previously performed, 39 observing volume of distribution of 28% to 114% of body weight for AFB1; notably, the 114% volume of distribution in rhesus monkey 39 led us to assume a similar pattern for humans. As such, the selected 10 ng AFB1/mL, ∼32 nmol/L, used in our current in vitro study might still preclude our concern of “very low in vivo dose,” especially in European and North American inhabitants. In vitro/in situ, we recently found that both ∼32 and ∼2 nmol/L (∼<16-fold) of AFB1 caused cytotoxicity of leukocytes and homeostatic alterations in bovine 6 and swine 29 models, leading to potential health risks in humans and animals. 2,3,6,21,29,41,42 Alarmingly, even ∼26 nmol/L AFB1 in the blood of swine workers could be detected in Europe. 26 Further, there is little knowledge on the effects of those levels of AFB1 on the molecular parts of innate immune system in humans. Researchers have even used 5 µmol/L AFB1, ∼1000-fold higher than our selected dose, in human lymphoblastoid Jurkat T-cell model. 43 Notwithstanding, nearly all individuals in developing countries are increasingly at risk of chronic exposure to even higher than the selected dose of AFB1, and globalization and harsh climate changes broaden AF-related issues; therefore, the concept of relevant dose of AFB1 in our study seems indisputable.
The TLR2 and TLR4 are pivotal PRR in humans and animals; they contribute to many cell signaling and various physiopathological phenomena, and their stimulation via, for example, AFB1 promotes secretion of many damaging molecules and proinflammatory cytokines and, eventually, modulates host immune responses. 17 –19 In our recent results on TLR4 and TLR2 and their interference with bovine, 44 canine, and porcine 29 DC functions in AFB1-exposed (2 hours) DC, we noticed how crucial the rapid activation of these 2 PRRs is. The TLR4 and TLR2 are expressed on the cell surface of all immune cells, peculiarly DC, in different organs like brain, intestine, lungs, skins, and so on of which mRNA and protein upregulation could trigger the inflammatory responses. That is why we focused on the effects of AFB1 on TLR2 and TLR4 in human DC. Our work evaluates for the first time the effect of AFB1 on TLR2 and TLR4 mRNA and protein abundance in human DC. In early hours of exposure, AFB1 modestly exhibited proinflammatory effect (increased secretion of IL-1β and IL-6) on DC possibly through TLR2 and TLR4. Selection of this single dose of AFB1 and exposure time in the current study were in accordance with our previous study in bovine neutrophils 6 and very recent findings on human, bovine, and porcine mononuclear leukocytes. 7,27,29,42 –44 Changes observed in cytokine formation would be part of the range of functional consequences that could be linked to any upregulation of TLR2/TLR4 expression. Generally, AFB1 damages DNA 41 and impairs the redox status of cells, 6,42 enhancing the risk of many infectious and noninfectious diseases, including cancer. 11,35 –37
This in vitro experiment suggests that AFB1 promoted expression of TLR2 and TLR4 genes and proteins in CD11c+ DC. The molecular pathways underlying these findings remain unknown. In vivo, we expect DC in contact with AFB1 to be activated, disarming the post-AFB1-exposed DC in the body. The concept of interplay between inflammation, immune regulation, and appropriate resolution of inflammation for proper protective immunity is a complex prototype in AFB1-exposed population to deal with (non)infectious diseases. Nonetheless, we posited that AFB1-exposed DC would respond inappropriately and inadequately to any invading microbes and thus failed to initiate proper antigen presentation and T-cell proliferation. 29 Repeated exposure to even naturally occurring level of AFB1 leads to the increased vulnerability of individuals to pathogens, their inappropriate immune response to vaccination, 3,5 and unnecessarily increased immune response against a number of other infectious and noninfectious diseases in humans and animals 3,5,11,36 ; this would, in part, be from DC incompetence. Further functional tests need to be performed in whole blood assays, as exemplified previously 9 or in generated organs’ APC, especially DC.
Albeit that limited functional DC tests were performed here, the observed changes in IL-1β and IL-6 formation by 2-hour, AFB1-exposed DC lend support to our contention that AFB1 can create proinflammatory milieu in situ. The increase in the levels of these 2 key cytokines might be due, in part, to the augmentation of TLR4/TLR2 expression in the DC. The DC cytokine analyses also indicated a lack of the effect on IL-10 secretion, further supporting the likely generation of a proinflammatory microenvironment in/around the AFB1-exposed DC. Interestingly, these outcomes were inconsistent with previous findings, 45 wherein AFB1 did not modulate IL-1β or tumor necrosis factor-α expression in swine alveolar macrophages. Based on the findings here and porcine DC, 29 the concept of little effect of AFB1 on IL-10 secretion by DC is, nonetheless, far from straightforward, mainly due to the presence of a only single concentration of AFB1 for 2 hours, requiring further experimentation on multiple AFB1 concentrations and additional time points. Further, the potential effects of AFB1 on the formation/release of other critical cytokines/chemokines by human DC warrant further investigation.
Based on the chemical structure and highly lipophilic properties of AFB1, it is clear that the AFB1 easily passes through the plasma membrane and converts into more or less toxic hydroxylated metabolites like AFB1 epoxide, aflatoxicol, AFM1, and so on causing potent oxidative stress in cells. 6,11,27 A similar disruption might happen in DC, especially with regard to the TLR signaling cascade. 12,46 Biochemically, these toxic metabolites can bind to almost all cell biomolecules—from surface proteins to DNA and nucleic acids—thereby deregulating and disturbing physioimmunological pathways. 47,48 Although DC are sensitive to the relevant concentration of AFB1, and TLR2 and TLR4 are involved in sensing AFB1, nevertheless, we are still in its infancy to conceivably answer whether there is a direct surface binding with AFB1 and which surface molecule(s) of DC are involved. In animal models, we have been working on the possible mechanism(s) of the effect of AFB1 on DC, mainly trying to elucidate whether there is a direct surface binding for AFB1 and whether it changes on AFB1 treatment. 29 Our previous findings in a bovine granulocyte in vitro model showed that AFB1 (at ∼20-fold lower than this study) induced substantial release of free radicals by the cells, providing a pro-oxidant microenvironment for these cells. 6 Therefore, although the DC switch their physiological status toward the inflammatory and oxidative status, a huge TLR signaling cascade occurs. 7,46 This status shares many similarities to what observed in the current study on TLR2 and TLR4 in DC.
Compared with their control counterparts, the magnitude of upregulation of TLR4 was higher than TLR2 at the mRNA and protein levels. In the early hours after AFB1 addition, the DC might have begun to metabolize the toxin into other reactive metabolites like AFB1 epoxide and aflatoxicol, thereby disturbing immune-surveillance molecules. Indeed, the scientific concept of a “danger” model and alarming status of immune response and potential release of “alarmins” 46,49 in relation to each TLR suggest that the DC partially respond to and signal the exogenous danger, AFB1, via TLR4 and TLR2. 37,50,51 Although the level of AFB1 used in our study seemed low, 6,29,42 –45 the DC nonetheless responded remarkably. Our current results would therefore support the notion that through TLR, DC can sense the presence of AFB1 and their derivatives as a danger molecule in vivo. Prolonged overexpression of TLR2/TLR4 on mRNA and protein levels in DC in response to a single apparently low dose of AFB1 and especially its outcome clearly needs further investigation.
To confirm our results of TLR2 and TLR4, we applied protein expression analyses for these immune sensor molecules. When exposed to AFB1 for 2 hours, the observed overexpression of each TLR in the DC was similar to what was observed in the flow cytometric analyses. Indeed, each TLR recognizes their ligands with cooperation of some other surface proteins like CD14 and MD2, for example, LPS recognition after heterodimerization of TLR4 with MD2. 13,14 Furthermore, TLR4 recognizes P/DAMP by CD14 as a mediator, and without MD2 corecognition, 13,14 revealing the concerns that the AFB1 has broader effects on different innate immune molecules as we normally see in inflammation and oxidative stress. Recent data suggest that AFB1 upregulates AhR expression in human hepatocytes. 52 In addition, activation of the AhR signaling pathway was detected by RNA-seq in the liver of AFB1-treated rats. 53 The main downstream targets of AhR are the xenobiotic metabolizing monooxygenases, CYP1A1 and CYP1A2, involved in the (de)toxification of AFB1. 27,52,54 Since AFB1 itself possesses pro-oxidant properties in immune cells in vitro/in situ/in vivo, 6,27,28,44,55,56 the observed augmented TLR4 and TLR2 at the mRNA and protein levels are in line with our scientific concept of pro-oxidant impact of AFB1 in immune cells, which should also be taken into consideration for the outcome of the AFB1 effects/toxicity in vivo. Apart from the observed effects of AFB1 on TLR2/TLR4 in human DC, in porcine in vitro model we also observed that AFB1 interferes with antigen-presenting capacity and maturation of DC. 29 It might therefore be likely that chronically AFB1-exposed people are at higher risk of (non)infectious diseases; this needs extensive epidemiological studies as well.
Concluding Remarks
In conclusion, considering the outcome of the upregulation of TLR2 and TLR4 genes and proteins in apparently relevant dose of 2-hour, AFB1-exposed DC, our novel finding would open a new door to the molecular aspects of AFB1 and PRR, inflammation, sepsis, and cancer in humans and animals. Although our work was inconsistent with others, for example, failure of showing upregulation of TLR4 mRNA in murine macrophage cell line by AFs, 56 this inconsistency might be due to a number of factors, such as cell type, animal species, and AF doses. The effectiveness of the AFB1 might therefore be different between nonruminant mammals and humans 29,39,40 and between cell types 6,43,56 and doses. 7,27,55 Our work was the first ever done in AFB1-exposed primary human DC, and we examined the current dose and time point, and further functional assays are in progress to explain the molecular mechanisms of this finding.
Footnotes
Acknowledgments
The authors gratefully acknowledge the bureau (area) for research and technology of Ferdowsi University of Mashhad. We also thank Ms N. Tabasi, Ms S. Zamani Tagizadeh Rabe, and Mr A. Bahari for their technical assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.