
Abstract
Dapagliflozin, a first-in-class, selective inhibitor of sodium-glucose cotransporter 2 (SGLT2), promotes urinary glucose excretion to reduce hyperglycemia for the treatment of type 2 diabetes. A series of nonclinical studies were undertaken to evaluate dapagliflozin in species where it was shown to have pharmacologic activity comparable with that in humans at doses that resulted in supratherapeutic exposures. In vitro screening (>300 targets; 10 μmol/L) indicated no significant off-target activities for dapagliflozin or its primary human metabolite. Once daily, orally administered dapagliflozin was evaluated in Sprague-Dawley rats (≤6 months) and in beagle dogs (≤1 year) at exposures >5000-fold those observed at the maximum recommended human clinical dose (MRHD; 10 mg). Anticipated, pharmacologically mediated effects of glucosuria, osmotic diuresis, and mild electrolyte loss were observed, but there were no adverse effects at clinically relevant exposures, including in the kidneys or urogenital tract. The SGLT2−/− mice, which show chronic glucosuria, and dapagliflozin-treated, wild-type mice exhibited similar safety profiles. In rats but not dogs, dapagliflozin at >2000-fold MRHD exposures resulted in tissue mineralization and trabecular bone accretion. Investigative studies suggested that the effect was not relevant to human safety, since it was partially related to off-target inhibition of SGLT1, which was observed only at high doses of dapagliflozin and resulted in intestinal glucose malabsorption and increased intestinal calcium absorption. The rigorous assessment of supra- and off-target dapagliflozin pharmacology in nonclinical species allowed for a thorough evaluation of potential toxicity, providing us with confidence in its safety in patients with diabetes.
Introduction
Worldwide, an estimated 347 million people have diabetes, of which 90% is type 2 diabetes mellitus (T2DM). 1 Because a high proportion of patients do not meet the recommended treatment goals with currently available therapies, 2,3 new therapies are needed. Inhibition of the sodium-glucose cotransporter 2 (SGLT2) is 1 new therapeutic option for the treatment of T2DM, 4,5 which has recently been approved in the case of dapagliflozin in the European Union, Australia, and Mexico. Dapagliflozin and other SGLT2 inhibitors are also under health authority review in many other locales. 6
SGLT2 is localized predominately in the kidney 7 and is a member of the SGLT family of proteins that is present in humans and other mammals. SGLT2 is highly conserved with rat and human amino acid sequences exhibiting 85% identity. 8 Pharmacologic inhibition of SGLT2 prevents glucose reabsorption from the proximal tubules in the kidney resulting in a significant increase in urinary glucose loss, also known as glucosuria. Investigations of dapagliflozin, a first-in-class, selective SGLT2 inhibitor, and other similar agents have demonstrated that this novel mechanism of action can lower blood glucose levels while minimizing the risk of hypoglycemia and also providing additional mechanism-related benefits. These benefits include weight loss as a result of caloric loss due to glucose excretion and lowered blood pressure potentially as a result of the osmotic diuresis due to the increased levels of glucose in the urine. 5
Also in the SGLT2 family is sodium-glucose cotransporter 1 (SGLT1), which has been located in several tissues, including intestine, trachea, kidney, heart, brain, testis, skeletal muscle, and prostate. 7,9 SGLT1 is the major glucose and galactose transporter in the intestines. 9 Mutations in SGLT1 can result in glucose–galactose malabsorption syndrome, which is characterized by severe diarrhea and dehydration when individuals are fed a glucose- or galactose-containing diet. 9 These data suggest that selectivity between SGLT2 and SGLT1 may be an important safety consideration.
In the context of T2DM, the kidney is prone to microvascular complications, and nephropathy is a commonly observed comorbidity. 10 Theoretical concerns regarding SGLT2 inhibition included the potential for undesirable effects in the kidney, including the osmotic diuretic effect on electrolyte balance and urine volume in a population 11 already at risk of kidney disease. Therefore, a comprehensive battery of toxicology screenings was undertaken in vitro and in animal models to investigate these and other potential safety signals that may be associated with SGLT2 inhibition. The studies presented here demonstrate the long-term safety of selective SGLT2 inhibition, and the methodology represents a case study in the evaluation of potential on- and off-target-related liabilities of any new class of drugs, including investigative follow-up to provide a mechanistic basis for aberrant findings.
Materials and Methods
Chemical Reagents
Dapagliflozin ((2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol; C21H25ClO6; formula weight ∼409) and its primary 3-O-glucuronide metabolite were synthesized by Bristol-Myers Squibb Company (New Brunswick, New Jersey). In early studies, the
Comprehensive In Vivo General Toxicology Testing Program for Dapagliflozin.
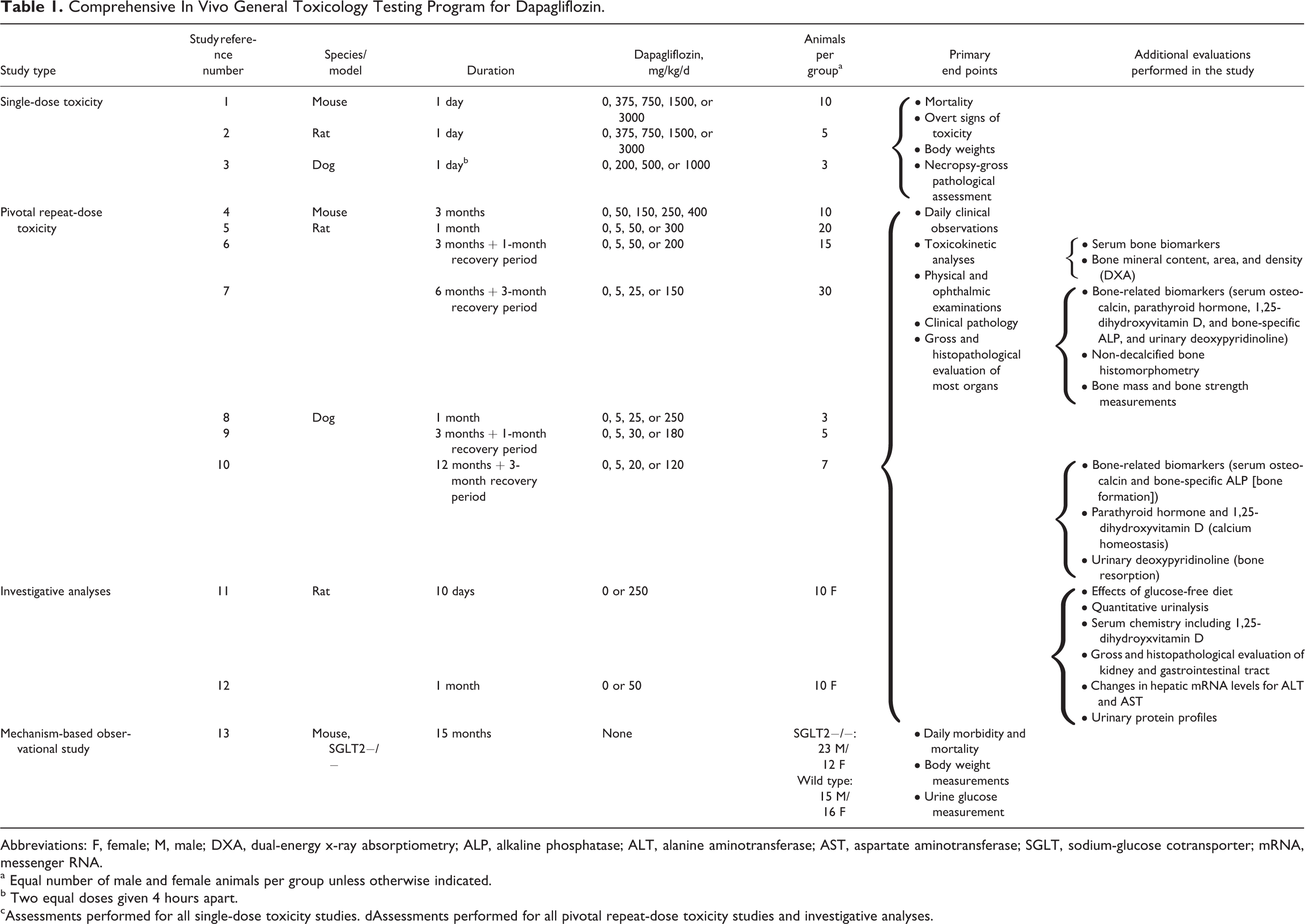
Abbreviations: F, female; M, male; DXA, dual-energy x-ray absorptiometry; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; SGLT, sodium-glucose cotransporter; mRNA, messenger RNA.
a Equal number of male and female animals per group unless otherwise indicated.
b Two equal doses given 4 hours apart. cAssessments performed for all single-dose toxicity studies. dAssessments performed for all pivotal repeat-dose toxicity studies and investigative analyses.
In Vitro Potency and Selectivity
The methodology used to evaluate the potency (half maximal inhibitory concentration [IC50]) of dapagliflozin against human and rat SGLT2 and SGLT1 cotransporters has already been described.
12
To evaluate the impact of dapagliflozin on murine and canine SGLT2 and SGLT1 transporters, the murine and canine homologs of these transporters were cloned and recombinantly expressed in Chinese hamster ovary (CHO cells; additional information in Supplementary Material). To evaluate inhibition, CHO cells expressing the relevant cotransporters were incubated in the previously described assay conditions
12
with a modified buffer that contained either 137 mmol/L NaCl (+Na) or 137 mmol/L N-methyl-
For the determination of potency of dapagliflozin versus other human homologs of SGLT2 (sodium myo-inositol cotransporter 1 [SMIT1], SGLT4, and SGLT6), CHO cell clones expressing the recombinant receptors (see Supplementary Material for details) were assayed as described earlier except that substrate concentration as well as compound concentration was varied in order to determine the inhibition constant (Ki) value for dapagliflozin inhibition of these cotransporters. For human SMIT1 and human SGLT6 assays, the substrate ( 3 H-myoinositol) concentration ranged from 0.5 to 500 μmol/L; for human SGLT4 assays, the substrate ( 14 C-AMG) ranged from 0.5 to 10 mmol/L. Dapagliflozin or phlorizin (as a positive control) was dissolved in DMSO and tested at concentrations ranging from 0.5 to 500 μmol/L (dapagliflozin) or 1 to 5000 μmol/L (phlorizin) at each substrate concentration, with the final DMSO concentration at 0.1% to 0.2%. Data were fit globally to f = Vmax × S/(S + Km × (1 + I/Ki)), where f is equal to the amount of AMG or myoinositol transported under the given substrate and inhibitor concentration conditions, where Km is the Michealis-Menton constant for the substrate, Vmax is the maximum uptake observed for the substrate in units of CPM/mg protein, S is the concentration of substrate, and I is the concentration of the inhibitor. All curve fitting was conducted using GraphPad Prism software.
The Ki values for dapagliflozin or phlorizin inhibition of human SGLT1 and SGLT2 were also determined using a nonradioactive method to detect cellular accumulation of AMG. Liquid chromatography–triple mass spectrometry (LC-MS-MS-MS) detection of AMG cellular concentration was performed on a Thermo-Fisher Accela uHPLC pump (Thermo Fisher Scientific, Waltham, MA) interfaced to an OrbiTrap Discovery mass (Thermo Fisher Scientific). Chromatography conditions were based on the previously published methods. 13 Briefly, mass spectrometry was performed in negative ionization mode using atmospheric pressure chemical ionization. The AMG was monitored using a mass-to-charge ratio (m/z) of 253.0 (acetate adduct) → m/z 193.0 (M-H) → m/z 101.1. Approximate concentration values were extrapolated from calibration curves generated using an AMG standard (Sigma-Aldrich, St. Louis, MO). The Ki was determined by varying both the concentration of substrate (AMG; 0.1-30 mmol/L) and the inhibitor (dapagliflozin; 0.003 nmol/L to 100 μmol/L or phlorizin; 0.01-1000 nmol/L). Data were fit globally to f = Vmax × S/(S + Km × (1 + I/Ki)), where f is equal to the amount of AMG or myoinositol transported under the given substrate and inhibitor concentration conditions, Km is the Michaelis-Menton constant for the substrate, Vmax is the maximum uptake observed for the substrate, S is the concentration of substrate, and I is the concentration of the inhibitor. All curve fitting was conducted using GraphPad Prism software.
The potential for dapagliflozin and its primary human metabolite, dapagliflozin 3-O-glucuronide (each at 10 μmol/L), to interact with >330 different receptors, enzymes, transporters, and/or ion channels was also evaluated to explore their potential for additional off-target effects via in vitro radioligand-binding and enzyme-binding assays (Ricerca Biosciences LLC, Taipei, Taiwan, and Cerep, Rueil-Malmaison, France).
In Vivo Nonclinical Studies
Study Design
All pivotal toxicology studies were conducted in compliance with the Good Laboratory Practice Regulations for Nonclinical Laboratory Studies of the United States Food and Drug Administration (21 CFR Part 58) and were approved by the Bristol-Myers Squibb Institutional Animal Care and Use Committee.
For toxicologic assessments, Crl: CD-1 (ICR) mice were obtained from Charles River Laboratories (Kingston, New York) and were approximately 5 weeks old at the beginning of the study. Sprague Dawley BR outbred albino rats were obtained from Harlan Laboratories (Frederick, MD) and were approximately 6 to 7 weeks old at the start of the study. Beagle dogs were obtained from Marshall Farms (North Rose, New York) and Covance Research Products (Cumberland, VA), and ranged from approximately 9 to 26 months of age for the 1-month study and 4 to 8 months for studies of longer duration at the start of the study.
A description of the methods used to generate and breed SGLT2−/− mice has been published previously. 14 Founder mice were supplied by Lexicon Pharmaceuticals, Inc (The Woodland, Texas) and back-crossed onto a C57BL/6J strain, which was then subsequently used as the wild-type (WT) control.
All studies were conducted using oral gavage administration, and a full listing of toxicology studies, dapagliflozin doses, and assessments is provided in Table 1. Single-dose toxicity was assessed in mice, rats, and dogs to determine the acute toxicity of dapagliflozin (Table 1, studies 1-3). Chronic toxicity was evaluated in a series of repeat-dose toxicity studies in mice for 3 months (Table 1, study 4), in rats for 1, 3, and 6 months (Table 1, studies 5-7), and in dogs for 1, 3, and 12 months (Table 1, studies 8-10). Postdose recovery periods ranging from 1 - 3 months were included in all chronic toxicity studies of ≥3 months' duration. In addition to these studies, targeted studies were conducted to assess the effects of dapagliflozin on calcium homeostasis, bone/tissue mineralization, proteinuria, and liver enzymes in rats (Table 1, studies 11-12). The safety profile of genetic SGLT2 ablation was also assessed in SGLT2 knockout mice for 15 months (Table 1, study 13).
Other study types, including those assessing reproductive toxicity and carcinogenicity potential, were conducted but are beyond the scope of this article and represent the topics of separate publications.
General Assessments
The systemic exposure to dapagliflozin and its metabolites was evaluated using blood samples collected via the orbital sinus or cardiac puncture prior to necropsy for mice and from tail bleeds, the cut tip of the tail, the aorta, or via the orbital sinus of rats. Blood was obtained from the external jugular veins of dogs. Toxicokinetic analysis of dapagliflozin was performed in samples taken from mice, rats, and dogs at 0.5, 1, 2, 4, 8, and 24 hours after dosing on the first (rats and dogs) and last (mice, rats, and dogs) days of the dosing periods. The blood samples were collected in tubes containing K3EDTA. All samples were stored at approximately −20°C until the time of analysis.
For toxicokinetic analyses, samples were analyzed by a validated LC-MS-MS method, and toxicokinetic parameter values were calculated using noncompartmental methods by eToolbox/Kinetica (Version 2.4; InnaPhase Corporation, Philadelphia, Pennsylvania). The standard curve range was 50 to 2000 ng/mL for each analyte. Values below the lower limit of quantitation (50 ng/mL) were treated as 0. Area under the plasma concentration–time curve (AUC) was calculated using the trapezoidal rule.
Animals in all studies were observed daily for clinical signs. Food and water consumption was measured for mice and rats (weekly) and estimated for dogs (daily). Physical and ophthalmoscopic examinations were performed by a veterinarian prior to dosing and during the last week of each study. The physical examinations included safety pharmacology evaluations of neurologic function, including cranial and peripheral nerve functions, and respiratory function. The ophthalmoscopic examination consisted of indirect ophthalmoscopy (American Optical 305 monocular indirect ophthalmoscope) using a mydriatic agent (tropicamide 1.0%).
Comprehensive assessments of hematology, coagulation, and standard serum chemistry and urinalysis (including urine volume) parameters were performed. In mice, serum chemistry and hematology were assessed prior to the end-of-study necropsy in the 3-month study. In rats, serum chemistry was determined at week 4 in the 1-month study, at weeks 6, 13/14, and 17/18 in the 3-month study, and at weeks 13, 25, and 38 in the 6-month study (Table 1, studies 5-7). In dogs, serum chemistry was determined at pretest and week 4 in the 1-month study, at pretest and weeks 4, 13, and 17 in the 3-month study, and at pretest and weeks 13, 25, 51, and at the end of the recovery period in the 12-month study (Table 1, studies 8-10). In rats, urinalyses were performed every week in the 1-month study, at weeks 3, 7, 12, and 17/18 in the 3-month study and at weeks 13, 26, and 38 in the 6-month study (Table 1, studies 5-7). In dogs, urinalyses were determined at week 4 in the 1-month study, at weeks 2, 7, 11, and 15 in the 3-month study, and at weeks 13, 25, and 51 in the 12-month study (Table 1, studies 8-10). In both the species, urinalysis was performed on urine collected over a 14 to 24-hour period.
Complete necropsies were conducted on all rodents and dogs at the time of death, or at the time of scheduled necropsies for all animals studied (study end) and included gross examination of organs and tissues. Organs weighed included adrenal glands, brain, heart, kidneys, liver, ovaries, pituitary gland, prostate gland, spleen, testes, and thyroid gland with parathyroids. Representative samples of each organ and tissue and all gross lesions were collected and fixed in 10% neutral-buffered formalin (except eyes, which were fixed in 5% glutaraldehyde [dogs only], and testes and epididymides, which were fixed in modified Davidson fixative). A comprehensive list of tissues was examined microscopically, including all key potential target organs. Tissues were processed, sectioned at approximately 6 μm, stained with hematoxylin and eosin, and examined by light microscopy for any drug-related or spontaneous lesions. Microscopic lesions were noted, and their incidences tabulated. Additional sections of testes and epididymides were stained with Periodic acid Schiff and hematoxylin, and kidney and stomach were stained with the von Kossa stain for mineralization. A full histopathological review of the collected tissues was conducted by board-certified veterinary pathologists, including peer review in each instance.
Investigative Studies
For assessment of bone health, several approaches were used. First, as part of the 3- and 6-month rat (Table 1, studies 6 and 7) and 12-month dog (Table 1, study 10) studies, a series of biomarkers of bone resorption and bone formation were evaluated, including osteocalcin, parathyroid hormone, 1,25-dihydroxy vitamin D, and bone-specific alkaline phosphatase (analyzed by Skeletech, Bothell, Washington). Second, additional measures of bone health were assessed, including bone volume by undecalcified histomorphometry analysis, bone mineral density and content by dual-energy x-ray absorptiometry (DXA) analysis, and bone strength by DXA and compression test analyses (analyzed by Skeletech).
For the assessment of the mechanism of calcium homeostasis changes in rats (Table 1, study 11), a dedicated study was conducted in which 1 group was fed a standard rodent chow diet and the other a glucose-free (fructose substituted) diet (Harlan Teklad Diet TD.04140; Harlan, Frederick, Maryland). Animals in both the groups received 250 mg/kg/d of dapagliflozin or PEG vehicle control for 10 days. Parameters for evaluation included toxicokinetics, clinical observations, food and water consumption, body weight, quantitative urinalysis, serum chemistry (including 1,25-dihydroxyvitamin D), cecal content analysis, and gross and histopathological examination of the kidney and gastrointestinal tract in all animals.
Investigative studies were also conducted to characterize dapagliflozin-induced increases in total urinary protein and minimal increases in serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activity levels after 1 month in rats (Table 1, study 12). Unique evaluations included (1) urinary protein profiles evaluated by polyacrylamide gel electrophoresis; (2) semiquantitative analysis of urinary α1-microglobulin as assessed by densitometry analysis following immunoblotting with a rabbit anti-rat urinary α1M polyclonal antibody (provided by Dr. Bo Akerstrom, Lund University, Lund, Sweden); and (3) quantitative polymerase chain reaction (qPCR) analysis of hepatic enzymes (ALT and AST messenger RNA [mRNA] levels; details provided in Supplementary Methods).
Assessment of SGLT2−/− Mouse
To evaluate the impact of genetic ablation as opposed to pharmacologic inhibition of the target, SGLT2−/− mice were directly compared with their C57BL/6J WT counterparts in a 15-month observational study. Mice were not subject to any therapeutic or pharmacologic interventions. In-life evaluations included daily morbidity and mortality observations, periodic body weight measurements, and a single urine glucose measurement (via Diastix urine glucose test strips; Bayer Corporation, Pittsburgh, Pennsylvania) near the end of the observational period. Necropsy was performed when animals were at the age of 15 months, and animals that were found dead before this time were not subject to necropsy. Criteria for evaluation at the end of study included body weights, clinical pathology, and gross and microscopic pathology. A full description of assessments is provided as Supplemental data.
Statistical Methods
Mean values from body weight and food consumption determinations, quantitative physical examination data, clinical laboratory tests, and absolute and relative (to body weight) organ weights of treated groups were compared with those of the control group by Dunnett test. Group comparisons for clinical observations and qualitative clinical pathology data and the incidence of gross findings and microscopic lesions in the treated groups were made using appropriate nonstatistical methods.
Results
In Vitro Selectivity for SGLT2 Relative to Related Family Members Across Species
Dapagliflozin inhibited SGLT2 at similar concentrations across species (Table 2). Although it is highly selective for SGLT2, selectivity differed among species, ranging from 130- to 1242-fold versus SGLT1 with greater fold selectivity in humans than rodents. The IC50 values were used to compare selectivity across species, since Ki values were not generated for dapagliflozin in species other than human. In vitro studies to more fully characterize the selectivity in humans using the more robust Ki value further demonstrated that dapagliflozin has >1400-fold selectivity for human SGLT2 relative to SGLT1, SMIT, SGLT4, and SGLT6 (Supplemental Table 1). Although the human selectivity value for SGLT2 versus SGLT1 inhibition varies somewhat depending on the values that are compared (IC50 or Ki), in both the methods, dapagliflozin is shown to be highly selective versus SGLT1. Dapagliflozin was also highly selective for SGLT2 (at least 100 000-fold) over glucose transporter (GLUT) 1, 2, and 4. 15
The IC50 Values for SGLT Inhibition by Dapagliflozin Across Species.
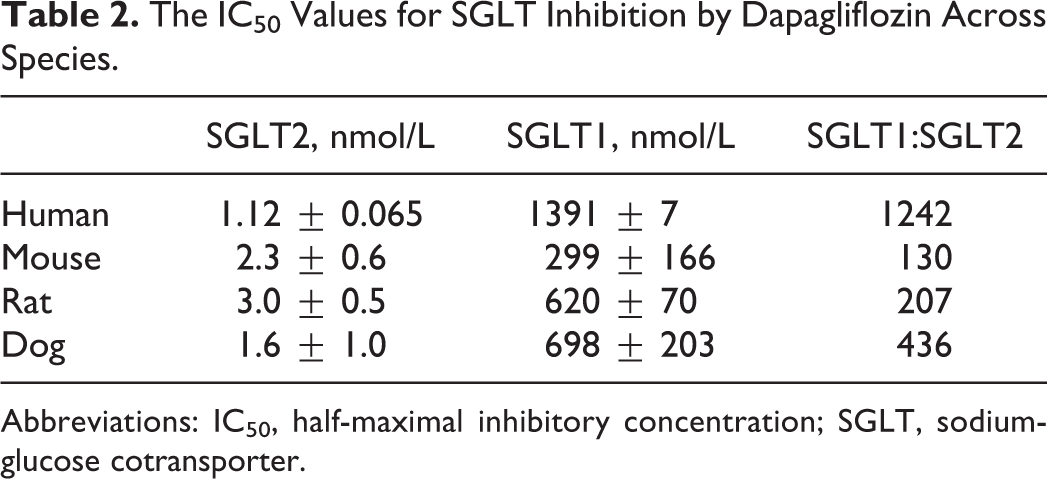
Abbreviations: IC50, half-maximal inhibitory concentration; SGLT, sodium-glucose cotransporter.
Dapagliflozin and its predominant metabolite, dapagliflozin 3-O-glucuronide, were also tested in a panel of >330 in vitro off-target liability assays at 10 μmol/L concentrations (based upon molecular weight: 5.03 μg/mL for dapagliflozin and 5.85 μg/mL for 3-O-glucuronide). No significant activity (defined as >50% inhibition) was detected in any of the assays.
Toxicokinetic Analyses
Systemic exposures were measured throughout all dapagliflozin safety studies to assess relative safety margins compared with clinical exposures in human patients. Exposure to dapagliflozin was dose related and approximately dose proportional (Supplemental Table 2). There was slight to no accumulation after repeated dosing. No significant sex-related differences were noted in exposure in dogs, but in rodents, exposure in females was generally greater than in males. In the chronic 3- and 6-month rat toxicity studies, doses ranged from 5 to 200 mg/kg/d with corresponding steady state AUC exposures ranging from 39 to 1440 μg h/mL. Similarly, in the 3- and 12-month dog toxicity studies, doses ranged from 5 to 180 mg/kg/d with steady state AUC exposures ranging from 59 to 2400 μg h/mL.
Two dapagliflozin metabolites were also assessed, the major (in humans) pharmacologically inactive metabolite dapagliflozin 3-O-glucuronide (a stable ether glucuronide) and an O-deethylated pharmacologically active dapagliflozin metabolite. The O-deethylated metabolite was measured in all studies; however, results are not reported here as it was later found to be a minor metabolite in humans and not associated with any observable toxicologic liabilities. Although systemic exposures (AUC) to the dapagliflozin 3-O-glucuronide metabolite were relatively low in rats and dogs, the amounts of dapagliflozin 3-O-glucuronide metabolite formed at the no observed adverse effect level (based on AUC) in both the 6-month rat and 12-month dog repeat-dose toxicity studies were estimated to be approximately equal to or greater than dapagliflozin 3-O-glucuronide exposures in humans at expected clinical exposures (maximum observed plasma concentration [Cmax] 0.196 µg/mL and AUC 0.837 µg h/mL at the maximum recommended clinical dose of 10 mg daily dose). As this is a pharmacologically inactive metabolite and is not associated with any toxicologic liabilities, results are not reported here.
Toxicologic Assessment of Dapagliflozin
Rat (rodent) and dog (nonrodent) were selected as the main species for nonclinical toxicologic evaluations, as they are standard models for toxicity assessments with a large historical database and are sensitive to the pharmacologic effects of dapagliflozin. Single oral doses up to 750 mg/kg in mice, 375 mg/kg in rats, and 1000 mg/kg in dogs were well tolerated, with no indications of toxicity. Lethality was observed at doses of 3000 mg/kg and 750 mg/kg in mice and rats, respectively, for single-dose studies. The cause of death was not identified in any of the single-dose studies, but at the doses tested, these effects present no relevance to human safety or risk assessment.
In repeat-dose toxicity studies, dapagliflozin treatment at all doses was associated with expected, pharmacologically induced increases in urinary glucose excretion (>350× relative to control), with compensatory responses including increased food consumption, water intake, and urine volume, and, in most studies, decreased urine osmolality (data not shown). Decreased body weights or decreased body weight gains were observed in dogs and rats, respectively, but were not associated with a decline in the overall health of the animals except at very high-exposure multiples relative to the maximum recommended human dose (MRHD; ≥2100× in rats and ≥4400× in dogs). No adverse effects were observed at supratherapeutic doses up to 50 mg/kg/d in rats and 180 mg/kg/d in dogs at >600 times the MRHD (Table 3). At higher doses and exposures, abdominal distension and diarrhea were observed in both rat (≥3000× MRHD) and dog (≥3200× MRHD), consistent with symptoms of glucose–galactose malabsorption as a result of SGLT1 inhibition. Additional changes in rats included distension, rales, dyspnea, decreased activity, and urine-stained coats.
Delineation of Exposure Margins Across Species and Study Durations for No Observed Adverse Effect Levels (NOAEL) and Lowest Observable Adverse Effect Levels (LOEL) in Pivotal Toxicology Studies.
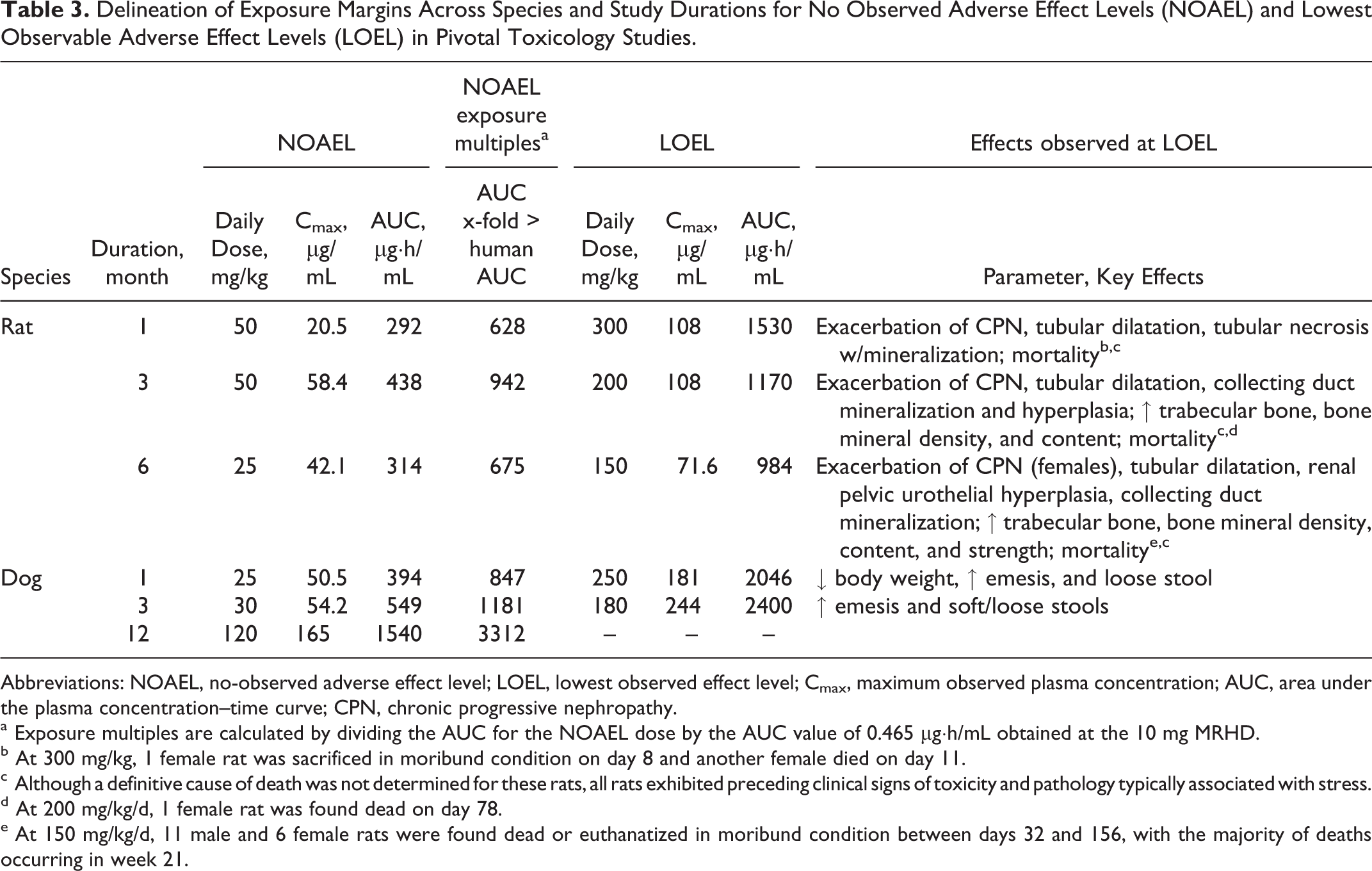
Abbreviations: NOAEL, no-observed adverse effect level; LOEL, lowest observed effect level; Cmax, maximum observed plasma concentration; AUC, area under the plasma concentration–time curve; CPN, chronic progressive nephropathy.
a Exposure multiples are calculated by dividing the AUC for the NOAEL dose by the AUC value of 0.465 µg·h/mL obtained at the 10 mg MRHD.
b At 300 mg/kg, 1 female rat was sacrificed in moribund condition on day 8 and another female died on day 11.
c Although a definitive cause of death was not determined for these rats, all rats exhibited preceding clinical signs of toxicity and pathology typically associated with stress.
d At 200 mg/kg/d, 1 female rat was found dead on day 78.
e At 150 mg/kg/d, 11 male and 6 female rats were found dead or euthanatized in moribund condition between days 32 and 156, with the majority of deaths occurring in week 21.
Drug-related increases were observed in urinary volume (1.2× to 14×) and urinary electrolytes in all animal studies. Increased urinary excretion of calcium was observed in all studies and at most doses in rats (1.6× to 15×) and dogs (1.8× to 25×; Table 4). Increased urinary sodium excretion was observed in rats and dogs after 1 and 3 months of treatment (1.3× to 6.0×) but not in studies of longer duration. Urinary phosphorus increases were also observed in rats after 6 months of treatment (1.6× to 3.2×) and in dogs in the 12-month study (1.3× to 2.4×). Despite increases in urinary electrolytes, however, there were no significant decreases in serum electrolytes in either the rat or the dog toxicity studies, suggesting that overall electrolyte balance was being appropriately maintained via expected physiologic mechanisms. Additional serum analysis showed increased cholesterol levels in rats after 1 month of treatment (1.1× to 1.2×) and in dogs at the highest dose after 1, 3, and 12 months of treatment (1.2× to 1.6×). Changes in urinary phosphorus and cholesterol are likely related to the increased food consumption.
Selected Changes in Clinical Pathology.

Abbreviations: M, male; F, female; PTH, parathyroid hormone.
a Increased total serum calcium (1.1×; in males, only in week 25); in females, increased ionized calcium (1.1×).
b Week 13, only in females; week 25, at 25 mg/kg/d, only in females.
c Week 13, at 5 and 20 mg/kg/d, only in females; in week 52, only at 20 mg/kg/d in males and 120 mg/kg/d.
d At 5 mg/kg/d, decreased only in males at week 12.
e At 5 and 50 mg/kg/d, only in males.
f Excluding males given dose of 300 mg/kg.
g Week 13, at 5 mg/kg/d, only in females.
Increases in urinary protein (1.8× to 5×) were noted at all doses in rats after 3 and 6 months of treatment and at high doses (≥120 mg/kg/d; ≥3300× human exposure) in dogs at 3 and 12 months. However, these increases in urinary protein were not considered indicators of toxicity based on an investigative study in rats (Table 1, study 12), which showed no qualitative differences in urinary protein profiles between control and dapagliflozin-treated rats. In addition, both control and dapagliflozin-treated rats had a similar amount of urinary α1M, a marker that increases with proximal tubular injury in humans. Other findings in 1-, 3-, and 6-month studies in rats included increased blood urea nitrogen (1.3× to 1.6×) at most doses, renal tubular mineralization and dilatation (Figure 1), and exacerbation of chronic progressive nephropathy (CPN; Figure 1) at high doses (≥2100× MRHD). No adverse dapagliflozin-induced renal histopathology was observed in repeat-dose mouse or dog toxicity studies (Figure 1) at any dose even at very high-exposure multiples (2300× and 3300× MRHD, respectively).
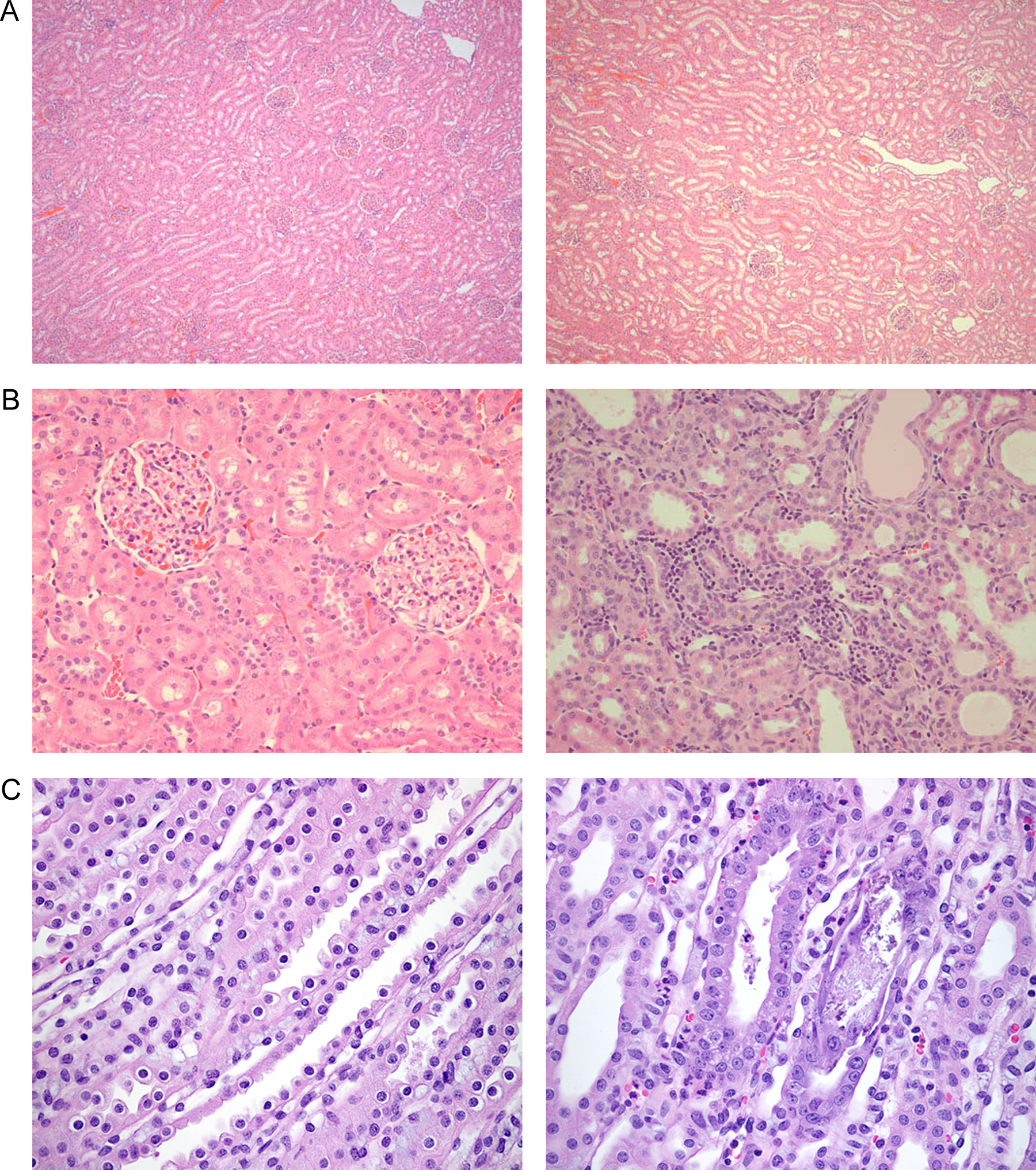
Histopathological sections of kidney in dapagliflozin and control animals. A, In dogs, no difference in kidney histopathology (40× magnification) between male control animals (left) and those receiving high doses of dapagliflozin (right) was seen. B, In rats, exacerbated chronic progressive nephropathy (CPN) was seen (200× magnification) in the kidney of animals receiving high-dose dapagliflozin (right) compared with control animals (left). On the right is the kidney cortex from a high-dose dapagliflozin-treated rat, with cortical tubules showing features of CPN including multifocal-dilated tubules filled with proteinaceous fluid, tubules with cytoplasmic basophilia and focally thickened tubular basement membranes, and foci of increased mononuclear-cell infiltrates in the interstitium. On the left is the control kidney cortex showing glomeruli and renal cortical tubules with no evidence of CPN. C, In rats fed with a standard diet (right), tubular degeneration with reactive hyperplasia and interstitial inflammation was seen compared with rats fed a fructose-free diet (left; 400× magnification).
A minimally increased incidence of lesions associated with urinary tract infections was noted in female rats and dogs treated with dapagliflozin for 1 month and 12 months, respectively. In the 1-month rat study (Table 1, study 5), effects occurred in 2 female rats and included acute inflammation of the renal pelvis; chronic active inflammation, necrosis, and bacterial aggregates in the urinary bladder and the kidney; and hydronephrosis in the kidney. These lesions were consistent with an ascending urinary tract infection. In the 12-month dog study (Table 1, study 10), minimal to slight suburothelial inflammation was present in the renal pelvis and/or papilla of 4 dapagliflozin-treated females (1 receiving 5 mg/kg/d, euthanized at month 6; 1 receiving 20 mg/kg/d, euthanized at 12 months; 2 receiving 120 mg/kg/d, euthanized at 6 and 12 months, respectively) but was not seen in any control females. In these dogs, there was an associated minimal to slight inflammation in the urinary bladder related to the urinary tract infections.
Other anatomic pathology changes included increased absolute and relative adrenal weights in studies from 1 to 6 months' duration in rats (≥350× MRHD) and in the 12-month study in dogs (≥130× MRHD). There were no histopathological correlates to the increased adrenal weights in rats or dogs except in the 6-month rat study, where slight exacerbation of hypertrophy/vacuolation of the zona glomerulosa of the adrenal cortex was observed. This specific finding was considered a physiologic compensatory response secondary to increased sodium excretion. 16,17 Increased levels of AST and ALT were also noted in the 1-, 3-, and 6-month studies in rats (1.2× to 1.8×). However, increases in AST and ALT were not considered indicators of hepatotoxicity due to the low magnitude of the increase and the lack of accompanying microscopic changes. These increases were further analyzed in a 1-month repeat-dose investigative study conducted in rats (Table 1, study 12), in which qPCR analysis showed statistically significant increases (P <.001) in hepatic AST and ALT mRNA levels (1.5× and 3.0×, respectively), suggesting an increased production within hepatocytes that would be released following normal hepatocellular turnover rather than any damage-related leakage. No significant dapagliflozin-related elevations in serum transaminases were observed in dogs.
Increases in bone formation and tissue mineralization, which were associated with increased serum calcium, were present in rats at doses ≥150 mg/kg/d (≥2100× MRHD; Figure 2) but not in dogs at up to the highest doses tested (>3000× MRHD). Increased bone formation was measured and observed in the 3- and 6-month studies in rats (Table 1, studies 6 and 7) and was characterized by an increase in the thickness of the primary trabeculae in the sternum (Figure 2) and femur due to the retention of cartilage and increased deposition of mineralized osteoid on the surface. The increase in bone formation observed by routine microscopic examination was accompanied by an increase in bone mass and strength as assessed by DXA, undecalcified histology, and histomorphometry and lumbar vertebra compression test analysis. Mineralization of soft tissue was also observed in the 1-, 3-, and 6-month studies in rats (Table 1, studies 6 and 7; Figure 2), in which minimal to moderate vascular mineralization was observed in the aorta and periaortic arteries, great vessels of the heart and myocardial arteries, kidneys, stomach, and many other tissues examined.
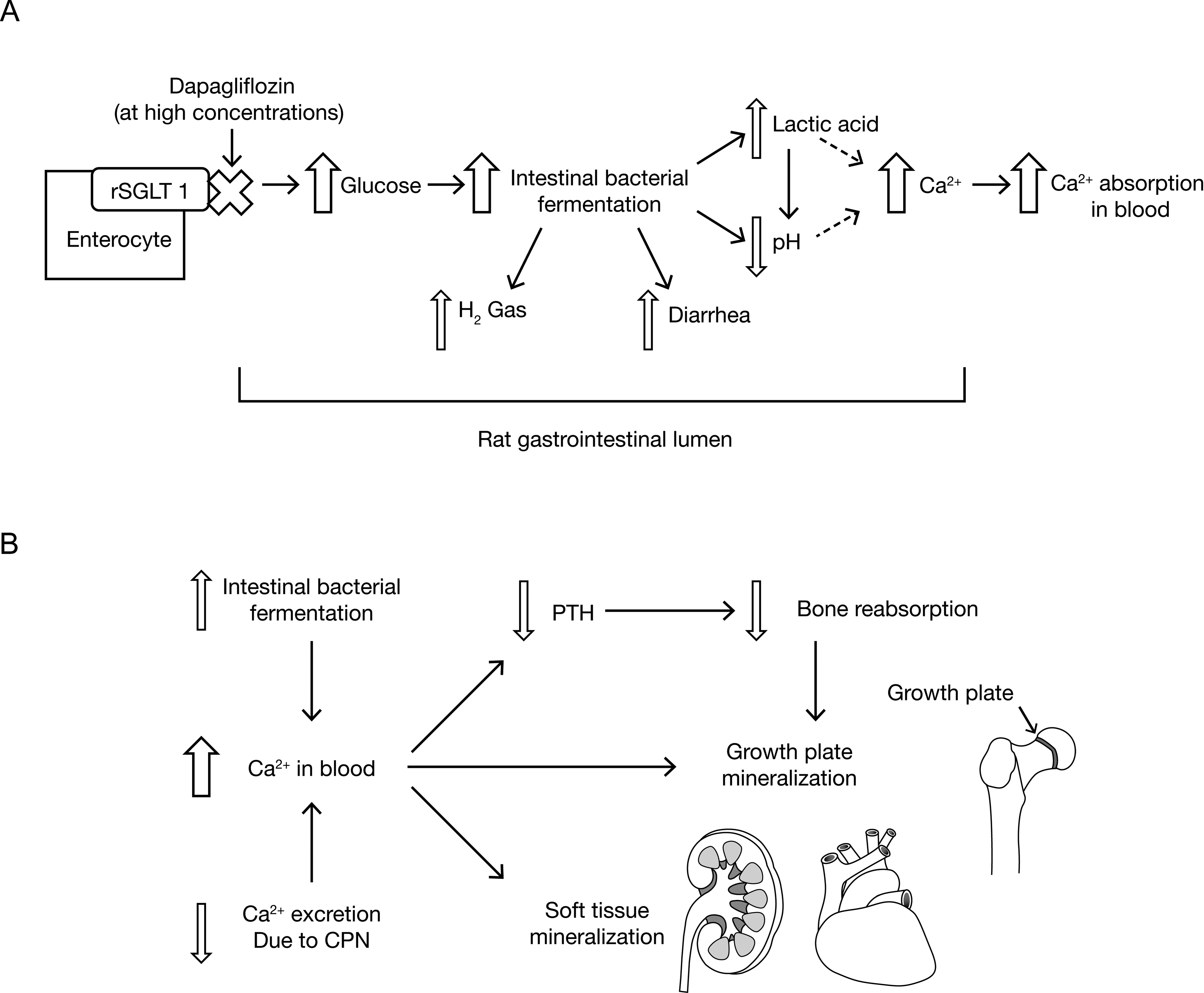
Proposed mechanism for dapagliflozin-related increases in trabecular bone in rats. A, High concentrations of dapagliflozin inhibit intestinal rat sodium-glucose cotransporter 1 (SGLT1) and increase glucose concentrations in the intestinal lumen resulting in increased intestinal bacterial fermentation, decreases in pH, and, subsequently, increased calcium absorption in the blood. B, Increased levels of blood calcium, due to intestinal bacterial fermentation, in conjunction with reduced urinary calcium excretion as a result of CPN specific to the rat, ultimately results in increased bone (C) and tissue mineralization (D). C, Histopathological images (40× magnification) showing increased trabecular bone formation in the tibia of female rats that received high-dose dapagliflozin (right) versus female control rats (left). D, Histopathological images (40× magnification) showing increased heart mineralization in myocardial and great vessels (blue-purple staining) in female rats that received high-dose dapagliflozin (right) compared with female control rats (left). E, Histopathological images (400× magnification) showing distinct area of calcification (black, von Kossa stain) in the gastric mucosa of dapagliflozin-treated rats fed a standard diet (right) in comparison with the gastric mucosa of dapagliflozin-treated rats fed a glucose-free diet (left).
Investigative studies were conducted to more fully assess the increased serum and urinary calcium levels and tissue mineralization observed in rats. Female rats were given a glucose-free diet and compared with rats receiving a standard diet to determine whether dapagliflozin-induced effects observed solely in the rat were associated with off-target inhibition of SGLT1 and reduced glucose absorption in the intestine (Table 1, study 11). Dapagliflozin exposures were similar between the 2 groups. After 10 days of dosing with dapagliflozin, principal dapagliflozin-related findings were generally similar to those observed in the previous studies in rats fed the standard diet. Findings that were unique to the rats fed the standard, glucose-containing diet were signs of loose, unformed, and or liquid feces; increased total serum (1.1× control [no dapagliflozin]) and ionized (1.1× control) calcium; and increased incidence and severity of single-cell necrosis in the cecal mucosa. Dapagliflozin-treated rats fed the glucose-free (fructose-substituted) diet had a smaller increase in cecal weight than those that received standard diet (31% vs 79%), and the cecal content contained a total soluble calcium concentration that was 2.1 times greater than control. Additionally, rats fed the glucose-free diet did not experience renal medullary tubular degeneration (Figure 1) and gastric mucosal mineralization (Figure 2) and had decreased urinary calcium levels compared with rats fed a standard diet.
Assessment of SGLT2−/− Mice
In addition to toxicologic assessments of pharmacologic SGLT2 inhibition, evaluations of SGLT2 genetic ablation were conducted in 15-month-old SGLT2−/− mice. As expected, marked glucose excretion (∼2000 mg/dL) was observed in SGLT2−/− mice, while WT mice did not exhibit glucose excretion (0 mg/dL). In both the groups, a similar number of mice survived to age 15 months (∼85%). No biologically relevant differences were observed between SGLT2−/− and WT mice in 15-month body weights or clinical pathology parameters. Microscopic evaluation of urinary bladder and kidney showed no difference between SGLT2−/− and WT mice. Similarly, evaluation of the liver, heart, pancreas, adrenal glands, thyroids, spleen, female reproductive tract, male sex glands, skin, brain, and skull did not reveal any adverse effect attributable to SGLT2 gene deletion.
Discussion
Safety liabilities are a leading factor in attrition for pharmaceutical research and development. 18 This places greater emphasis on the preclinical evaluation of drug safety to identify potential hazards and provide robust risk assessment. The studies presented herein with the SGLT2 inhibitor dapagliflozin serve as a case study for the evaluation of potential on- and off-target-related liabilities of any new class of drugs, including investigative follow-up studies that provide a mechanistic basis for aberrant findings.
One key first step was to evaluate the potential interactions with related proteins across species as well as a wide spectrum of unintended pharmacologic targets. Dapagliflozin was demonstrated to be highly selective for SGLT2 in comparison with proteins within the same family. Dapagliflozin was also shown to be highly selective for SGLT2 over other glucose transporters, specifically of GLUT 1, 2, and 4. 15 In vitro, large screening assays showed that dapagliflozin and its major human glucuronide metabolite did not significantly interact (defined as >50% inhibition) with >330 enzymes, transporters, ion channels, and receptors. Overall, these data are consistent with the in vivo studies showing that most effects were related to SGLT2 inhibition, although the potential for off-target effects at suprapharmacological doses cannot be excluded. The greater selectivity of dapagliflozin for SGLT2 over SGLT1 was demonstrated for humans and all animal models tested with the greatest selectivity observed in humans (Table 2). These results indicate that dapagliflozin is highly selective for SGLT2 compared with SGLT1, and thus in vivo treatment with dapagliflozin has a low potential for off-target adverse effects in humans.
In vivo, exaggerated pharmacology and off-target effects were assessed in mouse, rat, and dog in order to explore effects beyond the pharmacologic range of exposures and in the potential range where unintended effects might manifest. Assessments included dapagliflozin metabolites including dapagliflozin-3-O-glucuronide, the major metabolite in humans, and a pharmacologically active metabolite formed at high levels in animals but not humans. As a result of exposure to supratherapeutic levels of dapagliflozin, animals in the toxicology studies were exposed to levels of dapagliflozin-3-O-glucuronide that were equivalent (in rodents) or higher (in dogs) than exposures in humans. Toxicokinetic analyses of supratherapeutic doses demonstrated that repeated daily dosing resulted in little to no drug accumulation and that systemic exposure to dapagliflozin continued to increase with dose allowing exposures that were upward of 5000 times those observed at the MRHD. However, at supratherapeutic doses and with exposures to dapagliflozin and its metabolites that generally far exceeded human exposures, no significant toxicity related to SGLT2 inhibition was observed in studies of rats (up to 6 to month' duration) and dogs (up to 12 to month' duration) treated over the long term with dapagliflozin (Table 3).
Dapagliflozin effects at suprapharmacologic doses were mostly consistent with the anticipated pharmacologic action of the drug. Consistent findings for dapagliflozin in rat and dog either reflected pharmacology of the primary SGLT2 target (glucosuria) or were likely sequelae to the osmotic diuretic effect due to increased glucose excretion. Other effects also considered secondary to caloric loss due to urinary glucose excretion and its associated diuresis included increased food consumption, water intake, and urine volume and transiently increased sodium excretion. The observed changes in urine composition and increases in urinary electrolytes are consistent with the effects of osmotic diuretics, such as mannitol, which increase the urinary excretion of most electrolytes without deleterious effects. 19 Changes in urinary sodium excretion in rats and dogs were observed in studies of up to 3 months in duration but not in studies of longer duration, likely due to a physiologic compensatory response associated with increased aldosterone production. 20 Decreases in body weight were considered a secondary pharmacologic change pursuant to the markedly enhanced excretion of glucose and inability of the dogs to compensate for this loss of energy source due to their fixed dietary ration. The SGLT2−/− mice similarly showed marked glucosuria and increases in food and water consumption. 21 Given the relatively small magnitude of these effects, even at very large multiples of human exposures, these suprapharmacologic effects are unlikely to pose a safety risk to humans.
Careful assessment of the kidney, the target organ of dapagliflozin’s action, also showed no toxicity that would be relevant to humans (Figure 1). No renal histopathological findings were observed in dogs at exposure multiples >3000-fold those observed at the MRHD and over long-term exposure, and no renal toxicity was observed in SGLT2−/− mice after 15 months. In rats, effects on the kidney were observed only at the highest exposures and were likely related to exacerbation of CPN, a spontaneous disease that is unique to the aging laboratory rat (and not relevant to humans), 22 or off-target SGLT1 inhibition (discussed in greater detail below). However, interaction with other unidentified factors at suprapharmacologic concentrations in the rat cannot be definitely excluded. Although the presence of urinary protein was observed in both rat and dog studies, it was not correlated with histopathological changes; urinary protein patterns and biomarkers of proximal tubular injury showed no dapagliflozin-related changes; and investigative analysis showed that urinary protein excretion was likely related to profound diuresis. Similar increases in urinary proteins have been observed in long-term toxicity studies in rats with the osmotic diuretic erythritol. 23 Moreover, diuretics have a long history of safe use in humans. The species-specific effects and high-exposure multiple at which renal toxicity was observed along with the well-known safety profile of diuretic drugs suggest that dapagliflozin is unlikely to cause renal toxicity in humans at the MRHD.
Clinical trials of dapagliflozin have shown that at the MRHD, dapagliflozin is associated with a slight increase in urinary tract infections compared with placebo (4.3% vs 3.7%). 24 There was a slightly increased incidence of lesions associated with urinary tract infections in both rats and dogs; however, these lesions were rarely observed and not dose related, which generally supports the clinical data. Importantly, these effects were generally mild and are anticipated to be clinically manageable in routine clinical practice.
In addition to the primary effects anticipated from the mechanism of action, dapagliflozin was associated with some compensatory changes. For instance, increases in adrenal gland weights observed in rats and dogs were an expected physiologic response to likely increases in aldosterone production that would occur in response to a pharmacologically mediated excretion of sodium. 25 Although increases in adrenal gland weight in rats correlated with morphologic changes, there were no histopathological correlates observed in dogs. This finding is not considered to be adverse in either rat or dog. Similarly, some minor increases (<2-fold) in ALT and AST levels, which were demonstrated to be the consequence of increased gene expression (<3-fold), may be a result of dapagliflozin-induced nutritional fluctuations that lead to changes in gluconeogenesis, which has been shown to have an impact on ALT or AST gene expression. 26 An increase in gluconeogenesis would be expected in dapagliflozin-treated rats in repeat-dose toxicity studies given the substantial increase in the observed urinary glucose excretion and demonstrated gluconeogenesis increases observed in dapagliflozin pharmacology studies in rats. 27,28 Thus, the slight elevations in serum ALT/AST activities in rats more likely reflect higher than normal levels released into circulation following physiologically routine hepatocellular turnover. Importantly, there was no histologic evidence of any hepatotoxicity in mice, rats, or dogs treated chronically with dapagliflozin at exposures up to 5000-fold greater than those expected from 10 mg in humans.
Some unique dapagliflozin-related changes were observed only at high dapagliflozin exposures in rats, but not dogs, including disturbances in calcium homeostasis, tissue mineralization, and increased trabecular bone and renal medullary tubular degeneration, but hypothesis-driven investigations helped demonstrate that they were likely off-target effects and not a true safety concern for humans. Specifically, we hypothesized the effects to be the result of SGLT1 inhibition. This hypothesis is consistent with symptoms (primarily diarrhea) associated with glucose–galactose malabsorption. 9 The hypothesis is also consistent with the results of in vitro selectivity assessments that show a higher degree of dapagliflozin selectivity for SGLT2 over SGLT1 in dogs (436 ×) when compared to rats (207 ×) and may explain in part why rats are more susceptible than dogs to these observed effects. We propose the following mechanism (Figure 2) 29 –32 : inhibition of SGLT1 increases the levels of intestinal glucose, resulting in increased bacterial fermentation, a more acidic environment that increases ionized calcium, and, subsequently, increased calcium absorption from the gut into the blood. The CPN, which appears to be a rat-specific background lesion that is not relevant to humans, 33 would also have likely contributed to increased serum calcium levels by negatively affecting the ability of the kidney to properly excrete calcium in the urine. Increased systemic calcium would not only result in decreased levels of parathyroid hormone and decreased bone resorption 34 but would also serve as a substrate for increased calcium deposition and tissue mineralization. To that end, rats fed a glucose-free diet versus rats fed a normal rat diet containing glucose demonstrated only marginal or no increases in serum ionized calcium. Renal medullary tubular degeneration and gastric mucosal mineralization were also not observed in rats fed the glucose-free diet. These results support the overall hypothesis that increased serum calcium and tissue mineralization is a result of off-target inhibition of SGLT1. Importantly, off-target inhibition of SGLT1 by dapagliflozin is not expected to be a concern for humans due to the high exposures to dapagliflozin required for the observed calcium and bone effects as well as the much higher degree of selectivity of dapagliflozin for SGLT2 over SGLT1 in humans (>1400×) compared with rat (∼200×). It remains to be determined whether or not the same also applies to other SGLT2 inhibitors in development which have reported less selectivity versus SGLT1. 35
As part of the toxicologic assessments, bone health-related parameters were also specifically assessed, including bone serum biomarkers and functional assays for bone volume, mineral density and content, and strength. Several bone-related changes were observed in rats, including decreased levels of dihydroxyvitamin D, calcitonin, and parathyroid hormones and increased bone mass, strength, and mineralization. Unlike rats, bone-related changes were not observed in dogs. The rat-specific increases in trabecular bone accretion are therefore likely the result of increased serum calcium as a result of off-target SGLT1 inhibition at high doses of dapagliflozin. Moreover, the thickening of bone in rats was also likely attributed, at least in part, to the unique physiology of rats, in which bone growth plates are active into old age, which results in susceptibility to dapagliflozin-induced increases in trabecular bone formation versus other species such as mature dogs or humans, in which the growth plate closes at/near sexual maturity. 36
Based on the studies conducted, the dose ranges used, and the types of effects observed, the toxicity profile of the selective SGLT2 inhibitor dapagliflozin has been well characterized in the nonclinical species and has a profile consistent with that expected for its mechanism of action. These studies, which were conducted at exposures well above the expected therapeutic exposure in humans, support the conclusion that selective SGLT2 inhibitors can be safely and effectively used in appropriate human patient populations for long-term treatment of T2DM.
Footnotes
Acknowledgments
The authors would like to thank Dr Bo Akerstrom from Lund University, Lund, Sweden, for providing the α1M polyclonal antibody. The authors would also like to acknowledge many current and former members of the R&D organization at Bristol-Myers Squibb and our contract laboratory organization partners who actively contributed to the battery of preclinical studies disclosed in this publication.
Authors’ Note
Declaration of Conflicting Interests
All authors are employees of Bristol-Myers Squibb and shareholders of Bristol-Myers Squibb stock.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by AstraZeneca and Bristol-Myers Squibb. Medical writing support was provided by Alexandra Silveira, PhD, of PPSI (a PAREXEL company) under contract by AstraZeneca and Bristol-Myers Squibb.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.