
Abstract
Cigarette smoke condensate (CSC) has been reported to elicit morphological and transcriptional changes that suggest epithelial-to-mesenchymal transition (EMT) in cultured bronchial epithelial cells. The transdifferentiation potential of acute and prolonged CSC exposure alone or in combination with the β-catenin inhibitor, FH535, was investigated in the bronchial epithelial cell line, BEAS-2B, through assessment of cell morphology, transcript expression, protein expression, and protein localization. Changes in morphology, β-catenin translocation, E-cadherin expression, metalloproteinase expression, and fibronectin could be demonstrated independent of molecular or physiological evidence of EMT. FH535 was shown to increase CSC-induced cytotoxicity and depress β-catenin expression. However, FH535 effects were not limited to the β-catenin pathway as it also blocked the expression of early growth responsive protein 1 (EGR-1) target genes, fibronectin and phosphatase and tensin homologue, without affecting EGR-1 nuclear accumulation.
Introduction
Tobacco smoke is a complex mixture of chemicals, which is classified by the International Agency for Research on Cancer (IARC) as a group 1 human carcinogen. 1 While the epidemiology demonstrates a clear association between smoking and cancer, the underlying molecular mechanisms of disease causation and/or progression following smoke exposure are largely unknown. Further, efforts to identify a mechanism of action has been complicated by difficulties in generating rodent models of cigarette smoke-induced disease. 2 Epithelial-to-mesenchymal transition (EMT) is a transdifferentiation process whereby epithelial cells gain mobility and become more resistant to cell death, which has recently gained popularity as a possible cigarette smoke-induced carcinogenesis mechanism.3,4
During EMT, epithelial cells shed much of their epithelial identity and assume a mesenchymal phenotype. 5 Following EMT, epithelial cells downregulate proteins that function in the tight junction complex, have a limited ability to form nonpermeable barriers, gain the ability to reorganize and invade the basement membrane, become more resistant to apoptosis and anoikis and express many markers once thought specific only to cells of mesenchymal origin. 5 The role of EMT in carcinogenesis is still somewhat controversial, but many of the markers of EMT including loss of E-cadherin expression and translocation of β-catenin are the well-described prognostic markers of epithelial-derived cancers.6–7
β-Catenin translocation is such a common occurrence in EMT, that it is often considered a hallmark of EMT. 6 In nontransformed cells, β-catenin action is opposed by E-Cadherin-dependent sequestration at the cytoplasmic membrane or by the glycogen synthase kinase 3 (GSK3)-dependent targeting to the proteasome. 8 Upon stimulation of the classical WNT or noncanonical WNT/Ca2+ pathways, β-catenin increases in both the cytoplasm and nucleus due to the inhibition of GSK3 activity and insufficient degradation. 9 However, following EMT, reduced expression of E-cadherin is seemingly responsible for much of the changes to β-catenin signaling, although specific roles for the GSK3 pathway are also likely. 10
E-cadherin loss is the defining molecular event of EMT, but it is only an effecter protein of the pathway. Control of EMT lies with the transcription factor (TR) families SNAIL, ZEB, and TWIST, which depress E-cadherin messenger RNA (mRNA) and induce the expression of mesenchymal markers, like smooth muscle α-actin (α-SMA) or vimentin. 5 Recent independent reports have demonstrated a role for EGR-1 in EMT through SNAIL induction and have linked cigarette smoke condensate (CSC) to EGR-1 induction.11,12 No direct role for EGR-1 has been demonstrated in EMT, but multiple potential cross talk pathways exist. For instance, EGR-1 has been demonstrated to regulate multiple factors important to EMT, including PTEN, fibronectin, TP53, and β-catenin.13–15
In this work, the small molecule β-catenin inhibitor FH535 induces a mesenchymal phenotype, reduces mitochondrial membrane potential, and inhibits the expression of β-catenin, fibronectin, osteopontin, and PTEN in BEAS-2B cells. When combined with CSC, FH535 potentiates many of the cellular responses to CSC, including depression of mitochondrial membrane potential and inhibition of gene transcription targets. However, a small number of targets induced by CSC are specifically blocked by FH535 treatment and implicate the TR EGR-1 in cell survival following CSC. This is the first report to identify the potential of FH535 as an inhibitor of EGR-1 transcription and to demonstrate that CSC can induce loss of epithelial signals independent of mesenchymal marker induction.
Materials and Methods
Cell Culture and Treatment
The SV40 large T antigen transformed human bronchial epithelial cell line, and BEAS-2B were obtained from ATCC and were cultured in serum-free basal epithelial growth media supplemented with bovine pituitary extract, hydrocortisone, epidermal growth factor, epinephrine, insulin, triiodothyronine, transferrin, gentamicin, and retinoic acid (Lonza, Walkersville, Maryland) on collagen I (BD, Franklin Lakes, New Jersey) coated plates for routine culturing and experiments. BEAS-2B cells were between passage 40 and 55. CALU-3 cells were obtained from ATCC and were cultured in F12k media (Invitrogen, Carlsbad, California) supplemented with 20% fetal bovine serum ([FBS]; ATCC, Manassas, Virginia) and 1% penicillin/streptomycin (Sigma, St Louis, Missouri) on standard tissue culture plates. All cells were plated at 10 000 cells per well on a 96-well plate 24 hours prior to the addition of CSC.
Smoke Fraction Generation and Treatment
The CSC was generated by smoking 20 Kentucky Reference 3R4F cigarettes (University of Kentucky, Lexington) through a single Cambridge pad via a RM/CS 20 port rotary smoker (Borgwaldt Technik GmbH, Germany) under standard conditions, similar to the International Standards Organization smoking regime (2 seconds draw, 35 mL puff volume, 1 minute puff interval), followed with immediate extraction to a final concentration of 40 mg/mL in dimethyl sulfoxide ([DMSO]; Sigma) and storage at −20°C.
Thirty minutes prior to treatment with CSC, fresh media was applied to the cells containing 0.25% vol/vol DMSO with 700 nmol/L FH535 or 6 μmol/L GW9669 (EMD Chemicals, Temecula, California), except where specifically noted. All pretreatments were performed alone, in combination with a sham treatment of DMSO and in combination with CSC. Data from pretreatment alone and pretreatment combined with sham were equivocal in all experiments and only those combined with sham are presented here. The CSC was then applied to cells as a serial dilution from a maximum concentration of 100 μg/mL (0.25% vol/vol) for times indicated. For chronic treatments, the media and treatments were concurrently reapplied every 48 hours. Carbonyl cyanide 3-chlorophenylhydrazone (CCCP, Sigma) was added to the cells 30 minutes. prior to sample harvest in DMSO.
β-Catenin Translocation Assay
Nuclear and cytoplasmic β-catenin levels were quantified using the β-catenin activation kit on a Cellomics Arrayscan VTI (ThermoFisher, Pittsburgh, Pennsylvania), according to the manufacturer’s protocol. Briefly, β-catenin was detected by immunocytochemistry with a polyclonal rabbit antibody and fluorescently labeled secondary after fixation in 3.7% warm formaldehyde for 15 minutes, permeabilization in 1% Triton-X 100, and blockage in 2% FBS (Sigma) and blocking buffer.
Immunocytochemistry
At the completion of treatment, BEAS-2B were fixed at room temperature following addition of prewarmed 37°C, 3.7% formaldehyde-phosphate-buffered saline (PBS) for 15 minutes and washed twice with PBS. Cells were permeabilized with 1% Triton X100-PBS and blocked with 1% bovine serum albumin-PBS supplemented with 2% FBS prior to addition of antibodies or stains. Primary antibodies were used at 1:100 for 1 hour at room temperature. Secondary antibodies were used at 1:1000 for 1 hour at room temperature and were either Dylight (ThermoFisher) or Alexa Fluor (Invitrogen) tagged. Mitochondria were assessed by staining with Mitotracker Red, as previously reported (Cellomics Multiparameter Apoptosis Kit 16 ). Image capture and analysis were performed on the Cellomics Arrayscan VTI.
Focused Microarray
The focused microarray and RNA collection was performed by SA Biosciences (SA Biosciences, Frederick, Maryland) following cell collection and processing according to their recommended procedures. Briefly, 2.0 × 106 BEAS-2B cells were detached by trypsin, washed twice in ice cold PBS, spun at 4°C, and snap frozen in liquid nitrogen at termination of treatment. The cell pellets were stored at −80°C until shipment. SA Biosciences extracted the mRNA and analyzed the transcripts using the Epithelial to Mesenchymal PCR Array through the Pathway-Focused RT2 Profiler PCR Array Service. Genes of interest were identified through a 3-step assessment. First, all genes that exhibited a low expression value, assessed by a computed tomography score below 30 were excluded from the analysis. The remaining genes were assessed for significance, which was set at the P level of .05. Finally, the remaining genes were assessed for expression relative to the housekeeping gene HPRT. The genes presented were altered 1.5-fold or greater in the 15 and 50 μg/mL CSC-treated cells or 2.0-fold in either 15 or 50 μg/mL CSC-treated cells.
Statistics
Significance was routinely assessed by analysis of variance for trend and Tukey test to identify individual data points that were at the P ≤ .05 level as compared to vehicle control. All experiments were performed in triplicate with triplicate samples in each experiment.
Results
FH535 Potentiates CSC-Induced Cytotoxicity
Initially identified through a shotgun small molecule inhibitor high content screen (Supplement Figure 1), the β-catenin inhibitor FH535 increased the sensitivity of BEAS-2B cells to CSC depolarization of mitochondrial membrane potential (Ψ).
The CSC significantly depolarized mitochondria in a dose-dependent manner between 25 and 100 μg/mL but had no effect at 12.5 μg/mL when applied following a sham pretretment. This was in contrast to the 700 nmol/L FH535-pretreated cells, where 12.5 μg/mL CSC further reduced ψ and neared complete depolarization, as assessed by the mitochondrial uncoupler CCCP at 1 μmol/L (Figure 1A and B). However, cytotoxicity was evident in the FH535-treated cells in the absence of CSC (FH535 combined with 0 μg/mL CSC) so cytotoxicity was also assessed by cell counts. Cytotoxicity was evident in the groups treated with CSC, FH535 or their combination within 24 hours by cell count. However, potentiation of cytotoxicity was not evident in the combined CSC, FH535 treated cells until 6 days of continuous treatment (Figure 1C). Moreover, following 6 days of CSC exposure, 25 μg/mL CSC-treated cells recovered and grew to full confluence, but combining with FH535 resulted in a continual loss of viability that by 6 days of treatment neared complete culture loss (Figure 1C, Supplement Figure 2A). Complete loss of culture viability following the combination with FH535 was consistently observed as low as 6.25 μg/mL CSC, the lowest CSC dose tested (Supplement Figure 2B). To determine the specificity of the effects of FH535 on β-catenin, the pharmacologically similar GW9669 was used and did not alter cell viability at 24 hours or 6 days posttreatment. GW9669 is a pharmacologic analogue of FH535 that exhibits a similar inhibition of FH535 on the PPARs but works through a different site and does not affect β-catenin (Figure 1C, Supplement Figure 2A and B). 17
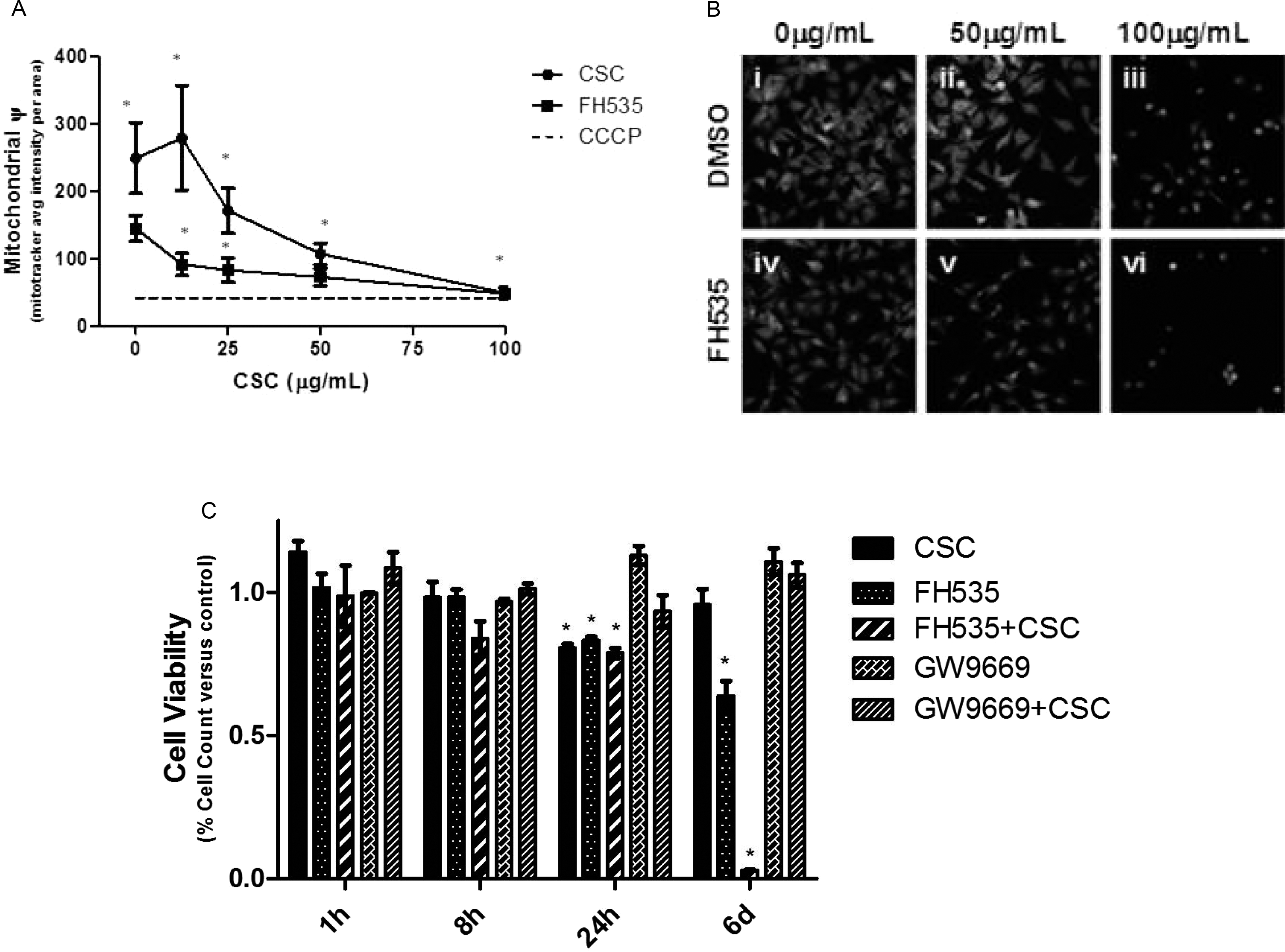
FH535 increased cytotoxicity of CSC treatment. Mitochondrial membrane potential (Ψ) was assessed by mitotracker staining in BEAS-2B cell following 24 hours of CSC exposure (1A, images in 1B). Cell counts were assessed by automated counting of stained nuclei (1C); 700 nmol/L FH535 and 6 μmol/L GW9669 were included in these experiments as a pretreatment to either DMSO (0 μg/mL) or CSC treatment for 30 minutes prior to agent addition. Significance from the vehicle control at the P value of .05 is denoted by asterisk. CSC indicates cigarette smoke condensate; DMSO, dimethyl sulfoxide.
The CSC Alters β-Catenin and Cell Morphology Through a Mechanism Distinct From Either FH535 or TGF-β
Since FH535 can impact pathways other than β-catenin at the doses used, the BEAS-2B cells were tested for confirmation of CSC-dependent activation of β-catenin. 17 Analysis of total β-catenin at 24-hour posttreatment demonstrated that nuclear β-catenin was increased significantly in the nucleus at 6.25 μg/mL CSC. Cytosolic β-catenin trended upward at low doses, but significance could not be demonstrated in the cytoplasm until the 50 μg/mL CSC concentration (Figure 2A, images in Supplement Figure 3A). By 6 days posttreatment, cytoplasmic β-catenin increased at CSC concentrations ≥12.5 μg/mL, but nuclear accumulation of β-catenin was not increased at concentrations of CSC below 25 μmol/L (Figure 2B). Moreover, CSC-induced β-catenin was not observed at 8-hour posttreatment, suggesting a lag in β-catenin activation following CSC exposure (Supplement Figure 4).
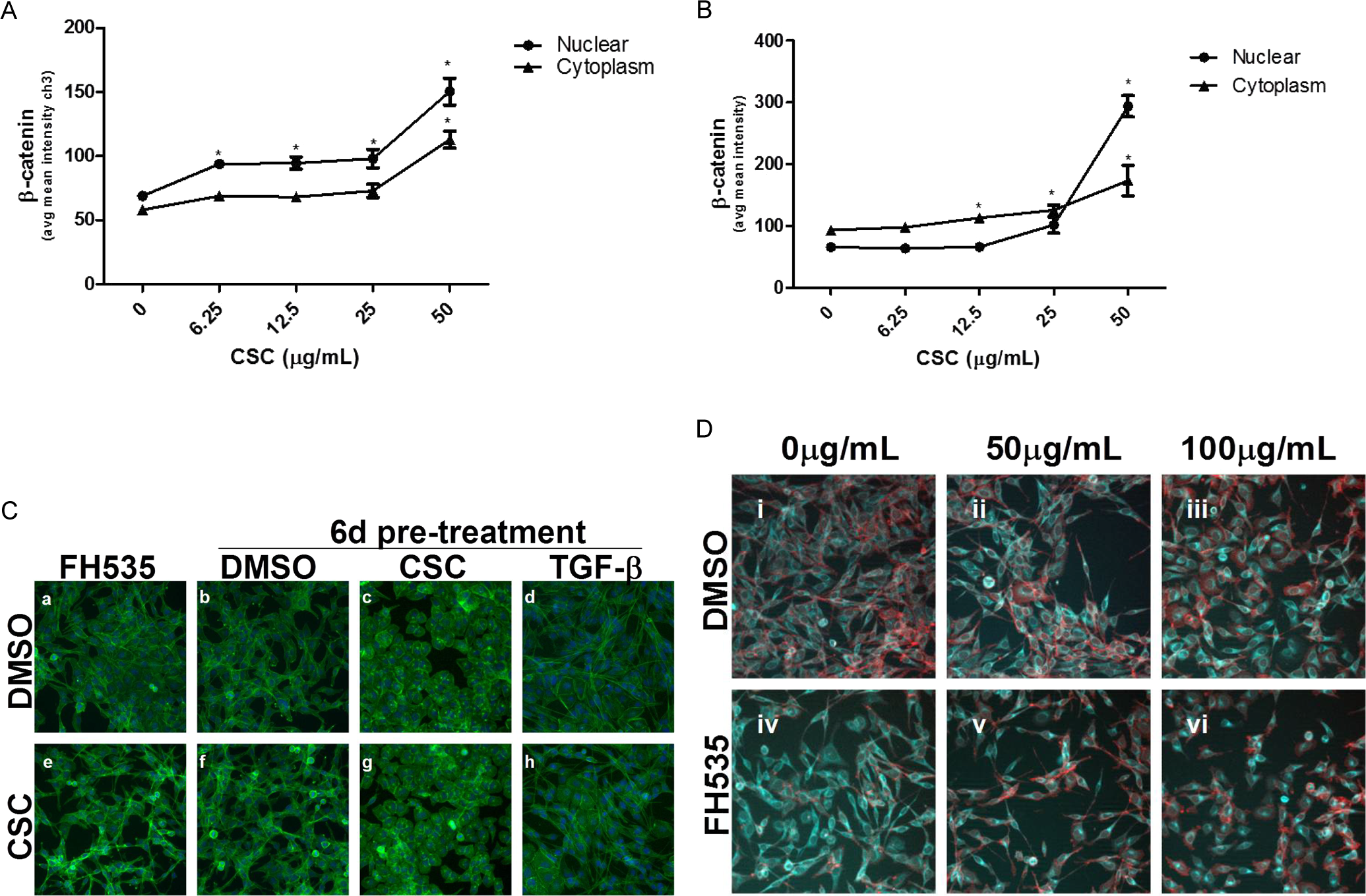
β-Catenin exhibited increased expression and nuclear translocation following CSC treatment. β-Catenin nuclear and cytoplasmic expression levels were assessed by ICC following 24 hours or 6 days of continuous CSC exposure (2A and 2B, respectively). Following 6 days of continuous treatment with DMSO, CSC, or TGF-β, the cells were either swapped over to DMSO for recovery or treated with CSC for an additional 24 hours prior to morphology assessment by phalloidin-FITC staining of actin (2C). Morphology was assessed after 24 hours following FH535 due to viability loss at longer timepoints (2C). β-Catenin was assessed by immunocytochemistry after a 24 hour cotreatment of 700 nmol/L FH535 with CSC (2D). Significance from the vehicle control at the P value of .05 is denoted by asterisk. CSC indicates cigarette smoke condensate; DMSO, dimethyl sulfoxide; TGF, transforming growth factor; FITC, fluorescein isothiocyanate.
Since nuclear accumulation of β-catenin is not a de facto EMT marker, morphologic assessment of the actin cytoskeleton was undertaken for confirmation of mesenchymal transition. Following 6 days of CSC treatment, BEAS-2B cells have reduced cell surface extensions, increased perinuclear actin, and loss of peripheral actin bundles which persisted for 24 hours after the swapping of CSC for DMSO (Figure 2C c,g). Similarly, β-catenin was lost from the periphery and localized to a cytoplasmic domain in many cells following 6 days of CSC exposure (Supplement Figure 3B). The transforming growth factor (TGF)-β induced a classic mesenchymal phenotype over the same 6 days treatment, which remained evident among a percentage of the cells following replacement of TGF-β with either CSC or DMSO for an additional 24 hours (Figure 2C d,h). FH535 alone (data not shown) or when combined with vehicle did not significantly alter cellular actin or cell morphology during acute treatment. However, use of FH535 as a pretreatment to CSC induced the loss of cell surface extensions and acquisition of a bipolar morphology resembling the classic mesenchymal phenotype (Figure 2C a,e). Moreover, FH535 treatment alone depleted β-catenin from the cell (Figure 2D iv). In initial experiments, it was recognized that CSC could partially rescue β-catenin expression, but the effect was modest when CSC was added 30 minutes after the FH535 pretreatment. Adding CSC simultaneously with FH535 greatly attenuated the FH535-dependent loss of β-catenin demonstrating that CSC and FH535 impact distinct intracellular pathways (Figure 2D v,vi). Thus, while CSC increased total and nuclear β-catenin through a mechanism that prevented FH535-dependent depletion of the protein, it did not result in mesenchymal transition.
The CSC Depletes E-Cadherin and Causes β-Catenin Translocation in CALU-3 Cells
β-Catenin is one component of the cell–cell tight junction complexes which are critical to tissue permeability and are important pathological markers in cancer progression. 9 In order to evaluate the robustness of the findings in BEAS-2B cells, the adenocarcinoma line CALU-3 cells were assessed following a 6-day CSC dose–response time course. In a similar fashion to BEAS-2B cells, both β-catenin and its binding partner E-cadherin are reduced at CALU-3 cell–cell junctions following 6 days of CSC exposure in a dose-dependent manner (Figure 3A-H). Furthermore, CALU-3 cells retained an epithelial-like morphology following this treatment (Figure 3A-D). While FH535 depleted β-catenin in some of the CALU-3 cells, the loss was heterogeneous with an accumulation of foci that retained β-catenin expression (Supplement Figure 6). Just as in BEAS-2B cells, CSC-treated CALU-3 cells had lost cell–cell junction proteins but did not progress to a mesenchymal phenotype.
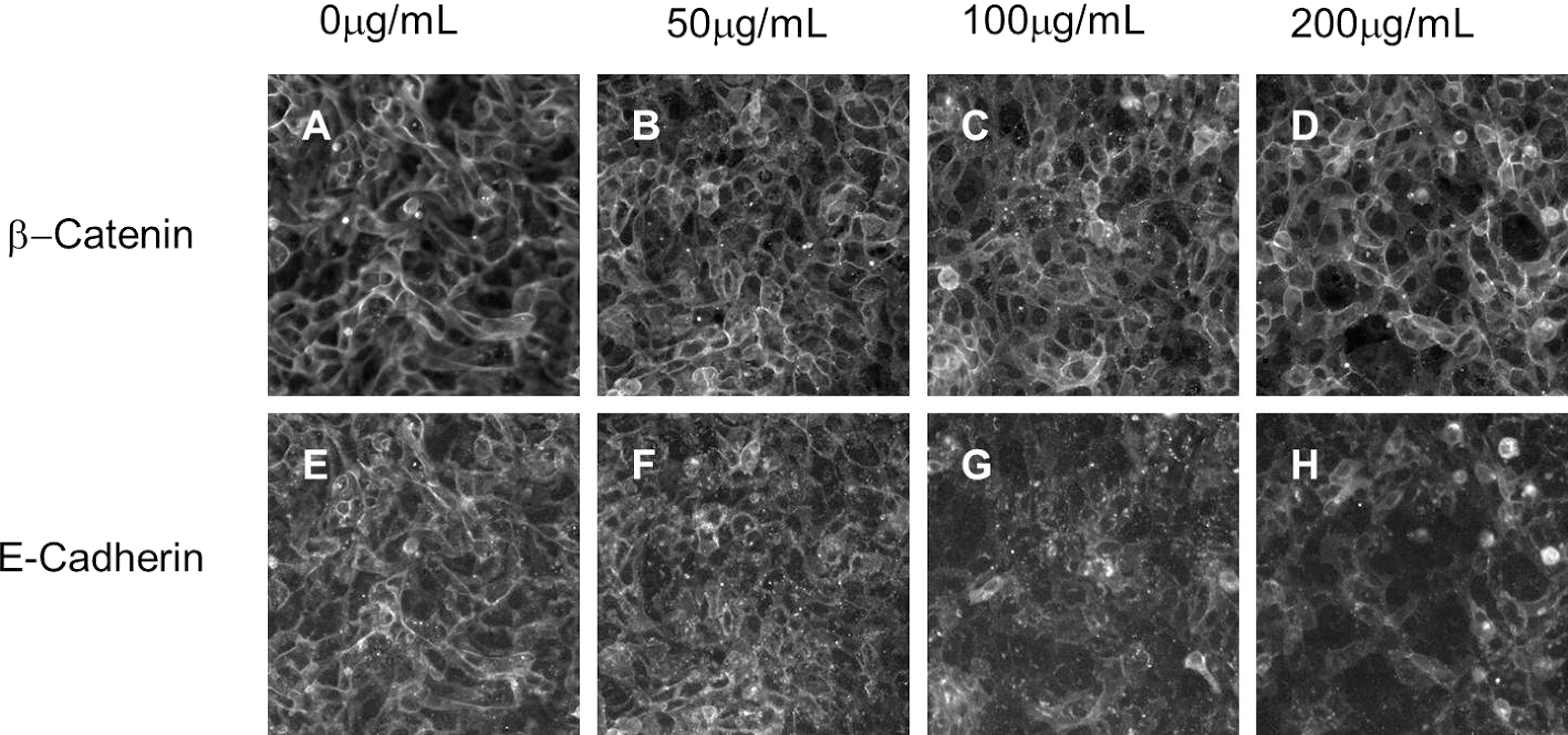
Tight junctions are reduced in CALU-3 cells following chronic treatment with CSC. β
EMT Markers Are Not Detected in BEAS-2Bs Following FH535 or CSC
Since BEAS-2B cells lose their ability to migrate in response to CSC and migration type is a determining factor in cell shape, lack of mesenchymal cell shape was not considered definitive evidence for transdifferentiation status. 16 Since many of the markers used in EMT have not been validated for global identification of EMT independent of tissue type, a focused microarray was used to identify and characterize the expression of critical EMT-associated genes in the BEAS-2B cells treated with FH535 and/or CSC for 8 or 24 hours. 6
Of 84 genes analyzed, 7 genes were significantly changed at least 1.5-fold by 15 and 50 μg/mL CSC treatment, and another 26 genes were significantly changed at least 2.0-fold by 50 μg/mL CSC (Tables 1 and 2). None of these genes analyzed were unique to the 15 μg/mL level at 2.0-fold. Moreover, FH535 cotreatment with CSC elicited equivocal or exaggerated effects in magnitude and direction to CSC alone in both gene analyses. There were 85% (6 of 7) and 96% (25 of 26) concordance between the genes identified in these analyses with the only transcript whose CSC trend was reversed by FH535 treatment being osteopontin (SPP1).
Genes Altered >1.5-Fold by CSC at 12.5 and 50 μg/mL a
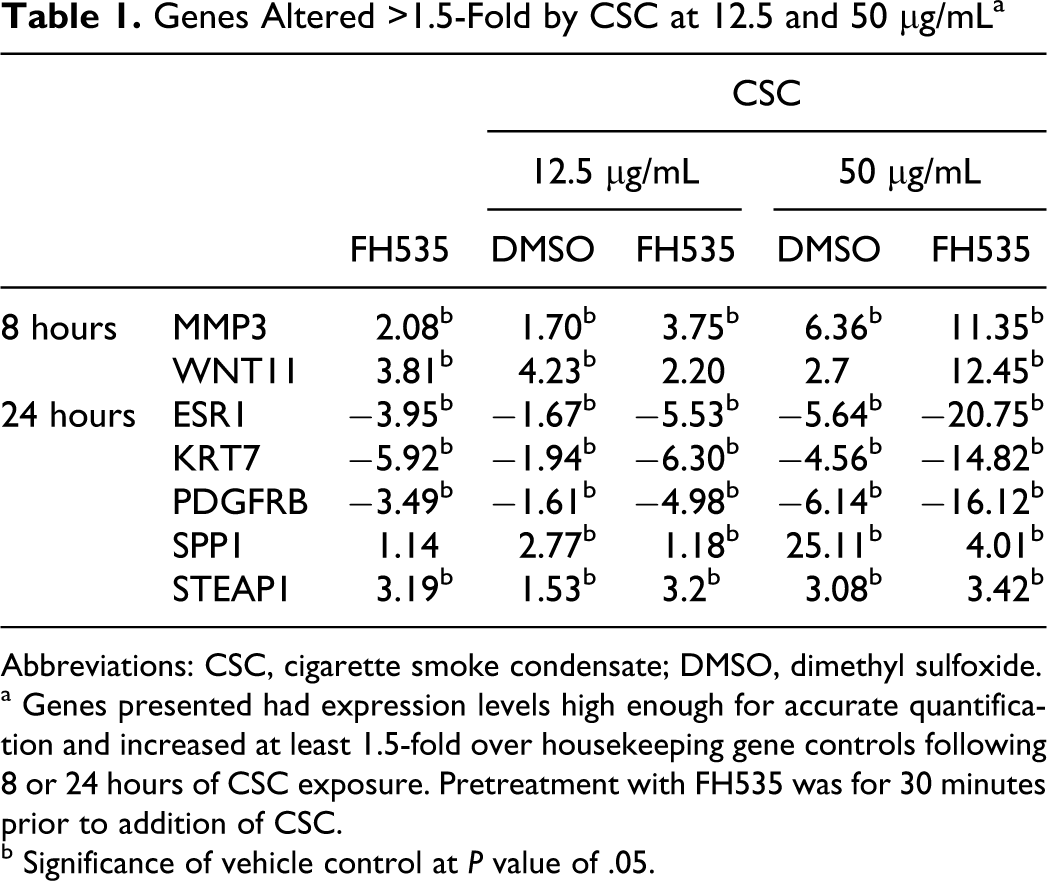
Abbreviations: CSC, cigarette smoke condensate; DMSO, dimethyl sulfoxide.
a Genes presented had expression levels high enough for accurate quantification and increased at least 1.5-fold over housekeeping gene controls following 8 or 24 hours of CSC exposure. Pretreatment with FH535 was for 30 minutes prior to addition of CSC.
b Significance of vehicle control at P value of .05.
Genes Altered >2.0-Fold at 50mg/mL
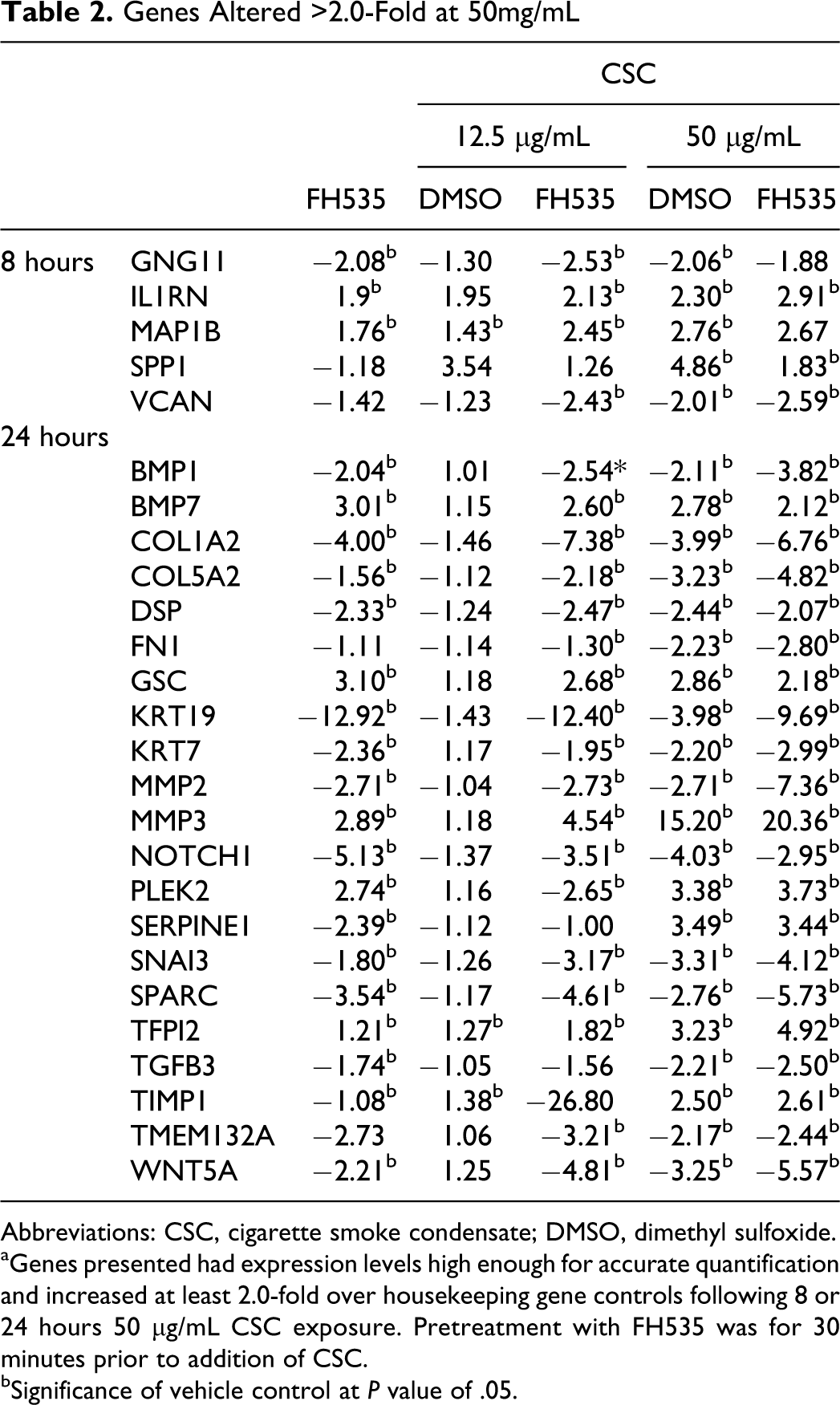
Abbreviations: CSC, cigarette smoke condensate; DMSO, dimethyl sulfoxide.
aGenes presented had expression levels high enough for accurate quantification and increased at least 2.0-fold over housekeeping gene controls following 8 or 24 hours 50 μg/mL CSC exposure. Pretreatment with FH535 was for 30 minutes prior to addition of CSC.
bSignificance of vehicle control at P value of .05.
Analyzing classic EMT genes revealed that a majority of genes associated with epithelial identity, including E-cadherin and keratin transcripts were depressed by both CSC and FH535 treatment, but classic inducers of mesenchymal transition, including TGF-β, SNAIL and TWIST, were either unchanged or were similarly depressed by treatment (Supplement Table 1). Furthermore, EMT surrogate markers that may suggest fibroblastic transformation, like collagen or fibronectin, did not indicate increased mesenchymal gene induction.6,7
Protein confirmation through immunocytochemistry was undertaken for the transcripts with the largest changes and for several EMT-specific markers that were not changed at the transcriptional level, in order to address the potential temporal nature of mRNA. Of the transcripts most changed by CSC at 24 hours, only fibronectin exhibited changes at the protein level that were consistent with the changes in mRNA within the individual FH535 and CSC treatments and their combination at 24 hours (Table 3, images in Supplement Figure 6). Osteopontin and osteonectin (SPARC) demonstrated less than a 10% change in protein expression at the doses tested (Table 3, images in Supplement Figure 6). Moreover, E-cadherin expression was not decreased by treatment (images in Supplement Figure 6). WNT5 and estrogen receptor were likewise not altered by CSC or FH535 treatment (Supplement Figure 7). In pilot experiments, the EMT-specific markers SNAIL, vimentin, N-cadherin, and FSP-1 were all analyzed but not detected following treatment with either CSC and/or FH535 but were detectable in positive control samples (data not shown). Mixed results were obtained with α-SMA, which yielded different background binding affinity with different antibodies but was equivocal with control across all treatments (data not shown). At 24-hour posttreatment, there was no evidence of acquisition of a mesenchymal phenotype or early markers of EMT in either the transcript or protein expression profiles examined.
Translational Analysis of Gene Hits Following CSC a
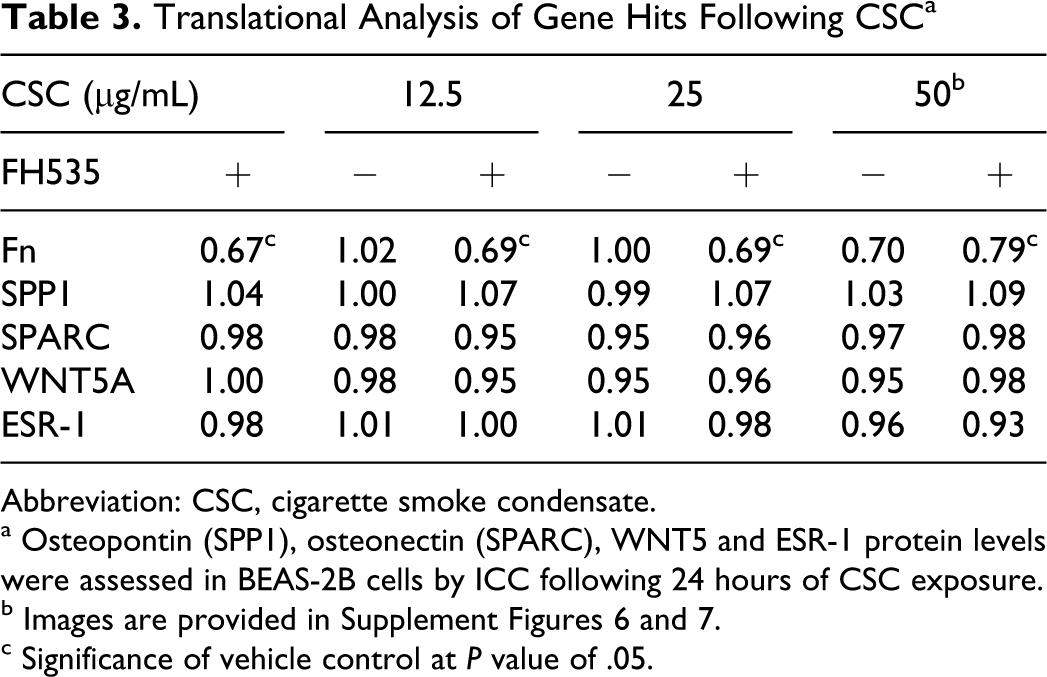
Abbreviation: CSC, cigarette smoke condensate.
a Osteopontin (SPP1), osteonectin (SPARC), WNT5 and ESR-1 protein levels were assessed in BEAS-2B cells by ICC following 24 hours of CSC exposure.
b Images are provided in Supplement Figures 6 and 7.
c Significance of vehicle control at P value of .05.
EGR-1 Outcomes Are Altered by CSC and FH535
Multiple outcomes of FH535 and/or CSC treatment identified in these studies, including fibronectin and osteopontin, are transcriptional targets of EGR-1.14,18 The EGR-1 accumulated in the nuclei following 24 hours of CSC, FH535 or combination CSC, FH535 treatment of BEAS-2B cells (Figure 4A and B). The CSC induced a dose-dependent nuclear accumulation of EGR-1, which was accompanied by a similar dose-dependent increase in the EGR-1 target gene PTEN (Figure 4C). 13 FH535 alone (data not shown) or when combined with vehicle increased nuclear EGR-1 expression but without inducing PTEN expression. Pretreatment with FH535 prevented CSC induction of PTEN, demonstrating that FH535 blocked EGR-1-associated transcriptional responses (Figure 4C).
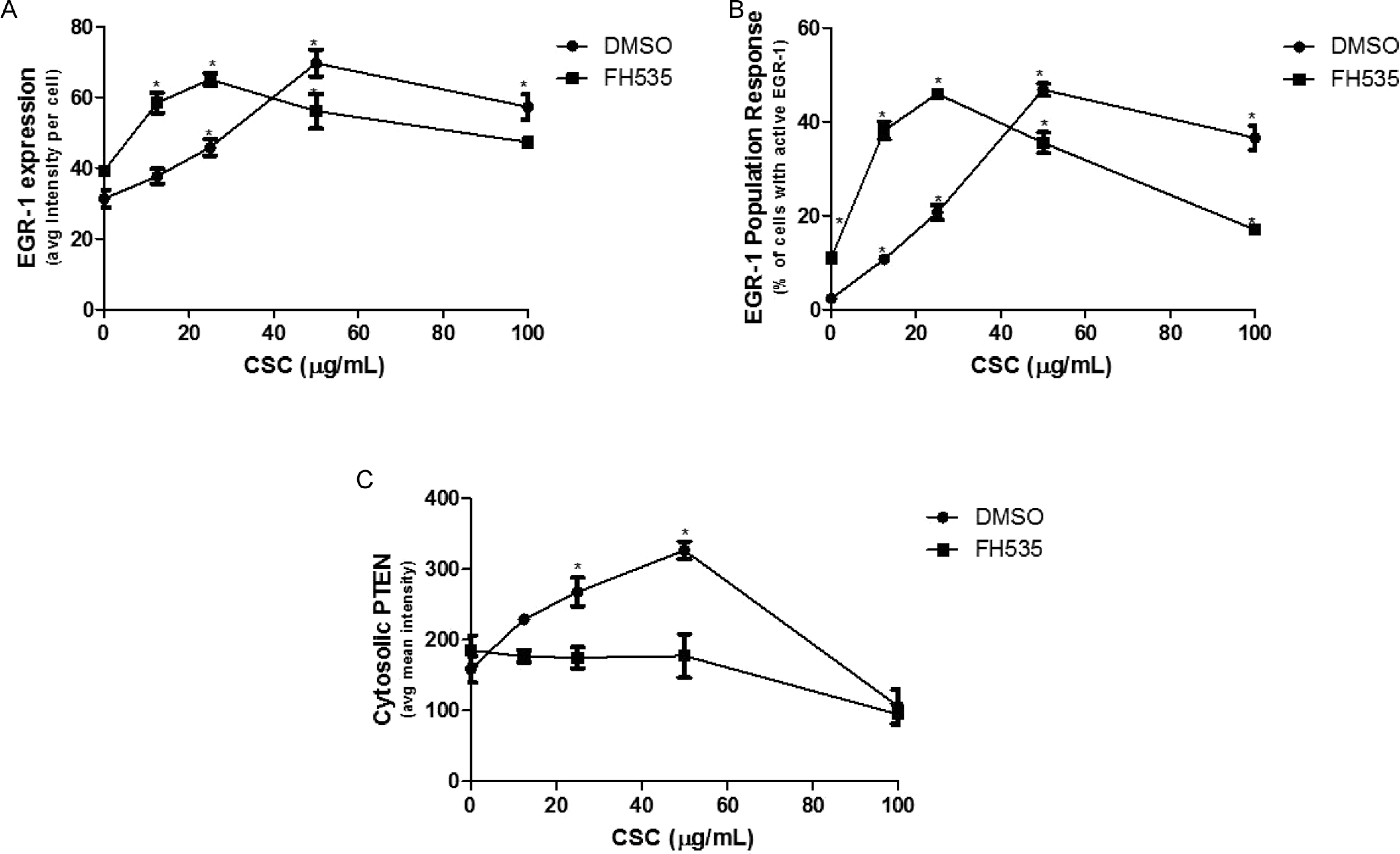
The EGR-1 signaling was altered by CSC and FH535. Average nuclear EGR-1, percentage population positivity of EGR-1 and PTEN expression was assessed by immunocytochemistry in BEAS-2B cells following 24 hours of CSC treatment (A, B, C, respectively). Significance from the corresponding vehicle control at the P value of .05 is denoted by asterisk.
Discussion
This study refines the current understanding of the in vitro model of cigarette smoke carcinogenesis by demonstrating that CSC-dependent generation of markers associated with an aggressive tumor phenotype can occur independently of the acquisition of the mesenchymal phenotype. This work also demonstrates that β-catenin protein expression and EGR-1 transcription are novel targets for the small molecule inhibitor FH535. And furthermore, FH535 potentiates the cytotoxic effect of CSC in the human bronchial epithelial cell line, BEAS-2Bs.
Multiple strengths of this model are evident. First, the demonstrated loss of E-cadherin and acquisition of MMPs is in keeping with what has been shown in clinical cases of non-small cell lung carcinoma (NSCLC).19–21 Moreover, these markers were generated in intermediate passage BEAS-2B cells, which have lower reported tumorogenesis potential than other cell types, including later passage cells of the same lineage. 22 Transformation of this weakly tumorogenic cell type demonstrates the potent carcinogenic nature of CSC as applied here and the strength of this model. Further evidence of the robustness of this model is exhibited in the similarity of response in the 2 cell lines used, which are of different ontogenies and exhibit different phenotypes prior to treatment. Furthermore, even with all the differences in this model as compared to previously reported, the signaling events demonstrated in Lemjabber et al and the gene induction demonstrated in Veljkovic et al are recapitulated here, which suggests that this model represents a continuum of effect consistent with those models.6,23
The inability to demonstrate mesenchymal transition in this model does not necessarily invalidate this model in assessing CSC-induced cellular dysfunction. Nor does it contradict earlier reports of CSC-induced EMT, but instead adds to the heterogeneous and apparent dose and condition-dependent nature of lung cell culture response to CS-components. Whereas EMT has been demonstrated at the cancer invasive front, it is not necessary for cellular invasiveness. 7,24,25 The EMT markers are inconsistently shown in diseased tissues and EMT represents only a single mechanism for cellular transformation, as compared to the more robust markers demonstrated here.5,7 The reasons for the inconsistency between the models are unclear, but this work differed from the previous reports in that (1) the dose of CSC was lower than the ED50 for cytotoxicity, (2) the recovery periods used were minimized, and (3) the total exposure time was less than the longest reported time. While no intentional alterations were made to the cells or to background culture conditions, variability in handling may account for the inability to generate mesenchymal markers. This interpretation would require a mechanism specific to CSC-induced EMT and not a general block on EMT processes, as 5 days of treatment with TGF-β readily induced EMT in these cells when assessed both by morphologically and by identification of molecular mesenchymal markers (data not presented). In this regard, the ability of TGF-β to induce EMT may be a clue as to the induction of EMT in models with a long recovery, as TGF-β is often part of inflammatory resolution and wound healing, as reviewed in Crosby et al. 26 If this was the case, it is of particular concern in the lungs of patients with chronic obstructive pulmonary disease where TGF-β expression has been demonstrated at higher levels. 27
It is also possible that the focus here on early timepoints and the use of a rigid a definition of EMT explain the apparent contradiction. Canonical EMT is induced by TFs of the SNAIL, SLUG, and ZEB families. Following TGF-β treatment, these TFs repress E-cadherin expression while inducing mesenchymal marker expression, which are simultaneous outcomes of the TF action. 5 The ability to distinguish between the 2 different steps of EMT here may represent a completely new EMT signaling mechanism from the canonical pathway or it may suggest that additional signals exist that can regulate the acquisition of the mesenchymal phenotype. If either explanation were to be favored, the data with FH535 would suggest the latter explanation, as 6 days treatment with FH535 reduces β-catenin expression, arrests cell proliferation and promotes the acquisition of mesenchymal morphology independent of mesenchymal marker induction. Invoking the FH535 data to justify calling CSC-induced dysfunction in this model EMT, even in a nascent form, still stretches all known definitions of EMT though, as treatment with FH535 or CSC induce completely different cell shapes, β-catenin distribution, and growth characteristics at the doses tested.
The contrast between the cellular responses to FH535, CSC, or their combination proved valuable for identifying critical events in this model beyond the effect on EMT. For most end points, CSC and FH535 exhibited similar directionality and trends, whereas the differences in response allowed for identification of critical CSC-related events. For example, EGR-1 was identified as a transcriptional target of interest solely because its targets trended differently in CSC- and FH535-treated cells, whereas most gene responses were similar in direction. Moreover, the difference in morphology following treatment coincided with the depression of β-catenin protein.
β-Catenin has been considered a strong target for chemotherapeutical interventions, owing to its disruption in multiple cancers and its action as a proto-oncogenic TR in cultured cells.18,28 Since FH535 was originally identified as a transcriptional inhibitor for β-catenin from a blind screen, the identification of nonspecific effects on β-catenin expression and EGR-1 was unsurprising, but insightful. 17 The work here with FH535 demonstrates that depletion of β-catenin from the cytoplasmic face through prolonged FH535 treatment was associated with disruption of tight junctions and acquisition of a mesenchymal cell shape. This suggests strongly that β-catenin depletion from the cell–cell junction may contribute to acquisition of the most invasive tumor phenotypes, like spindle cell morphology independent of β-catenin’s transcriptional activation. Thus, loss of tight junction maintenance may be an unappreciated contribution of β-catenin translocation during tumor promotion.
Prolonged treatment with FH535 may also be a useful model for future examination of the 2 separate processes in circumstances where gene knockdown is difficult due to the necessity of β-catenin in cell cycle, as experienced in this work or in models where transfection is difficult, like immunocytes. Similarly, FH535 may be a useful tool for studying EGR-1. As no other small molecule inhibitor for EGR-1 is commercially available, FH535s inhibition of the EGR-1 transcription response is a promising tool for moving forward in studying chemotherapeutic intervention of EGR-1-dependent tumors. The lack of specificity in this case could prove effective in tumors dependent on EGFR/EGR-1 and β-catenin signaling for survival and metastasis. The lack of specificity of FH535 should be considered in experimental design though, as nonspecific inhibition of EGR-1 transcription led to inhibition of PTEN expression, a critical tumor suppressor. 29
At the molecular level, better dissection of the β-catenin and EGR-1 pathways following CSC is needed. Use of genetic techniques for β-catenin modulation, reporter systems for rapid monitoring of the kinetics of β-catenin and EGR-1 transcriptional activity, and expansion of the cell types used for validation should all provide refinements to these data and are needed to attribute cellular outcomes to a specific pathway. The ability to separate the effect of these 2 pathways will be critical to understand how cross talk between β-catenin and the EGR-1 effectors, like P53, can impact the outcome of a transformed cells and possibly metastatic potential. It is anticipated that further refinement of this model and these processes will prove a useful avenue for defining those interactions following CSC-induced cellular disruption.
Footnotes
![]() , but genes presented were selected based on relevance to EMT. Significance from the corresponding vehicle control at the P value of .05 is denoted by asterisk. EMT, epithelial to mesenchymal transition; CSC, cigarette smoke condensate; PCR, polymerase chain reaction; TGF, transforming growth factor.
, but genes presented were selected based on relevance to EMT. Significance from the corresponding vehicle control at the P value of .05 is denoted by asterisk. EMT, epithelial to mesenchymal transition; CSC, cigarette smoke condensate; PCR, polymerase chain reaction; TGF, transforming growth factor.
Acknowledgments
Special thanks to Dr Charleata Carter for manuscript review and discussion.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article: funded in whole by Lorillard Tobacco Company of Greensboro, NC.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.