
Abstract
Prasugrel, a thienopyridine ADP receptor antagonist, is an orally administered prodrug requiring in vivo metabolism to form the active metabolite that irreversibly inhibits platelet activation and aggregation mediated by the P2Y12[sub 12] receptor. A comprehensive nonclinical safety assessment including genotoxicity and carcinogenicity studies supported the chronic use of prasugrel in patients with atherothrombotic disease. In addition, a special assessment of the potential for prasugrel to enhance tumor growth was undertaken to address regulatory concerns relating to increases in human cancers. Prasugrel demonstrated no evidence of genotoxicity and was not oncogenic in a 2-year rat carcinogenicity study. In the 2-year mouse study, an increase in hepatocellular adenomas was considered secondary to enzyme induction and not relevant to human safety. Further, the absence of any increase in common background tumors at any other organ site in either rodent study indicated a lack of tumor promoting activity (apart from the CYP450 induction-related increase in mouse liver tumors). Cell culture studies with 3 human tumor cell lines (lung, colon, prostate) demonstrated that exposure of serum-starved cells to prasugrel's active and major circulating human metabolites does not increase cell proliferation relative to starved cells stimulated to proliferate by addition of 10% FBS. Prasugrel also did not increase tumor growth relative to vehicle controls in nude mice implanted with 3 human tumor cell lines. Thus, traditional genotoxicity and 2-year bioassays as well as specially designed tumor growth enhancement studies in human tumor cell lines and mouse xenograft models clearly demonstrated prasugrel's lack of tumorigenic potential.
Introduction
Prasugrel is a novel member of the thienopyridine class of antiplatelet agents that includes ticlopidine and clopidogrel. Prasugrel is an orally administered prodrug requiring in vivo metabolism to form a thiol-containing active metabolite (R-138727). Prasugrel’s active metabolite specifically and irreversibly antagonizes the P2Y12 class of adenosine diphosphate (ADP) receptors on platelets and thus inhibits ADP-mediated platelet activation and aggregation. Prasugrel is indicated for the reduction of thrombotic cardiovascular events (including stent thrombosis) in patients with acute coronary syndrome and is approved as Ef(f)ientTM in several countries (including the United States, European Union, and Canada).
Following oral administration, prasugrel is rapidly hydrolyzed in vivo by esterases including carboxylesterases to a thiolactone which is subsequently metabolized by several cytochrome P450 (CYP) enzymes to the thiol-containing active metabolite, R-138727 (Figure 1). R-138727 is further metabolized and inactivated by S-methylation to R-106583, the major circulating metabolite in humans. The metabolic pathways of prasugrel in mice, rats, and humans are generally similar. 1
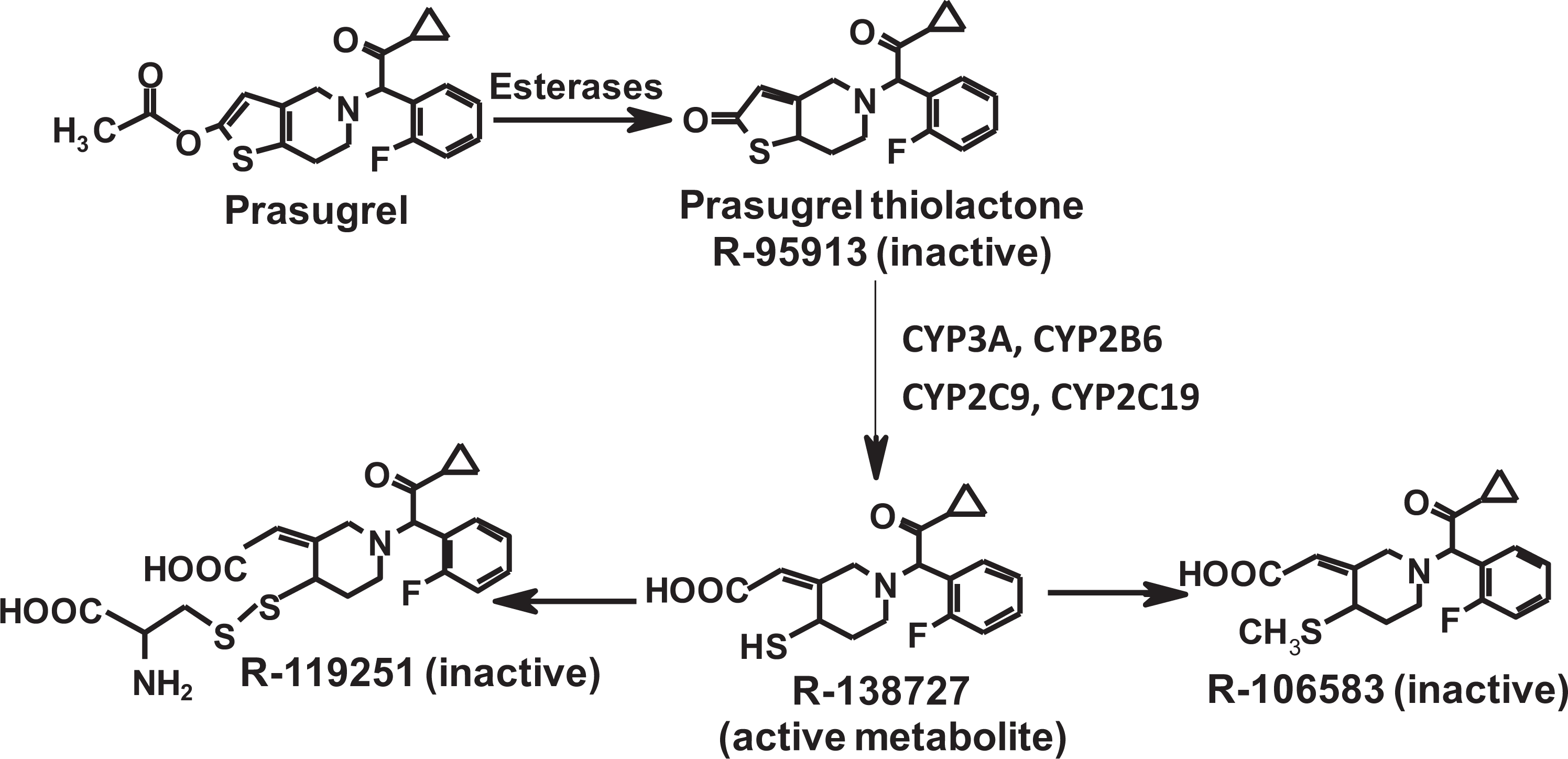
Metabolic pathway and structures: Following oral administration, the prodrug, prasugrel is rapidly hydrolyzed to the inactive thiolactone intermediate R-95913 which is subsequently metabolized by the CYP 450 system to the active metabolite, R138727, which in turn is converted to the major inactive metabolite, R-106583.
A comprehensive nonclinical safety assessment including genotoxicity and carcinogenicity studies supported the chronic use of prasugrel in patients with atherothrombotic disease. 1,2 Due to regulatory concerns about increased malignancies in a phase 3 clinical trial with prasugrel and the comparator clopidogrel, 3 additional nonclinical studies to examine the potential of prasugrel to accelerate tumor growth were undertaken. These studies included both in vitro evaluations of human tumor cell lines in culture and in vivo nude mouse xenograft models of lung, colon, and prostate origin, collectively allowing an evaluation of diverse human tissue types highly relevant to human disease. The results presented herein support the conclusion that prasugrel exhibits neither carcinogenic nor tumor growth enhancement activity.
Methods
The nonclinical testing strategy for prasugrel employed a standard pharmaceutical development pathway that supported the chronic administration of an orally administered therapeutic agent. Carcinogenic risk specifically was assessed in a traditional battery of genotoxicity studies and in conventional, lifetime bioassays in male and female mice and rats. These studies were performed in accordance with Good Laboratory Practice regulations and were consistent with the Organisation for Economic Cooperation and Development and Japanese Ministry of Health, Labour, and Welfare standards in effect at the time. Applicable guidance documents in effect at the time (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use [ICH], European Committee for Medicinal Products for Human Use, and US Food and Drug Administration [FDA]) were also referenced. In addition to the traditional nonclinical safety assessment package, focused in vitro and in vivo studies were designed and implemented to further explore the potential of the active and major circulating inactive metabolites of prasugrel to enhance tumor cell proliferation and tumor growth.
Test Article and Vehicle
Prasugrel hydrochloride (prasugrel) is a thienopyridine identified chemically as (±)-2-[2-acetyloxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl]-1-cyclopropyl-2-(2-fluorophenyl)ethanone hydrochloride, has a molecular formula of C20H20FNO3S. HCl (MW 409.90), and exists as a white to light brown solid. The chemical structure of prasugrel is shown in Figure 1. The vehicle utilized in the in vivo studies was 0.5 w/v% tragacanth solution.
Genotoxicity Studies
Prasugrel was evaluated for genetic toxicity in a standard battery of studies. 1 In vitro, prasugrel was tested for its potential to induce bacterial mutation in the Ames test and to induce chromosome aberrations in Chinese hamster lung (CHL) cells. These in vitro tests were conducted without and with metabolic activation using a hepatic S9 fraction prepared from phenobarbital- and 5,6-benzoflavone-induced rats. Consistent with ICH Guidance, 4 the highest dose represented either a limit dose (eg, 5000 µg/plate for the Ames test) or that which caused up to 50% suppression of the mitotic index (chromosome aberration study). Appropriate positive controls were used in each test to verify the sensitivity of the test systems.
In vivo, prasugrel was tested for induction of micronuclei in the bone marrow of male ICR mice (6 weeks of age, SPF, Charles River Laboratories, Japan), which received a single intraperitoneal injection of vehicle or prasugrel suspended with 0.5% tragacanth gum/saline at doses up to a maximum tolerated dose (MTD) of 1000 mg/kg (20 mL/kg).
Rodent Carcinogenicity Studies
Carcinogenicity studies were conducted in male and female Crj;B6C3F1 mice and F344/DuCrj rats (55 per sex per group, started at 6 weeks of age). In these studies, prasugrel was orally administered daily for 2 years at dosage levels of 0 (0.5 w/v% tragacanth solution), 30, 100, and 300 mg/kg (mice) and 10, 30, and 100 mg/kg (rats). The highest doses caused 9% to 13% decreases in body weight relative to controls and thus represented a MTD. 5 Plasma exposures to R-138727 (active metabolite) and R-106583 (major circulating metabolite in humans), clinical signs, body weight, macroscopic observations at necropsy, and histopathology were evaluated.
Statistical analyses of tumor incidence employed conventional techniques. 6 Increasing trend of incidence to dose level and pairwise comparison between the control group and each dose group was evaluated using survival-adjusted Peto’s test 7 for tumors with high incidence (10 and more animals in total for each sex) or using exact test for those with low incidence (<10). For incidental tumors, the analysis intervals were weeks 0 through 52, 53 through 78, 79 through 92, and 93 until termination of the live phase. Analysis of positive trend in incidence was conducted at the significance levels of .005 (1-tailed level) for common tumors and .025 (1-tailed level) for rare tumors. Pairwise comparison was conducted at the significance levels of .01 (1-tailed level) for common tumors and .05 (1-tailed level) for rare tumors. Common tumors were defined as those with a historical incidence in controls of 1% or more (>1%) and rare tumors as less than 1% (<1%).
Enhancement of Tumor Growth Studies
In vitro cell proliferation
The active (R-138727) and major circulating human (R-106583) metabolites of prasugrel were evaluated for their ability to enhance in vitro cellular proliferation of serum-starved human tumor cell lines NCI-H460 (non-small-cell lung carcinoma), HCT-116 (colon carcinoma), and PC-3 (prostate carcinoma). The tumor types were selected based on tumors of interest in the phase 3 clinical trial. 3 Cells were serum-starved for 24 or 48 hours prior to treatment of 24 or 48 hours with the metabolites or 10% fetal bovine serum (FBS) as a positive control for enhanced cellular proliferation. Experimental details are provided below.
Cell lines were obtained from the American Type Culture Collection (Manassas, Virginia) and propagated in a humidified 5% CO2 incubator at 37° C in the following media: HCT-116 colon carcinoma, McCoy 5A medium containing
Cells (4000 per well for HCT-116 and PC-3; 2000 per well for NCI-H460) were plated in Costar (#3596) 96-well tissue culture plates in 100 μL complete media and allowed to adhere overnight. Following the overnight incubation, the 10% FBS-containing media was removed and serum-free media (or media containing 10% FBS where noted) was added back for a 24- or 48-hour starvation period. Prasugrel metabolites were added after 24 or 48 hours of starvation in serum-free media at levels that corresponded to approximately 1× (70 ng/mL) or 10× (700 ng/mL) of human plasma exposures (Cmax) observed in patients receiving a 10-mg dose of prasugrel. Compound treatment was for 24 or 48 hours as indicated. Positive controls for enhanced proliferation of serum-starved cells consisted of media containing 10% FBS added to cells that had been starved for 24 or 48 hours. Cells treated with compound for 48 hours were given a fresh aliquot of prasugrel metabolites after the initial 24-hour incubation for a total of 2 doses of 70 or 700 ng/mL.
Cell proliferation was analyzed by the WST-1 cell proliferation assay (#1644807; Roche) whereby the stable tetrazolium salt, WST-1, is converted to a soluble formazan by a bioreduction mechanism that is largely dependent on the glycolytic production of NAD(P)H in viable cells. Therefore, the amount of formazan dye formed correlates directly with metabolically active cells. The cells were incubated with the ready-to-use WST-1 reagent for approximately 2 to 4 hours, and the amount of formazan dye formed was quantified with a scanning multi-well spectrophotometer (ELISA reader 420 to 480 nmol/L). The measured absorbance correlates directly with the number of viable cells.
Statistical analysis of the results was performed by calculating the median raw signal for each treatment condition per plate. These medians were analyzed across runs using a mixed effect model with a random run effect and a fixed treatment effect. Compound treatments and 10% FBS added-back positive controls were compared with the 0% FBS-starved control group using Dunnett test. The median data were converted to percent activity within each experiment by defining 0% FBS treatment as 0% activity and 10% FBS add-back as 100% activity. Each experiment was repeated 3 separate times with 6 replicates per data point.
In vivo mouse xenograft studies
Daily oral doses of prasugrel (1 and 10 mg/kg/d) were administered for up to 28 days to nude mice harboring human tumor xenografts derived from lung, colon, and prostate human tumor cell lines (described above). The approximate 3-week dosing term was based on the anticipated growth rates of the tumor xenografts. Previous experience with these models suggests that tumors will achieve a tumor volume of approximately 300, 500, or 1000 mm3 for the prostate, colon, and lung models, respectively, at the end of a 21-day treatment cycle (data on file). Treatment was initiated when tumors reached approximately 100 mm3. This enabled the evaluation of prasugrel effect on tumor growth for models that represent clinically relevant tumor types as well as models that have variable growth rates in vivo while avoiding undue stress to the animals due to excessive tumor size. Tumor growth rates of the xenografts were compared with vehicle control treated animals. Plasma levels of the R-138727 and R-106583 metabolites were evaluated in satellite animals on days 1 and 14. Experimental details are provided below.
Cell lines were obtained and propagated as described above. Cells were harvested, washed, and suspended (1:1 v/v) in serum-free media-containing matrigel (Becton Dickinson, Bedford, Massachusetts). Athymic nude mice (female and male, 24-26 g per mouse, Harlan Laboratories, Indianapolis, Indiana) were implanted subcutaneously in the rear flank with 0.2 mL of cell suspension as follows: NCI-H460, female mice, 2 × 10 6 cells; HCT-116, female mice, 5 × 10 6 cells; PC-3, male mice, 5 × 10 6 cells. Tumors were allowed to grow to approximately 100 mm3 prior to the initiation of compound treatment. Animals were treated daily for 13 (NCI-H460 xenografts), 26 (HCT-116 xenografts), or 28 (PC-3 xenografts) days with tragacanth vehicle or doses of 1 or 10 mg/kg prasugrel (0.2 mL dose volume). Tumors were measured twice weekly by calipers and tumor volume (mm3) was estimated from the formula l × w2 × 0.536, where l is the larger and w is the smaller of perpendicular diameters. One day following the last dose of vehicle or compound, tumors were carefully excised and weighed in a blinded fashion to determine the accuracy of the estimated tumor volume to actual tumor weights at study termination. Subsequent to weighing, tumor tissue from each animal was fixed in either zinc-tris fixative or neutral-buffered formalin for possible histological examination.
Tumor measurement data were captured and analyzed (2-way analysis of variance [ANOVA]) using the KRONOS tumor measurement system. Tumor volume data were transformed to a log scale to equalize variance across time and treatment groups. The log volume data were analyzed with a 2-way repeated measures ANOVA by time and treatment using the MIXED procedure in SAS software (version 8.2). The correlation model for the repeated measures was spatial power. Treated groups were compared with the control group at each time point. The MIXED procedure was also used separately for each treatment group to calculate adjusted means and standard errors at each time point. Both analyses account for the autocorrelation within each animal and the loss of data that occurs if animals with large tumors are removed from the study early. A 1-way ANOVA was used to compare average tumor weights at the end of the study, as well as end point tumor volumes, between groups with Dunnett’s test to compare treated groups versus the control. End point tumor weights and tumor volume measurements were also examined for their correlation with each other. Statistical significance was ascribed at P < .05.
Exposure in tumor-bearing mice
Blood samples were collected on days 1 and 14 from a separate group of NCI-H460 tumor-bearing mice to determine exposure after daily oral administration of 1 and 10 mg/kg of prasugrel. The concentrations of the active metabolite (R-138727) and the major inactive metabolite (R-106583) were determined in plasma using a validated liquid chromatography with tandem mass spectrometry (LC/MS/MS) method described previously. 8 The validated sensitivity range was 0.5 ng/mL to 125 000 ng/mL.
Results
Gentoxicity Studies
Prasugrel was negative in the bacterial reverse mutation assay at concentrations up to 5000 mcg/plate in 4 Salmonella typhimurium strains (TA98, TA100, TA1535, and TA1537) or in Escherichia coli strain WP2uvrA. There were no significant changes in the incidence of the CHL cells possessing abnormal chromosomes exposed to prasugrel concentrations up to 0.833 mmol/L for 24- and 48-hour treatments without S9 metabolic activation and for 6 hours (with and without S9). No micronucleus-inducing activity was observed in the in vivo mouse bone marrow micronucleus test following single intraperitoneal doses up to 1000 mg/kg.
Rodent Carcinogenicity Studies
Mouse
The high dose of 300 mg/kg in the 2-year mouse carcinogenicity study yielded systemic exposures (AUC) of R-138727 and R-106583 that were >650-fold greater than clinical exposures at the 10-mg maintenance dose of prasugrel. An increased incidence of hepatocellular adenoma, considered related to enzyme induction, was observed in male mice in the 300 mg/kg group and in females in the 100- and 300-mg/kg groups (Table 1).
Regarding non-tumor findings, the incidence of eosinophilic altered cell foci in the liver tended to increase in both sexes in the 100 and 300 mg/kg groups, and centrilobular hypertrophy of the hepatocytes was observed in males in the 100 and 300 mg/kg groups. Altered cell foci are considered progenitor lesions from which hepatocellular neoplasia might arise, and some of the benign hepatocellular neoplasia would progress to malignant hepatocellular neoplasia. 9 In addition, the incidence of hypertrophy and pigmentation of the thyroid follicular cells tended to increase in both sexes in the 300 mg/kg group. These morphologic changes are consistent with enzyme induction and were observed in repeat-dose studies in mice, rats, and dogs as increases in liver weight, centrilobular hepatocellular hypertrophy, and increased smooth endoplasmic reticulum and with liver enzyme induction data (Table 2).
Effect of Prasugrel on Liver Enzyme Induction-Related Hepatocellular Tumors in Mice (2-year Carcinogenicity Bioassay)
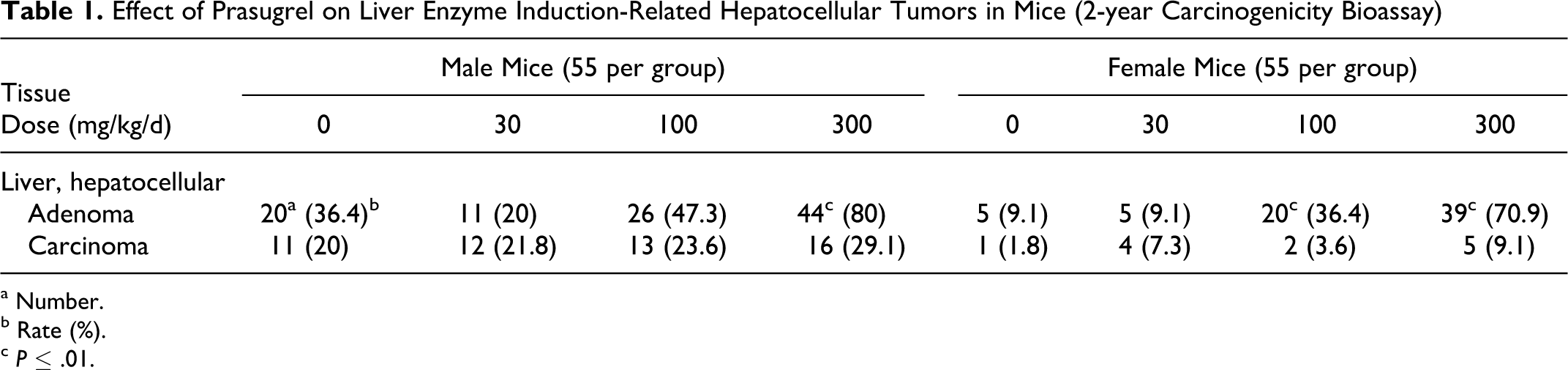
a Number.
b Rate (%).
c P ≤ .01.
Effect of Prasugrel on Hepatic Drug Metabolizing Enzymes in Male Rats Administered Once-Daily Oral Doses of Prasugrel for 7 Days

Abbreviations: UDP, uracil-diphosphate glucose; NS, no change; not siginificant from vehicle control; ↑, increased relative to vehicle control; NADPH, nicotinamide adenine dinucleotide phosphate-oxidase.
The association of enzyme induction and tumor development in the rodent liver is well documented and is not considered relevant for human risk (see Discussion).
Rat
The high dose of 100 mg/kg in the 2-year rat carcinogenicity study yielded systemic exposures (AUC) of R-138727 and R-106583 which were >1000- and >70-fold, respectively, greater than clinical exposures associated with the approved prasugrel maintenance dose of 10 mg. Hepatocellular hypertrophy, considered the result of microsomal enzyme induction, was observed in the 100-mg/kg group. Prasugrel did not increase the incidence of any type of neoplasm. Regarding non-tumor findings, diffuse hypertrophy of the hepatocytes was observed in both sexes in the 100-mg/kg group and increased severity of eosinophilic foci in the liver was observed in males in the 100-mg/kg group.
Enhancement of Tumor Growth Studies
In vitro cell proliferation
Prasugrel’s active and inactive metabolites at 70 and 700 ng/mL did not increase tumor cell proliferation in human lung, colon, or prostate tumor cells in vitro (Figure 2). The data also demonstrate that the assay conditions employed in these studies maintained the ability of the cells to respond to mitogenic stimuli as shown by the response to FBS.
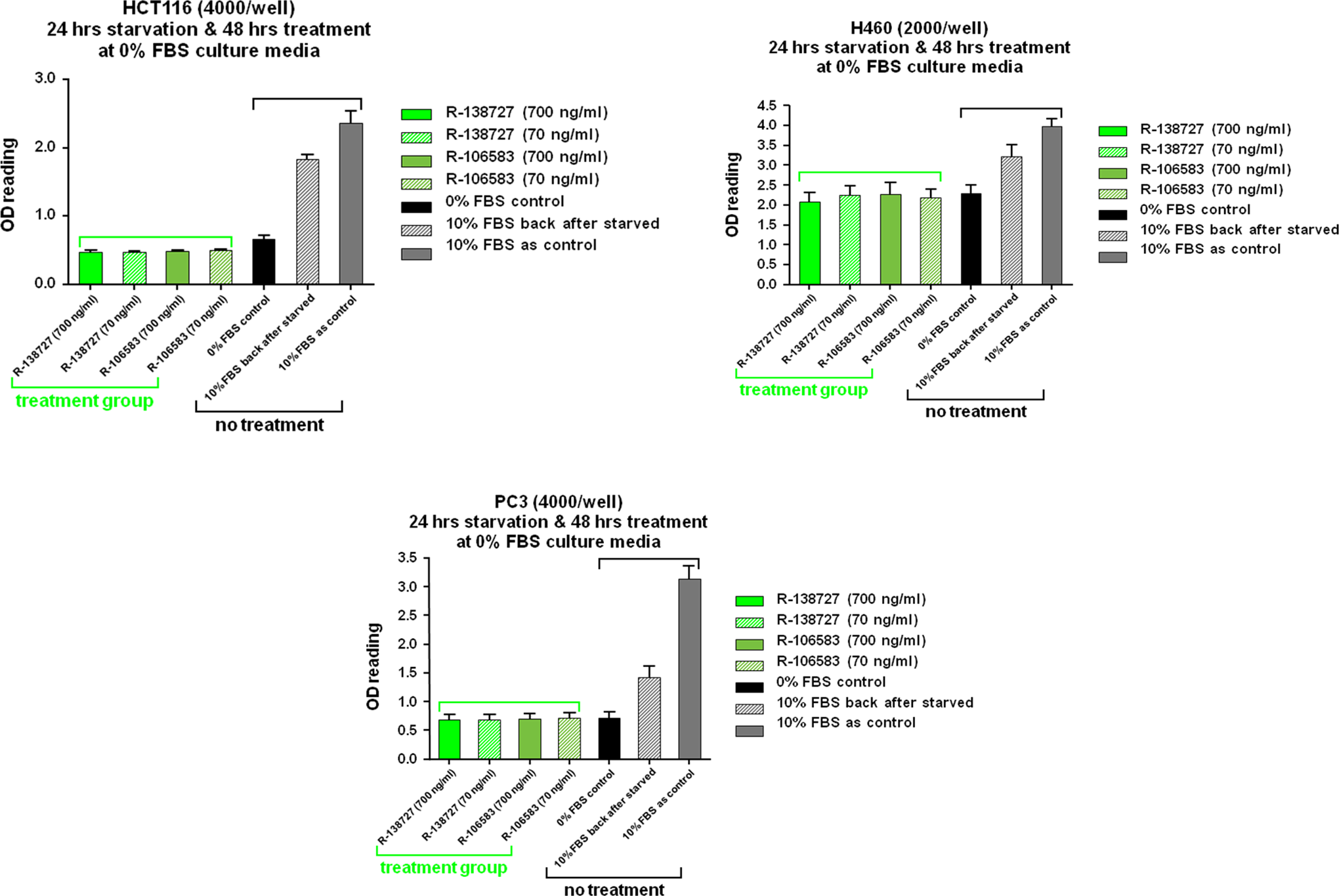
Prasugrel metabolites had no discernable effect on in vitro cell proliferation of human tumor cell lines (HCT-116, colon carcinoma; NCI-H460, non-small-cell lung carcinoma; PC-3, prostate carcinoma). FBS indicates fetal bovine serum; OD, optical density.
Mouse xenograft studies
The exposures (AUC) in the tumor-bearing nude mice to both R-138727 and R-106583 at a dose of 1 mg/kg of prasugrel were similar to or higher than the exposure of humans administered 10-mg maintenance doses of prasugrel. The mean R-138727 AUC on day 14 was 135.5 and 2237.1 ng × h/mL for the 1 and 10 mg/kg doses, respectively, and the mean R-106583 AUC on day 14 was 253.8 and 6641.3 ng × h/mL for the 1- and 10-mg/kg doses, respectively. Exposures at 10-mg/kg doses in the tumor-bearing nude mice were approximately 34-fold higher than the exposure to R-138727 and 22-fold higher for the exposure to R-106583 in humans administered 10-mg maintenance doses.
There was no significant difference in mean terminal body weights for control versus treated animals for each tumor type. Prasugrel treatment (1 or 10 mg/kg/d) did not increase tumor cell proliferation in human colon, lung, or prostate tumor cells in vivo (Figure 3). End point tumor volumes and weights were highly correlated (correlation coefficients ranging 0.90-0.96). Regression analyses that compared xenograft growth rates (in terms of either the slope of log [volume] vs time, or doubling time) did not yield any statistically significant differences between treated versus control mice. When all data were combined—tumor weights (at necropsy) for the 3 tumor types and the 2 doses of prasugrel—there was no statistical difference between mean tumor weight for prasugrel-treated animals compared with that of control.
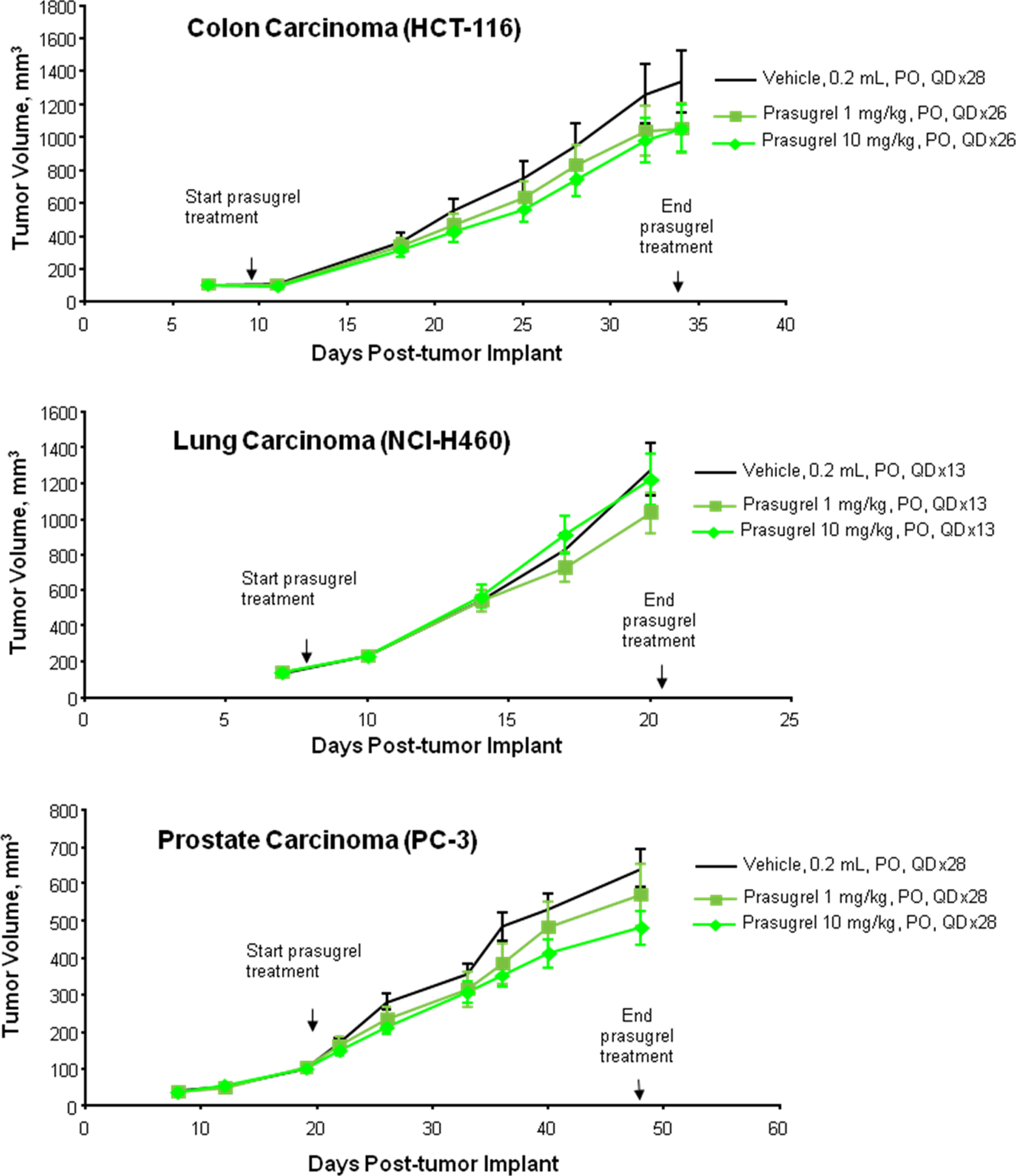
Prasugrel had no effect on the growth of human tumor xenografts in mice (HCT-116, colon carcinoma; NCI-H460, non-small-cell lung carcinoma; PC-3, prostate carcinoma).
Discussion
Lifetime bioassays in rodents are employed as the “gold standard” to assess the potential of a compound to be carcinogenic. The lifetime nature of these studies allows for realization of all the stages of tumor development: initiation, promotion, and progression. Results from these studies have been shown to identify virtually all known human carcinogens. 10 Prasugrel was adequately tested in chronic bioassays using both rats and mice. When tested at exposures up to 74-fold (inactive metabolite) to 1081-fold (active metabolite) higher than the clinical exposure during 10-mg/d maintenance dosing, prasugrel was negative in a 2-year rat carcinogenicity study. Statistically significant increases in hepatocellular adenomas were seen in a mouse 2-year carcinogenicity study at a dose 250-fold the clinical exposure. These tumors are common in mice and are most likely related to chronic CYP450 enzyme induction which occurs in laboratory animals. It is well-known that a variety of nongenotoxic xenobiotics can induce hepatocellular tumors in rodents, and drug-metabolizing enzyme inducers, such as phenobarbital are representative. 11,12 The tumor and nontumor lesions in the livers of prasugrel-treated mice were considered related to hepatic drug-metabolizing enzyme induction.
Prasugrel is not genotoxic and has been shown to induce hepatic drug-metabolizing enzymes in rats and mice. Morphologic changes consistent with enzyme induction were observed in repeat-dose studies in mice, rats, and dogs as increases in liver weight, centrilobular hepatocellular hypertrophy (ground glass appearance), and proliferation of smooth endoplasmic reticulum. Thyroid follicular cell hypertrophy observed in mice at the highest dose of 300 mg/kg in the carcinogenicity study was also consistent with hepatic drug-metabolizing enzyme induction. Drug-metabolizing enzyme inducers are known to elevate serum thyroid-stimulating hormone level via exaggerated metabolism of thyroxine in the liver; prolonged stimulation of the thyroid results in thyroid follicular cell hyperplasia and/or tumor. 13
The association between enzyme induction and tumor development in the rodent liver is well documented. 14 –17 Importantly, the mechanism by which compounds with a phenobarbital-like induction profile cause rodent liver tumors is not considered relevant to humans. 16,17 In addition, extensive epidemiologic studies in humans with phenobarbital and other anticonvulsants at relatively high doses for long periods of time have not shown any association with the development of liver tumors or an increased incidence of neoplasms. 18 –20 Therefore, the increase in liver tumors with prasugrel administration in mice is not considered a relevant human risk.
Other data also add to the weight of evidence indicating that prasugrel does not pose a carcinogenic risk in humans: Prasugrel was tested and found to be without activity in an ICH-compliant battery of genetic toxicology tests including a bacterial mutation assay, chromosomal aberration assay in cultured mammalian cells, and in vivo chromosomal damage assay in mice. In silico structure activity assessment suggested that prasugrel is not carcinogenic. There were no proliferative signals (eg, hyperplasia) in the rodent carcinogenicity studies or in chronic studies in rats (6 months) or dogs (9 months; data not shown.)
The relatively short duration (median of 15 months) of the prasugrel phase 3 clinical trial and the early emergence of many of the tumors in that clinical study also argues against de novo induction of the cancers by prasugrel treatment
3
; even the shortest latency periods are significantly longer than the duration of the clinical trial. If, indeed, the excess in cancers seen in the clinical trial were considered related to drug treatment, one could hypothesize that prasugrel stimulates growth of existing tumors to become clinically recognizable. To this point, rats and mice used in chronic bioassays express spontaneous tumors at a number of organ sites after 18 to 24 months of age. These sites frequently display preneoplastic lesions and would be targets of tumor promoters following chronic exposure. Classic tumor promoters, when given alone, will increase the incidence of tumors at their target sites in the chronic bioassay because of their activity on the spontaneously induced proliferative lesions. The classic liver tumor promoter, phenobarbital, produces increased number and incidence of liver adenomas and carcinomas, but not at other sites in rats and mice in chronic bioassays. Classic skin tumor promoters, such as the phorbol ester 12-O-tetradecanoylphorbol-13 acetate, will increase the incidence of skin tumors when chronically applied to the skin. Similar patterns are produced in tumor promoters’ targets for other sites of spontaneous tumors (eg, bladder, lung, thyroid, and mammary tissue).
21
In the prasugrel carcinogenicity studies, there were no increases (statistically significant compared to concurrent controls or compared with historical control data) in tumor incidence for either species at any site except for the increased incidence of tumors in the mouse liver. The lack of an increase in common background tumors at any other organ site in either the rat or the mouse indicates that prasugrel does not have tumor promoting activity (except for possibly the CYP450 induction-related increase in mouse liver tumors).
Thus, the weight-of-evidence assessment of the data suggests that prasugrel is neither a “complete carcinogen” nor a “tumor promoter.” Nevertheless, the FDA requested that in vitro and in vivo studies of enhanced tumor growth potential by prasugrel be performed to help address concerns regarding potential for drug-induced acceleration of tumor growth. The results of the studies presented in this report showed no growth-stimulating signal in either the in vitro cell proliferation studies or the human tumor xenograft mouse models.
One might question whether there is any plausible biological basis for antithrombotic agents such as prasugrel to be carcinogenic. Examination of the literature reveals that, for many decades, research has, in fact, suggested the opposite. It is generally accepted that prohemostatic or prothrombotic pathways, namely the coagulation cascade and platelet activation and aggregation are pro-carcinogenic. Accordingly, preclinical studies have documented the tumor-inhibitory activity of both anticoagulants and antiplatelet agents. Recently Chatterjee et al 22 demonstrated that the activation of platelets via ADP receptors, including P2Y12 the target of prasugrel, stimulates the release of pro-angiogenic factors. These results are consistent with the lack of effect (or even apparent decrease in tumor growth, eg, for colon and prostate carcinomas) of prasugrel in the in vivo models uitilized in the present studies. Regarding the role of platelets and antiplatelet agents, several studies in the literature attest to the effect of platelet activation, and P2Y12 receptor stimulation in particular, on tumor growth, 23 –26 including one study in a mouse colon tumor xenograft model in which tumor growth was inhibited by blocking the Platelet-derived growth factor (PDGF)-R signaling pathway. 27 Several reports have concluded that the co-aggregation of platelets with tumor cells provide a means for tumor cells to travel to distal sites and metastasize and to avoid immune surveillance. ADP specifically has been implicated in the formation of platelet-tumor cell aggregates, 28 and agents that interrupt the activity of ADP reduce the formation of such co-aggregates.
Conclusion
Based on an assessment of all available nonclinical data—gentoxicity, carcinogenicity, and in vitro and in vivo tumor growth enhancement studies, prasugrel consistently demonstrates a lack of tumorigenic risk.
Footnotes
Acknowledgments
The authors thank the following colleagues for helpful discussions: Drs Gerald Long and Chuck Mahrt (perspective regarding carcinogenicity bioassays); Dr Atsushi Kurihara (toxicokinetic data); Rebecca Lynch, Spring Weir, Andrew Capen, Phil Schwier, Steve Hatch, and Lei Yan (technical aspects of the tumor growth studies); Keri Ann Poi (technical writing), and Drs William Macias and Jeffery Reismeyer (clinical perspective).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.