
Abstract
2-Chloroethyl ethyl sulfide (CEES) or half-mustard gas, a sulfur mustard (HD) analog, is a genotoxic agent that causes oxidative stress and induces both apoptotic and necrotic cell death. Sodium pyruvate induced a necrosis-to-apoptosis shift in HaCaT cells exposed to CEES levels ≤ 1.5 mmol/L and lowered markers of DNA damage, oxidative stress, and inflammation. This study provides a rationale for the future development of multicomponent therapies for HD toxicity in the skin. We hypothesize that a combination of pyruvates with scavengers/antioxidants encapsulated in liposomes for optimal local delivery should be therapeutically beneficial against HD-induced skin injury. However, the latter suggestion should be verified in animal models exposed to HD.
Introduction
Sulfur mustard (2, 2’-dichlorodiethyl sulfide [HD]) is a vesicant and chemical weapon with both acute and devastating long-term effects. 1 The exact molecular mechanisms of HD toxicity are not yet fully elucidated and countermeasures for HD toxicity are still being optimized. Numerous studies show that HD and its monofunctional analog, 2-chloroethyl ethyl sulfide (CEES), induce massive DNA damage, genotoxic and oxidative stress, inflammation, and lead to cell death pathways. 2
Inside the cell, DNA damage derived from rapid alkylation of guanines by mustard sulfonium ions is the main direct chemical impact of alkylating agents, such as HD or CEES. 2,3 It is very likely in such cases that the activity and effectiveness of cellular DNA repair machinery defines whether the cell survives and also determines the mode of cell death when the cell is terminated. Poly (adenosine diphosphate [ADP]-ribose) polymerase (PARP), a key DNA repair enzyme, plays a central role in modulating cell death pathways induced by genotoxic stress. 4 –7 PARP appears overexpressed in response to genotoxic or physiological stressors; it rapidly catalyzes the massive conversion of nicotinamide adenine dinucleotide (NAD+) into polymers of ADP-ribose required for the DNA repair mechanisms. The event causes rapid NAD+ depletion and consequently disrupts normal function of mitochondria by inhibiting pyruvate biosynthesis and thereby decreasing mitochondrial adenosine triphosphate (ATP) production. As a result, excessive activation of PARP due to genotoxic stress leads to a “regulated form” of necrosis. 6 The hallmark of necrosis, in general, is ATP depletion, whereas apoptosis requires sufficient presence of intracellular ATP. Hinshaw et al 8 have found that the exposure of keratinocytes to HD (≥ 500 μmol/L) activates PARP and lowers both NAD+ and ATP levels. Recent work by Kehe et al 9 have shown that the inhibition of PARP activity shifts the mode of cell death in HD-treated HaCaT keratinocytes from necrosis to apoptosis. Kehe et al 9 observed ATP depletion in HaCaT cells exposed to HD; the effect was blocked by a PARP inhibitor.
In accordance with the “regulated” necrosis mechanism, 6 exogenous pyruvate is capable of preventing NAD+ and ATP depletion, thus protecting mitochondria function and switching the mode of cell death pathway to apoptosis. 6 Indeed, ethyl pyruvate, for example, has been shown to induce a shift from necrosis to apoptosis in glucose-deprived A549 lung adenocarcinoma cells. 10 In addition, ethyl pyruvate inhibited the release of high-mobility group box protein 1 (HMGB1), 10 a proinflammatory nonhistone chromatin protein that acts as a cytokine when it is released by necrotic cells.
It is generally accepted that necrosis promotes inflammatory processes after the cell burst and plasma membrane rupture with leakage of the cytosolic contents into the extracellular space. In contrast, cells undergoing apoptosis produce apoptotic bodies that are phagocytized by macrophages generally without inflammation. Therefore, as it was suggested by other investigators, a switch from necrosis to apoptosis and consequent downregulation of inflammation should be highly beneficial for skin and other tissues exposed to HD. 8,11
In the work presented here, we test the hypothesis that pyruvate influences the mode of cell death in human keratinocytes exposed to a genotoxic agent. We propose that exogenous pyruvate could help to maintain mitochondrial ATP production in HaCaT cells exposed to CEES and thereby shift the mode of cell death from necrosis to apoptosis. We measured cell viability as well as markers of apoptosis, necrosis, and inflammation in HaCaT cells exposed to CEES in the presence or absence of sodium pyruvate.
In this study, we used the human keratinocyte cell line HaCaT as a substitute model for primary human keratinocyte cells. Immortalized nontumorigenic HaCaT cells and normal human epidermal keratinocytes (NHEKs) both display distinct keratinocyte morphology and functionality. 12 –14 In organotypic co-cultures, HaCaT cells form well-organized and differentiated epithelia in the presence of human or mouse fibroblasts. 15,16 However, certain differences in the biology of HaCaT cells and primary human keratinocytes have been described. 17,18 Kehe et al have shown recently that in HaCaT cells, HD induces DNA damage followed by necrotic and/or apoptotic changes. 9 The downstream events, such as PARP-1 activation with subsequent ATP depletion or caspase 3 activation without ATP depletion occur in the HaCaT cells in a similar manner as in proliferating keratinocytes in the basal layer of the skin. 19,20 Thus, we assume that our results obtained with HaCaT cells would allow a generalized extrapolation to proliferating keratinocytes.
Materials and Methods
Chemicals and Reagents
Stemline keratinocyte medium II (serum-free, calcium-free, without
Cell Culture and Treatments
Spontaneously immortalized human keratinocytes HaCaT were purchased from Cell Lines Service (Eppelheim, Germany). HaCaT cells were cultured at 37°C in a humidified incubator with 5% CO2 in Stemline keratinocyte medium II (without
Propidium Iodine Assay
The PI assay was performed by a slight modification of the method described previously.
21
Briefly, at the end of each experiment, cultured cells in 96-well dark plates with clear bottom (200 µL of medium per well) were incubated with PI (final concentration 2 µg/mL) at 37°C for 30 minutes. Propidium iodine fluorescence was measured using a Fluostar Galaxy microplate reader (BMG, Germany) using an excitation wavelength of 485 nm and an emission wavelength of 650 nm. The results were calculated using the formula: viability (%) = 100 × (1– [(test sample − low control)
Caspase 3 Activity and ATP Assays
Caspase 3 activity and intracellular ATP were assayed in the cell lysates using a Caspase-3 assay kit and a Sigma Luciferase ATP Determination kit, respectively (Sigma-Aldrich Inc) according to manufacturer’s instructions. To prepare cell lysates, HaCaT cells (about 2 × 106 per flask) were scraped from a flask and placed in 1.5 mL microcentrifuge tubes. A volume of 100 µL of somatic cell ATP-releasing reagent (Sigma-Aldrich Inc) was mixed with the cell pellet in each tube, and the tubes centrifuged at 4°C for 30 minutes at 16 000g. The cell lysates were transferred to fresh plastic tubes and placed on ice until assayed. All assays were performed immediately upon preparation of cell lysates.
Poly (ADP-Ribose) Polymerase Activity Assay
HaCaT cells in 96-well plates were prepared and exposed to CEES and 1.0 mmol/L sodium pyruvate as described above (see Cell Culture and Treatments). Medium was then replaced by PARP reaction buffer containing 0.01% digitonin and 10 µmol/L biotinylated NAD+, incubated for 30 minutes at 37°C, and processed further as described in the method optimized by Bakondi et al. 22 The optical density at 450 nm was measured with a Spectramax Plus 384 microplate spectrophotometer (Molecular Devices, Sunnyvale, California). Data were expressed as mean ± SD of triplicate samples.
Western Blotting
Cellular protein lysates were prepared as described above. Cell medium (Hank FBS) was filtered through a 0.21 μm filter and concentrated using Amico Ultra-4 tubes with molecular weight (MW) cutoff 3000 Da (Millipore, Ireland). Total protein concentrations were determined by the bicinchoninic acid (BCA) protein assay (Pierce Chemical Co, Rockford, Illinois) and 20 μg of protein of each sample was applied per lane and separated with a 4% to 20% gradient sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride (PVDF) membrane. Western blotting was performed with anti-TNF-α (AB1441, rabbit, anti-human) polyclonal antibodies from Chemicon International (Temecula, California), with anti-PARP-1 (sc-80280, rat, anti-human) monoclonal antibodies from Santa Cruz Biotechnology (Santa Cruz, California) or with anti-β-actin (PA1-21167, rabbit, anti-human) polyclonal antibodies from Thermo-Fisher Scientific (Rockford, Illinois) for loading control. The protein was detected using an enhanced chemiluminescence kit from Amersham Life Science Company (Arlington Heights, Illinois).
Fluorescence Microscopy
The cell density was adjusted to 2 × 105/mL, and a 100 µL aliquot of the cell suspension in Stemline Keratinocyte Medium II was placed in each well of an 8-well Lab-Tek chamber glass slide (Nunc, Rochester, New York). The cells were incubated overnight at 37°C in the medium, and nonadherent cells were then removed. Prior to experiments, the cells were preincubated in pyruvate-free Hank FBS for 6 hours. 2-Chloroethyl ethyl sulfide or vehicle (1% DMSO) was added simultaneously with sodium pyruvate, and the chamber slides were incubated for 6 hours (reactive oxygen species [ROS] monitoring) or 12 hours (chromatin condensation or caspase 3 staining) at 37°C in 5% CO2.
For the oxidative stress monitoring, the cells were incubated as indicated above or with 0.1 mmol/L TBHP as a positive control. At the end of the treatment a stock solution of carDCFH-DA in DMSO was added (20 µmol/L final concentration), and the slides were incubated for additional 20 minutes at 37°C. The cells were washed with cold phosphate-buffered saline (PBS) twice, observed and digitally photographed using a Motic AE31 inverted phase contrast fluorescence microscope (Martin Microscope, Easley, South Carolina) equipped with a standard fluorescein isothiocyanate (FITC) filter (Chroma Technology Corp, Rockingham, Vermont) and a Nikon Coolpix E4300 4-megapixel camera (Martin Microscope).
For the live cell caspase 3 monitoring, the cells were incubated as indicated above (except Z-VAD-FMK at final concentration of 2 µmol/L was added simultaneously with pyruvate if indicated) or with 5 µmol/L staurosporine as a positive control. At the end of the treatment a Live Cell NucView 488 Caspase-3 Reagent was added (5 µmol/L final concentration), and the slides were incubated for 25 minutes at 37°C. The cells were washed with cold PBS twice, observed and digitally photographed as described above.
For the chromatin condensation assay, the cells were incubated as indicated above or with 5 µmol/L staurosporine as a positive control. At the end of the treatment Hoechst 33342 dye was added (20 µmol/L final concentration), and the slides were incubated for an additional 20 minutes at 37°C. The cells were washed with cold PBS twice, observed and digitally photographed as described above (except a standard 2-(4-Amidinophenyl)-6-indolecarbamidine [DAPI] filter was used). Three representative pictures for each treatment were chosen, and the numbers of apoptotic nuclei per 200 cells were determined by visual examination.
Statistical Analysis
Data were analyzed by analysis of variance (ANOVA) followed with the Scheffe test for significance with P < .05 using SPSS 15.0 for Windows (Chicago, Illinois). Results were expressed as the mean ± SD. In all the figures, mean values marked with asterisks are significantly different (P < .05). The mean values and standard deviations of at least 3 independent experiments are provided in all the figures.
Results
The Influence of Sodium Pyruvate on Cell Viability, Apoptosis, and Necrosis
Ethyl pyruvate, a stable lipophilic pyruvate derivative, has been shown to promote the necrosis-to-apoptosis switch 10 ; it drastically reduces necrosis by protecting intracellular ATP content and preventing oxidative stress in vitro. 10,23,24 We examined whether sodium pyruvate has a similar effect on apoptosis and necrosis in human keratinocytes and is therefore capable of modulating the cell death pathway.
We initially determined the effect of sodium pyruvate on the viability of HaCaT cells exposed to CEES for 24 hours. Percentage of viable cells were calculated as (100 − N), where N is percentage of dead cells determined by staining of cell nuclei with PI (see the Method section). Propidium iodine is an indicator of cell death as it stains nuclei only in cells with ruptured cellular membranes; both necrotic and apoptotic (at later stages) cells can be stained with PI. Figure 1A shows the influence of 1.0 mmol/L sodium pyruvate on cell viability for cells treated with various levels of CEES (0.5-5.0 mmol/L). Sodium pyruvate is typically present in commercially available keratinocyte media in 0.5 mmol/L concentration (ie, Stemline Keratinocyte Medium II from Sigma-Aldrich, Basal Keratinocyte Medium from Invitrogen). In order to avoid the presence of exogenous pyruvate in our control samples, we used Hank buffer (pyruvate free) supplemented with 10% FBS. Pyruvate was found to slightly increase the cell viability at 1.0 mmol/L CEES level; however, the protection was not evident at all other CEES concentrations. In this experiment, we also measured caspase 3 activity, a marker of apoptosis in HD-exposed keratinocytes, 25 –27 and intracellular ATP levels that should be depleted if the cells undergo necrosis. 9 Figure 1B (cell lysate measurements) and Figure 2 (live cell analysis) show that the presence of 1.0 mmol/L pyruvate enhanced caspase 3 activity shifted the peak of caspase 3 activity toward decreased CEES concentrations (from 3 mmol/L to 1.5 mmol/L CEES). In addition, Figure 3 illustrates the level of apoptosis monitored by chromatin condensation. In both Figure 2 and Figure 3, sodium pyruvate increased apoptosis in the cells exposed to 1.5 mmol/L CEES.
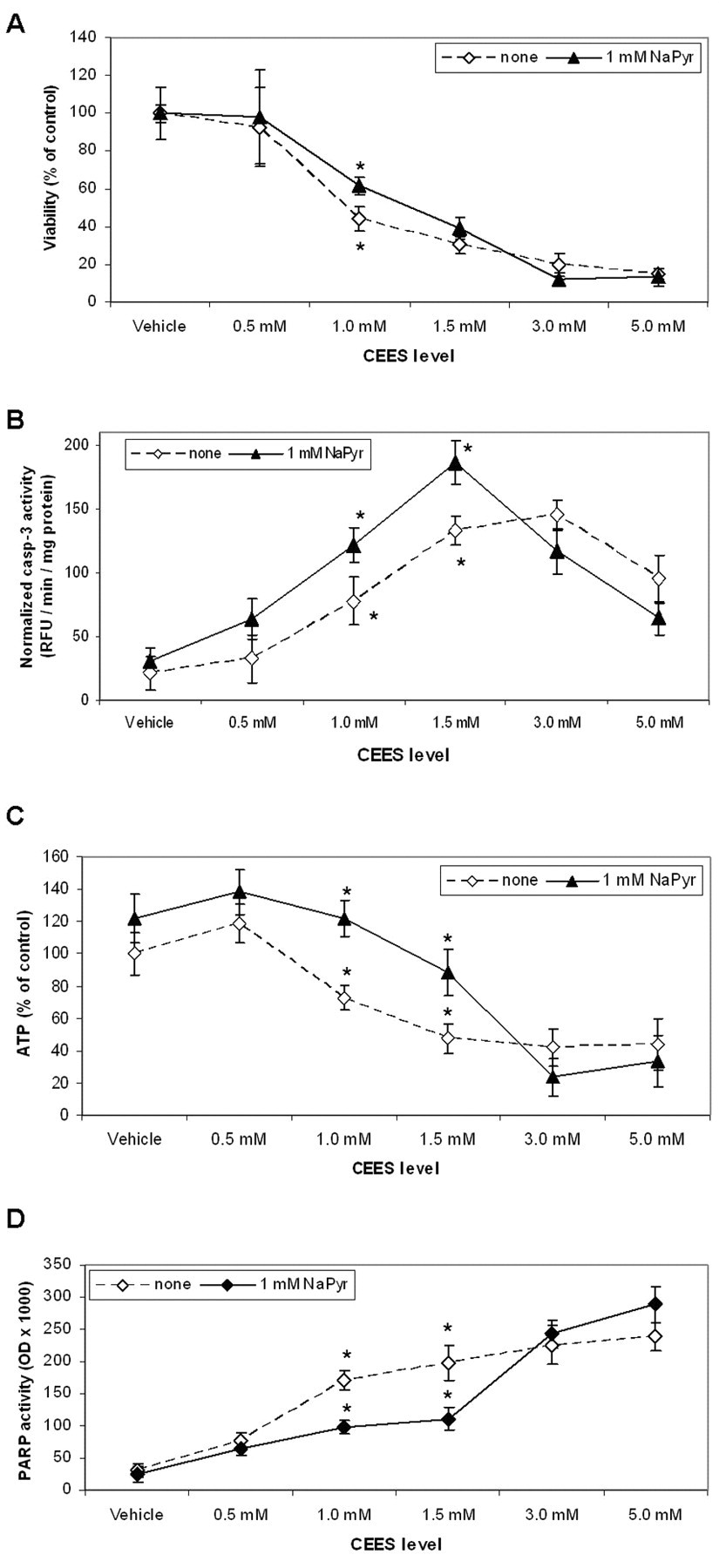
Sodium pyruvate influences CEES toxicity in HaCaT cells. HaCaT cells were preincubated in pyruvate-free Hank Buffer with 10% FBS for 6 hours. Sodium pyruvate (1.0 mmol/L) in the Hank Buffer was then applied to the cells simultaneously with CEES (as indicated) or vehicle (1% DMSO). Cells were incubated at 37°C for 24 hours. A, Cell viability was measured using the PI assay. B, Caspase 3 activity was measured in the cell lysates and normalized to the total protein. C, ATP levels were measured in the cell lysates and normalized to the total protein. D, Cells were incubated at 37°C for 1 hour. PARP activity was measured in the cell lysates using biotinylated NAD+ substrate (see Materials and Methods). Mean values marked with asterisk are significantly different (P < .05). ATP indicates adenosine triphosphate; CEES, 2-chloroethyl ethyl sulfide; DMSO, dimethyl sulfoxide; NAD+, nicotinamide adenine dinucleotide; PARP, poly (ADP-ribose) polymerase.
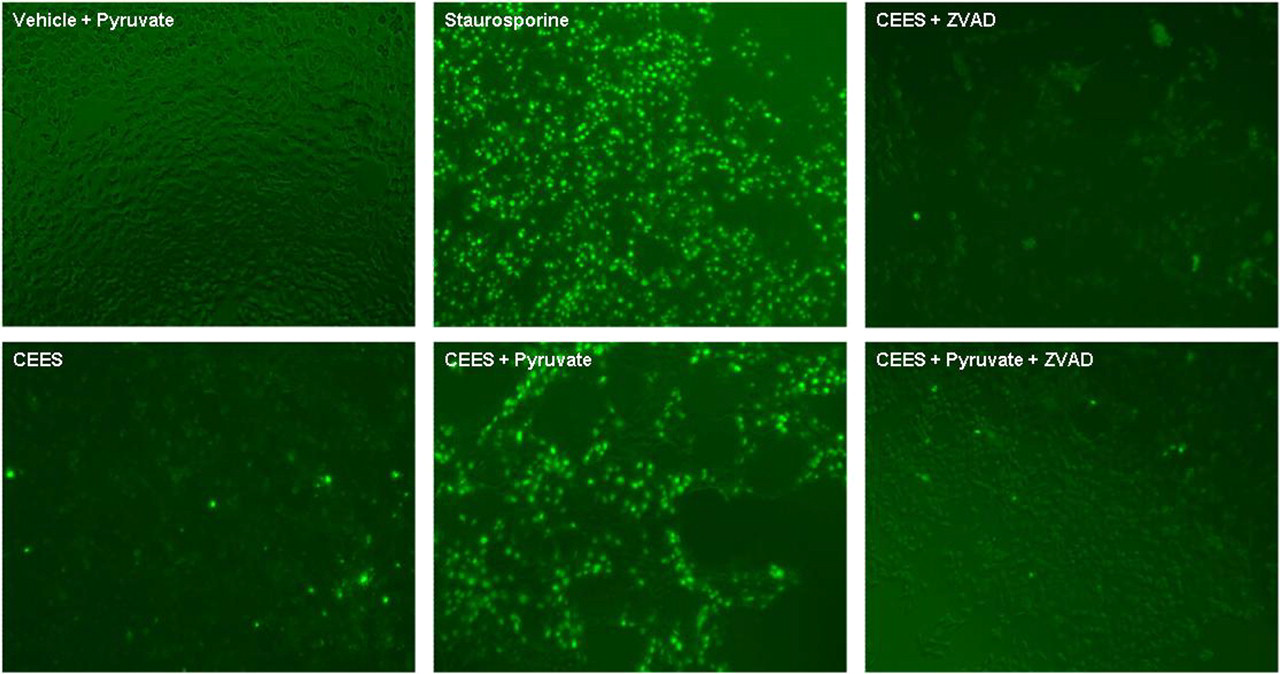
Effect of sodium pyruvate on apoptosis in live cells exposed to CEES. HaCaT cells were preincubated in pyruvate-free Hank Buffer supplied with 10% FBS for 6 hours. CEES (1.5 mmol/L) or vehicle (1% DMSO) was applied to the cells in the presence or absence of sodium pyruvate (1.0 mmol/L) and/or caspase inhibitor Z-VAD-FMK (2 µmol/L) in the Hank Buffer (as indicated). Cells were incubated at 37°C for 12 hours. Caspase 3 activity was monitored using Live Cell NucView 488 Caspase 3 assay kit under fluorescent microscope (×200 magnification) equipped with a standard FITC filter. Staurosporine (5 µmol/L) was used as a positive control. CEES, 2-chloroethyl ethyl sulfide; DMSO, dimethyl sulfoxide; FBS, fotal bovine serum; FITC, fluorescein isothiocyanate; PARP, poly (ADP-ribose) polymerase; Z-VAD-FMK, N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethyl ketone.
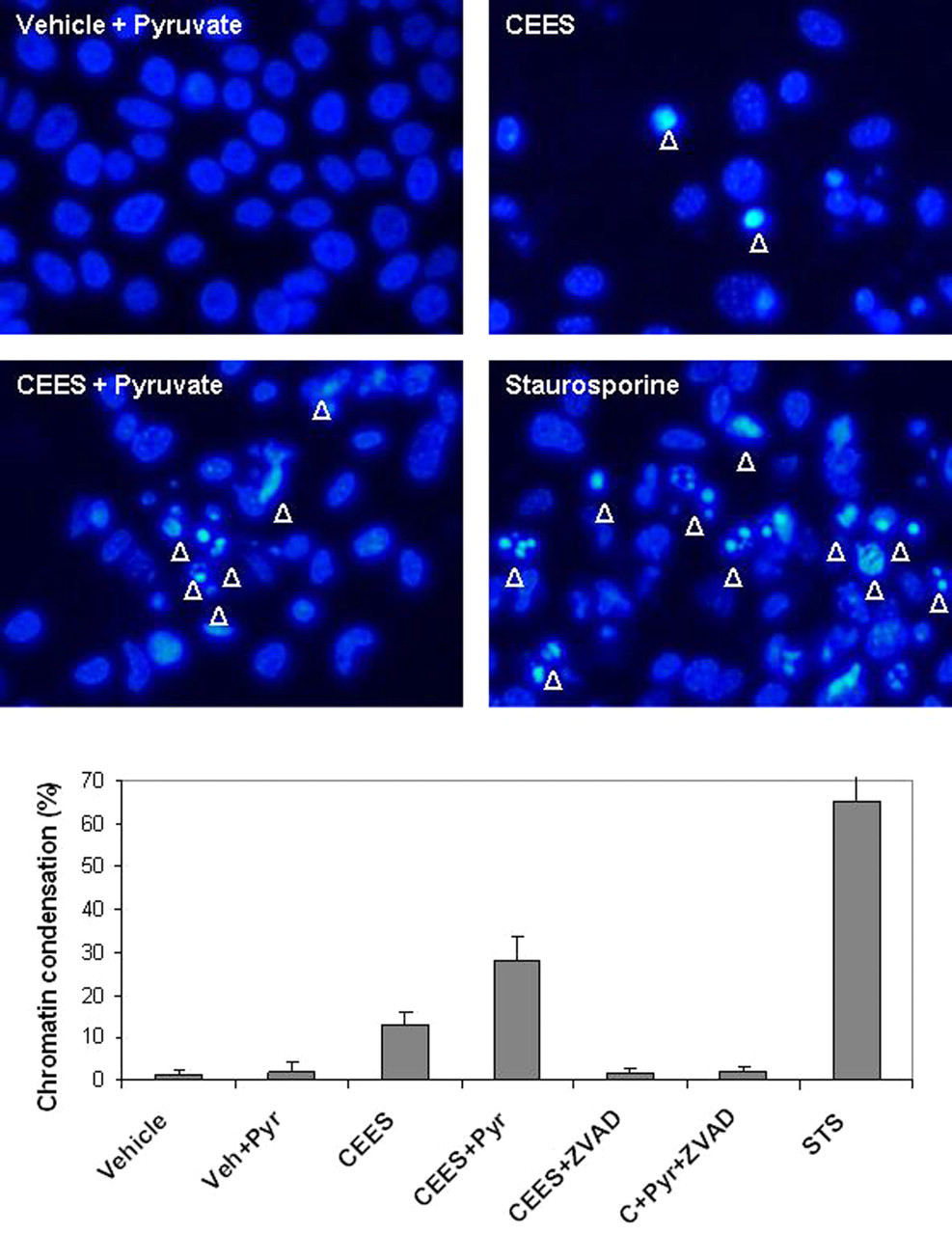
Effect of sodium pyruvate on chromatin condensation in HaCaT cells exposed to CEES. HaCaT cells were preincubated in pyruvate-free Hank Buffer supplied with 10% FBS for 6 hours. CEES (1.5 mmol/L) or vehicle (1% DMSO) was applied to the cells in the presence or absence of sodium pyruvate (1.0 mmol/L) and/or caspase inhibitor Z-VAD-FMK (2 µmol/L) in Hank Buffer with 10% FBS (as indicated). Cells were incubated at 37°C for 12 hours. Chromatin condensation was monitored after staining with Hoechst 33342 dye (20 µmol/L) for 20 minutes under fluorescent microscope (×200 magnification) equipped with a standard DAPI filter. Staurosporine (5 µmol/L) was used as a positive control. Representative photographs show the cell nuclei with chromatin condensation (indicated with arrows). The graph represents average count of apoptotic nuclei per 200 cells for various treatments (as indicated). CEES, 2-chloroethyl ethyl sulfide; DAPI, 2-(4-amidinophenyl)-6-indolecarbamidine; DMSO, dimethyl sulfoxide; FBS, fotal bovine serum; Z-VAD-FMK, N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethyl ketone.
Notably, the increase of apoptotic cells did not promote a reduction of viable cells (Figure 1A and B). Therefore, we suggest that sodium pyruvate not only stimulated apoptosis at this level of CEES but simultaneously decreased the number of necrotic cells. Indeed, Figure 1C indicates that sodium pyruvate decreased the utilization ATP in the cells exposed to 1.0 mmol/L or 1.5 mmol/L CEES levels, which showed upregulated apoptosis in the presence of pyruvate (Figures 1B, 2 and 3). Figure 1C also showed that 1.0 mmol/L sodium pyruvate is sufficient to block ATP depletion in HaCaT cells at 1.0 mmol/L or 1.5 mmol/L CEES levels but not at higher CEES concentrations. Overall, we suggest that apoptosis was the primary mode of cell death in keratinocytes exposed to CEES ≤ 1.5 mmol/L, whereas necrosis was the primary mode of cell death in the cells exposed to CEES ≥ 3.0 mmol/L. These data are similar to the observations made by Kehe et al 9 in HaCaT cells exposed to HD. Taking into account the lower toxicity of CEES compare to HD, 1.0 mmol/L sodium pyruvate is very likely able to reduce necrosis and to stimulate apoptosis at the same time. The caspase 3 activity and ATP depletion data were self-consistent as they showed that at 1.0 mmol/L or 1.5 mmol/L CEES level, exogenous pyruvate blocked ATP depletion and simultaneously increased caspase 3 activity. Thus, these data provided resolute evidence that exogenous pyruvate is capable of shifting the cell death pathway from necrosis to apoptosis.
Sodium Pyruvate Reduces PARP Activity, Oxidative Stress, and Inflammation
DNA damage derived from rapid alkylation of guanines by mustard sulfonium ions represents the main direct chemical impact of alkylating agents inside the cell. 2,3 PARP-1, the key enzyme of the DNA repair machinery, is activated in CEES/HD exposed keratinocytes. 8,28 PARP-1 is only mildly active in the cells under normal conditions, and its activation in CEES/HD exposed cells may serve as a marker of DNA damage. At the same time, PARP-1 cleavage by proteases is a marker of apoptosis; PARP-1 inhibition by chemical inhibitors promotes upregulation of apoptosis over necrosis in CEES/HD exposed cells. 29 Thus, an increase of PARP-1 activity in CEES/HD exposed keratinocytes will reflect the degree of DNA damage and indicate indirectly the mode of cell death. We measured PARP activity in CEES exposed HaCaT cells in the presence and in the absence of pyruvate using biotinylated NAD+ substrate. 22 The activity was measured after 1 hour; it was shown previously that in keratinocytes, PARP activity reaches its maximum within 1 hour after HD exposure. 26 Sodium pyruvate sufficiently reduced PARP activity in HaCaT cells exposed to 1.0 mmol/L or 1.5 mmol/L CEES levels; the effect was not detected at higher CEES concentrations (Figure 1D). Western blotting experiment indicated PARP-1 enzymatic cleavage, a widely used marker of apoptosis; the cleavage was noticeable in the cells exposed to 1.0 mmol/L CEES in the presence of sodium pyruvate (after 12-hour incubation) but not in the absence of sodium pyruvate (Figure 4 ), suggesting a shift of apoptosis in the presence of pyruvate. These data combined with the ATP measurements (Figure 1C) suggest that pyruvate is able to lower immediate DNA damage and decrease PARP activation and ATP depletion consequently shifting the mode of cell death from necrosis to apoptosis in keratinocytes exposed to CEES ≤ 1.5 mmol/L.
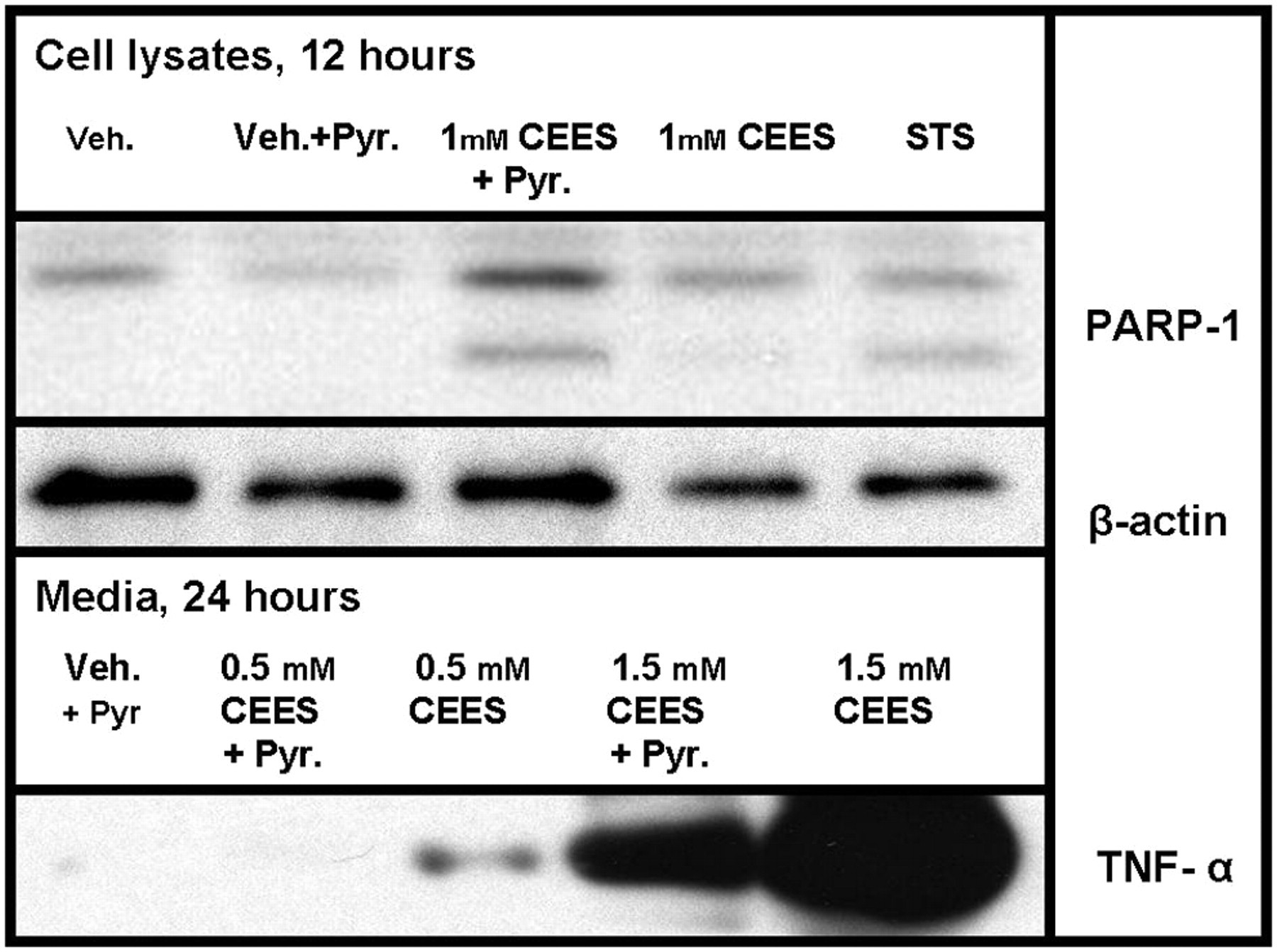
Sodium pyruvate accelerates PARP-1 cleavage and reduces proinflammatory factors in HaCaT cells exposed to CEES. HaCaT cells were preincubated in pyruvate-free Hank Buffer supplied with 10% FBS for 6 hours. Sodium pyruvate (as indicated) in the Hank Buffer was applied to the cells simultaneously with CEES (as indicated) or vehicle (1% DMSO). STS, staurosporine (5 µmol/L) was used as a positive control for apoptosis. Cells were incubated at 37°C for 12 or 24 hours and cell lysates or media (as indicated) were analyzed by Western blotting (see Materials and Methods) using anti-human antibodies to PARP-1, β-actin (loading control) or TNF-α (as indicated). CEES, 2-chloroethyl ethyl sulfide; DMSO, dimethyl sulfoxide; FBS, fotal bovine serum; PARP, poly (ADP-ribose) polymerase; TNF-α, tumor necrosis factor-α.
We also determined whether sodium pyruvate was able to decrease the expression of proinflammatory markers in HaCaT cells exposed to CEES. During necrosis, a number of inflammation factors are expressed and released outside the cell due to the rupture of the cellular membrane. 30 –33 In particular, proinflammatory cytokines are released after exposure of human keratinocytes to HD. 28,34 –36 Using Western blots, we examined the effect of sodium pyruvate on CEES-induced expression and release of tumor necrosis factor-α (TNF-α). Sodium pyruvate alone did not induce TNF-α release; however, it did reduce TNF-α release induced by 0.5 mmol/L or 1.5 mmol/L CEES (Figure 4). These results provide strong evidence that pyruvate attenuates CEES-induced inflammatory response in HaCaT cells, while the mode of cell death is shifted from necrosis to apoptosis (see the above described experiments).
Next, we utilized fluorescence microscopy in order to determine whether pyruvate reduces oxidative stress in HaCaT cells exposed to CEES. Oxidative stress and inflammation are the most common events that accompany necrotic cell death. 30,32,33,37 –39 Genotoxic stress disrupts mitochondrial function, as a consequence of NAD+ and ATP depletion, and induces massive ROS production. 6 We stained HaCaT cells with 6-carboxy-2’,7'-dichlorodihydrofluorescein diacetate (carDCFH-DA), a membrane-permeable fluorescent probe for oxidative stress that is sensitive to intracellular ROS. The influence of 1.0 mmol/L sodium pyruvate on ROS production in HaCaT keratinocytes exposed to 1.5 mmol/L CEES for 6 hours is shown in Figure 5 . Reactive oxygen species generation was drastically enhanced after the CEES treatment; however, in the presence of sodium pyruvate, ROS production was attenuated. Tert-butyl hydroperoxide, a stable chemical analog of H2O2 and potent inducer of oxidative stress, was used as a positive control. Overall, these experiments showed that pyruvate is able to effectively reduce markers of necrosis, inflammation, and oxidative stress, in human keratinocytes exposed to CEES ≤ 1.5 mmol/L.
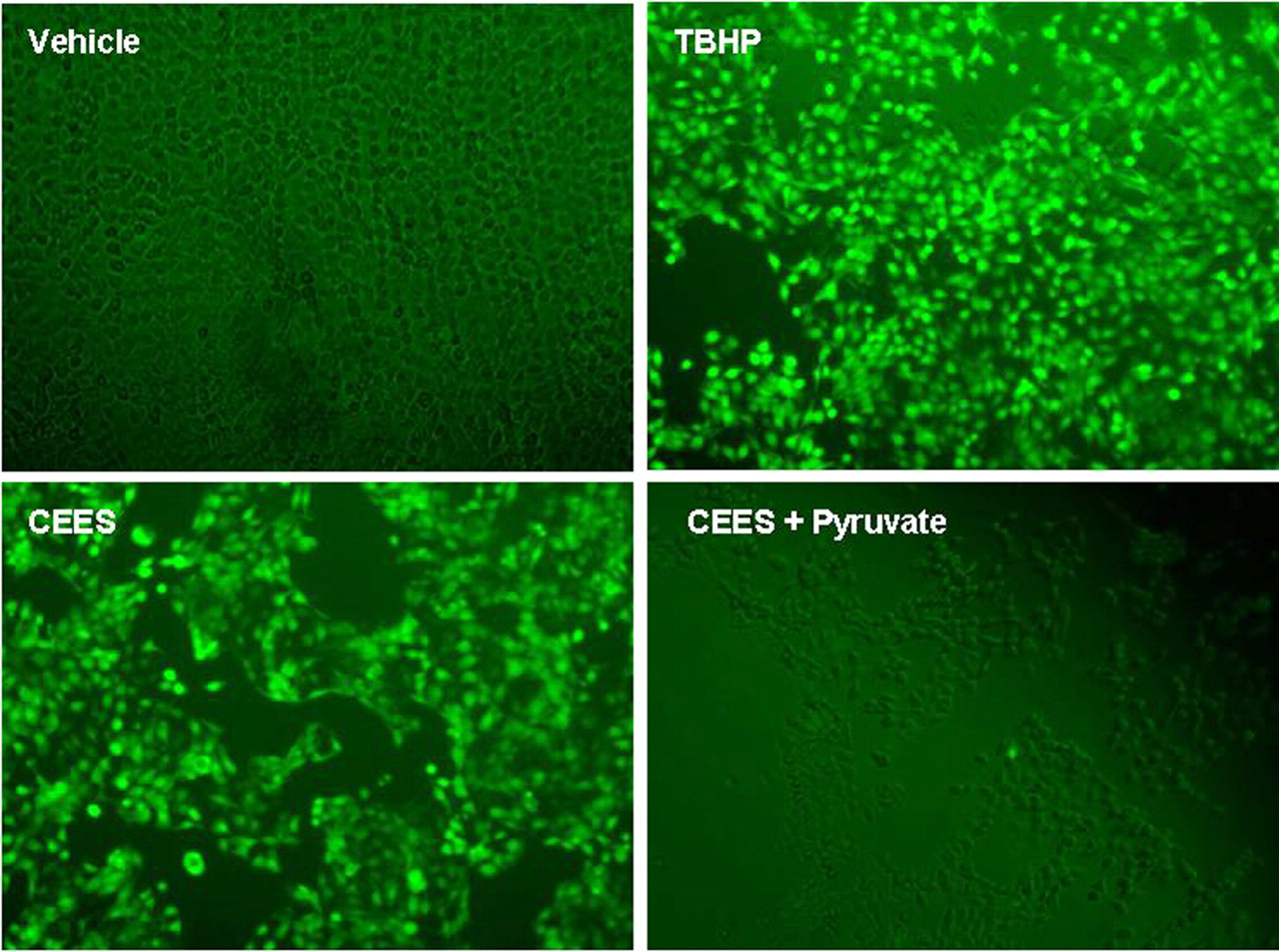
Sodium pyruvate reduces ROS production in HaCaT cells exposed to CEES. HaCaT cells were preincubated in pyruvate-free Hank Buffer with 10% FBS for 6 hours. Sodium pyruvate (1.0 mmol/L) in the Hank Buffer was then applied to the cells simultaneously with 1.5 mmol/L CEES or vehicle (1% DMSO). Cells were incubated at 37°C for 6 hours, and then stained with ROS-sensitive carDCFH-DA dye (20 µmol/L) for 20 minutes and examined under fluorescent microscope (×200 magnification) equipped with a standard FITC filter. Tert-butyl hydroperoxide, TBHP (0.1 mmol/L) was used as a positive control. CEES, 2-chloroethyl ethyl sulfide; DMSO, dimethyl sulfoxide; FBS, fotal bovine serum; PARP, poly (ADP-ribose) polymerase; ROS, reactive oxygen species.
Discussion
Pyruvates, including sodium, methyl, and ethyl derivatives of pyruvic acid, have been described as potent anti-inflammatory agents in vitro and in vivo. 40 –42 In various in vitro models, pyruvates suppress expression of various proinflammatory factors, in particular TNF-α. 43 A previous report indicated that pyruvate-containing mixture attenuates CEES-induced damage to rabbit cornea. 44 In the work presented here, we tested the hypothesis that exogenous pyruvate could modulate the mode of cell death in human keratinocytes exposed to sulfur mustard analog, CEES. We found that 1 mmol/L sodium pyruvate was able to lower PARP activation and ATP depletion (markers of excessive DNA damage and necrosis) and reduce TNF-α and ROS generation (markers of inflammation and oxidative stress, respectively) and increase simultaneously caspase 3 activity, chromatin condensation, and PARP-1 cleavage (markers of apoptosis) without further loss of cell viability in keratinocytes exposed to CEES ≤ 1.5 mmol/L. These effects are very likely due to the shift in the mode of cell death from necrosis to apoptosis. However, it is likely that high levels of CEES/HD and longer times of the incubation, in general, should promote faster pyruvate depletion, consequently increasing overall number of necrotic cells in vitro even when the cells are exposed to CEES/HD in pyruvate-containing media. Therefore, in our experiments, the effect of 1.0 mmol/L pyruvate was negligible at CEES levels ≥ 3.0 mmol/L. Cumulatively, these data confirm our hypothesis that exogenous pyruvate can modulate the mode of cell death in human keratinocytes. The addition of pyruvate did not, however, markedly alter the overall viability of HaCaT cells treated with CEES (Figure 1A). Nevertheless, a shift from necrosis to apoptosis in the tissues exposed to genotoxic agents is expected to be therapeutically beneficial due to reduction of inflammation. 8,11 Our finding that sodium pyruvate reduces TNF-α release (Figure 4) supports this point of view. We further suggest that pyruvate is able to reduce proinflammatory factors in other skin cells, such as macrophages and mast cells (unpublished data), which drastically reduce inflammation due to CEES/HD exposure.
The molecular mechanisms responsible for the toxicity of HD and its monofunctional analog, CEES, are still under active investigation. Advances in this area will help to promote the future design of optimal countermeasures. The initial stages of the CEES/HD-induced dermal damage have been well documented in vitro. In human keratinocytes and human skin models, HD or CEES induce alkylation of macromolecules, genotoxic stress, oxidative stress, fragmentation of extracellular matrix, and inflammation. 2 Genotoxic stress is thought to be particularly critical since HD is capable of chemically modifying DNA by alkylation, cross-linking, and single-strand breaks. Extensive DNA damage leads to an overexpression and excessive activation of PARP, a key DNA repair enzyme. 5,7,45 While moderate PARP activation promotes cell survival by DNA repair mechanisms, excessive activation of PARP can be fatal to the cell as originally proposed in the “suicide hypothesis” by Berger. 4 Poly (ADP-ribose) polymerase is not only a trigger for “necrotic suicide,” but also a “switch-molecule” that defines (by its activity) the mode of cell death in genotoxic stress. 46 Poly (ADP-ribose) polymerase plays a key role in HD toxicity, 19,20,29 and our observations are in agreement with this point of view. Overall, our data, including PARP activity measurement, demonstrate that apoptosis prevails at CEES ≤ 1.5 mmol/L, whereas necrosis is the primary mode of cell death in the cells exposed to CEES ≥ 3.0 mmol/L. Pyruvate is able to influence the mode of cell death by reducing necrosis and altering apoptosis (without further loss in cell viability), especially at CEES ≤ 1.5 mmol/L. This effect of pyruvate was explained earlier by Chiarugi. 6 Considering this mechanism, it is logical to assume that exogenous pyruvates would present a useful alternative to PARP inhibitors in the development of therapies for HD injury in skin and other tissues. While PARP inhibitors effectively restore intracellular NAD+ and ATP levels increasing cell viability after HD injury in vitro and especially in vivo, they suppress DNA repair, subsequently increasing genomic instability and chromosome damage that may lead to mutagenesis/cancerogenesis. 29 We speculate that sufficient pyruvate intervention during first hours after the HD exposure (when PARP activation is accelerated) would help to restore intracellular NAD+ and ATP levels without negative influence on the DNA repair machinery. This possibility should be further explored in skin models exposed to HD or CEES.
Notably, even in apoptotic cells, the mode of the cell death pathway is strictly determined by the PARP status. If PARP cleavage is blocked in apoptotic cells (ie, CD95/Fas inhibition; p53, bax, bak signaling disruption; caspase 3 inhibition), active PARP triggers the “suicidal” chain of events that lead to necrosis instead of apoptosis. 33,37,46 In the absence of PARP (PARP−/− cell lines) or if PARP activity is inhibited, massive DNA damage leads to apoptosis but not necrosis. Thus, PARP−/− human fibroblasts exposed to HD undergo apoptosis, whereas PARP+/+ fibroblasts undergo necrosis 47 in accordance with this mechanism. Surprisingly, necrosis has been documented in PARP−/− fibroblasts only; in keratinocytes, HD induces apoptosis in both PARP−/− and PARP+/+ cells. 47 This controversy can be explained, at least in part, by the presence of sodium pyruvate in the keratinocyte culture medium and its absence in the fibroblast medium. In our experiments, exogenous pyruvate suppressed necrosis and shifted the mode of cell death toward apoptosis in HaCaT keratinocytes. Commercially available culture media for human keratinocytes are often proprietary with no explicit compositional details, but typically contain 0.5 mmol/L sodium pyruvate. The presence of pyruvate in the culture media could be an uncontrolled variable for in vitro experiments studying cell death pathways in human keratinocytes.
In summary, present in vitro work showed that exogenous pyruvate lowers necrosis and shifts the mode of cell death to apoptosis, consequently reducing biomarkers of oxidative stress and inflammation in human keratinocytes. The effect, at least in part, was due to the protection of intracellular ATP content and the downregulation of ROS production. Although sodium pyruvate did not remarkably increase the survival of CEES-treated HaCaT cells, it did induce a necrosis-to-apoptosis shift that would be beneficial in treatment of HD toxicity in vivo. This study provides a rationale for the future development of multicomponent therapies for HD toxicity in the skin. Phospholipid-based liposomes possess an ability to effectively deliver both lipid-soluble chemical antioxidants (in the lipid bilayer) and water-soluble chemical antioxidants (in the aqueous phase). 48,49 Moreover, liposomes rapidly penetrate the dermal barrier making the delivery of water-soluble compounds as effective as lipid-soluble drugs. 50 We speculate that a combination of pyruvates with scavengers/antioxidants encapsulated in the liposomes for optimal local delivery will be therapeutically beneficial against HD-induced skin injury. We intend to explore the effectiveness of pyruvate containing antioxidant liposomes in animal models exposed to HD.
Footnotes
The author(s) declared no potential conflicts of interests with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research of this article: United States Army Medical Research Command (USAMRMC) Grants – “Topical Application of Liposomal Antioxidants for Protection against CEES Induced Skin Damage,” Contract No. W81XWH-05-2-0034, and “A Proteomic Approach for Studying the Therapeutic Use of Antioxidant Liposomes,” Contract No. W81XWH-06-2-044.