
Abstract
Peroxisome proliferator-activated receptor α (PPARα) agonism in ocular inflammation has not been thoroughly investigated. The objective of this investigation was to determine the effect of WY-14 643, a selective PPARα agonist, on inflammatory cytokine release in human ocular cells. Stimulation of primary human corneal epithelial cells, keratocytes, and retinal endothelial cells with 1 to 10 ng/mL interleukin 1β (IL-1β) resulted in a significant increase in numerous inflammatory cytokines, including IL-6, IL-8, and tumor necrosis factor α (TNF-α); and dexamethasone was able to significantly inhibit these effects. However, WY-14 643 did not effectively block IL-1β-induced cytokine release in ocular cells; rather, significant increases in IL-1β-induced inflammatory cytokines were observed in these cells but not in aortic smooth muscle cells. WY-14 643 also significantly upregulated vascular endothelial growth factor (VEGF) expression in corneal epithelial cells and keratocytes. These studies demonstrate for the first time that PPARα agonism may be proinflammatory and proangiogenic in a variety of ocular cells and suggest that therapeutic applications of such agents in ophthalmology may be limited.
Introduction
Activation of inflammatory pathways is a common tissue response to perturbations in homeostasis, and inflammatory processes have been implicated in a wide variety of ocular disease states, including dry eye, 1 allergy, 2 diabetic eye disease, 3 and age-related macular degeneration. 4 The role of nuclear receptors in mediating inflammatory processes is well known, 5 in fact, nuclear receptor modulators such as glucocorticoid are among the most widely prescribed ocular therapeutics. However, due to the lack of transrepressive specificity of current glucocorticoid receptor modulators, 6 their use in ophthalmology is often associated with unwanted side effects, including cataract formation and elevation of intraocular pressure. 7 Therefore, identification of new agents without such undesirable effects continues to represent a substantial unmet medical need. Another nuclear receptor family member, the peroxisome proliferator-activated receptor (PPAR) comprising 3 members (PPARα, PPARβ/δ, and PPARγ), may offer such therapeutic promise.
The role of PPAR in ocular inflammation has been less well studied, but a substantial body of evidence exists, particularly for PPARγ. Peroxisome proliferator-activated receptor γ agonists inhibit vascular endothelial growth factor (VEGF)-induced proliferation and tube formation in retinal and choroidal endothelial cells in vitro, inhibit laser-induced choroidal neovascularization in rat and monkey, 8,9 and protect against retinal leukostasis and retinal vascular permeability in diabetic rats. 10 However, a comprehensive evaluation of the effect of PPARα agonism on inflammatory mediators in key ocular tissues has not been performed. Therefore, the objective of this investigation was to determine the effect of PPARα agonism on important proinflammatory cytokines in vitro in human corneal epithelial cells (HCEpiCs), human keratocytes (human corneal fibroblasts), and human retinal endothelial cells (HRECs). Furthermore, the effects of PPARα agonism on primary human aortic smooth muscle cells (HASMCs) were studied as a systemic benchmark, and the expression of PPARα in these various tissues was determined.
Materials and Methods
Materials
Tissue culture reagents were from Invitrogen (Carlsbad, California), Cascade Biologics (Portland, Oregon), ScienCell (San Diego, California), and Cell Systems (Kirkland, Washington). Dexamethasone and WY-14 643 were from Sigma (St Louis, Missouri). Interleukin 1β (IL-1β) was from R&D Systems (Minneapolis, Minnesota). alamarBlue solution was from Biosource (Camarillo, California). Human cytokine multiplex Luminex kit was obtained from Millipore (Billerica, Massachusetts). Polymerase chain reaction (PCR) reagents were from Qiagen (Valencia, California), Invitrogen, Bio-Rad (Hercules, California), and Stratagene (La Jolla, California). Inventoried Taqman human PPARα primers and probes for real-time quantitative PCR were from Applied Biosystems (Foster City, California).
Cells and Cell Treatments
Primary HCEpiCs were purchased from Cascade Biologics and maintained in EpiLife medium containing human corneal growth supplement (Cascade Biologics). Primary human keratocytes were purchased from ScienCell and maintained in fibroblast medium containing fibroblast growth supplement and 2% fetal bovine serum (FBS; ScienCell). Primary HRECs were purchased from Cell Systems and maintained in CS-C complete medium (Cell Systems). Primary HASMCs were purchased from ScienCell and maintained in smooth muscle cell medium containing growth factor supplement and 2% FBS (ScienCell). These cells were cultured in the above media containing 100 U/mL of penicillin and 100 µg/mL of streptomycin at 37°C in a humidified incubator with 5% CO2. Prior to treatments, cells were seeded in 24-well plates and cultured until confluence. Cells were pretreated with WY-14 643 or dexamethasone for 2 hours, and then further treated with vehicle (0.1% dimethyl sulfoxide [DMSO]), IL-1β, WY-14 643, dexamethasone, or their combinations in the basic medium for 18 hours. Each treatment was performed in triplicate, and appropriate dilutions were prepared to deliver a constant amount of the vehicle to each well. Media were collected for Luminex cytokine assays and cells for metabolic activity assay.
Multiplex Luminex
Cytokine content in the culture medium was analyzed using multiplex Luminex technology. 11 Eighteen cytokines (IL-1α, IL-1β, IL-6, IL-7, IL-8, IL-12(p40), IL-15, Eotaxin, Fractalkine, granulocyte colony-stimulating factor (G-CSF), granulocyte macrophage colony-stimulating factor (GM-CSF), interferon-inducible protein-10 (IP-10), monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-1β (MIP-1 β), transforming growth factor-α (TGF-α), tumor necrosis factor α [TNF-α], normal T-cell expressed, and secreted (RANTES), and VEGF) were measured according to the manufacturer's instructions. Samples were analyzed using Luminex 200 (Luminex, Austin, Texas) and Beadview software v1.0 (Upstate Cell Signaling Solutions, Temecula, California). Standard curves of known concentrations of recombinant human cytokines were used to convert median fluorescence intensity (MFI) to cytokine concentration in pg/mL. Only the linear portions of the standard curves were used to quantify cytokine concentrations, and in instances where the fluorescence reading exceeded the linear range of the standard curve, an appropriate dilution was performed to ensure that the concentration was in the linear portion of the curve.
Real-Time Quantitative PCR
Total RNA was prepared using RNeasy Plus Mini kit (Qiagen) and quantified using Quant-iT RNA Assay kit (Molecular Probes). First-strand complementary DNA (cDNA) was prepared using Affinity Script QPCR cDNA Synthesis kit (Stratagene). Real-time quantitative PCR was performed in the Mx3005P (Stratagene) using Brilliant II quantitative PCR (qPCR) Master Mix (Applied Biosystems). 12 Relative quantities of PPARα messenger RNA (mRNA) levels in different cell types were determined in comparison to HASMCs, whose expression level was set equal to 1. Polymerase chain reaction products (147 bp) of PPARα for each cell type were also determined by agarose gel electrophoresis.
Cellular Metabolic Activity
Cellular metabolic competence was determined by the alamarBlue assay. 13 Briefly, after removal of the medium, cells were incubated with 1:10 diluted alamarBlue solution for 3 hours at 37°C in a humidified incubator with 5% CO2. The plate was read fluorometrically by excitation at 530 to 560 nm and emission at 590 nm. Relative fluorescence units (RFUs) were used to determine the cellular metabolic activity, an index of cell viability.
Data Analysis and Statistics
All cytokine concentrations (pg/mL) were expressed as mean ± SD. Statistical analysis was performed using a 1-way analysis of variance (ANOVA) with the Dunnett post hoc comparison test using either control or IL-1β treatment as references. For all assays, P ≤ .05 was predetermined as the criterion of statistical significance.
Results
Effects of WY-14 643 and Dexamethasone on IL-1β-Induced Cytokine Release in HCEpiCs, Human Keratocytes, and HRECs
Three primary human ocular cells, HCEpiCs, human keratocytes, and HRECs, were pretreated with the test agents, WY-14 643 or dexamethasone, for 2 hours and then further treated with vehicle, IL-1β, or IL-1β plus test agents in the medium without growth factors and serum for 18 hours. No statistically significant effect on cellular metabolic activity, as measured by the alamarBlue assay, was observed with these treatments. Media were collected and used for determination of cytokine content using an 18-cytokine Luminex kit. Under basal conditions (without IL-1β stimulation), cytokine levels in the conditioned medium were very low, with almost all the cytokines studied below the limits of detection by Luminex, and no statistically significant effect of either WY-14 643 or dexamethasone on cytokine release was observed.
In HCEpiCs, 11 out of 18 cytokines tested were detected (G-CSF, GM-CSF, IL-1α, IL-6, IL-7, IL-8, MCP-1, RANTES, TGF-α, TNF-α, and VEGF) and 8 out of 11 cytokines detected were induced by 10 ng/mL IL-1β (all except IL-1α, TGF-α, and VEGF). IL-1β was excluded from analysis because it was the stimulus. As anticipated, dexamethasone significantly inhibited IL-1β-induced IL-6 and TNF-α release at 100 nmol/L, 1 µmol/L, and 10 µmol/L and significantly inhibited GM-CSF release at all doses tested (10 nmol/L, 100 nmol/L, 1 µmol/L, and 10 µmol/L; Figure 1). However, rather than inhibiting cytokine release, WY-14 643, a selective PPARα agonist, 14 significantly increased GM-CSF, IL-6, MCP-1, and TNF-α levels at 50 and/or 150 µmol/L (Figure 1). No statistically significant effects of dexamethasone or WY-14 643 on other cytokines were observed.
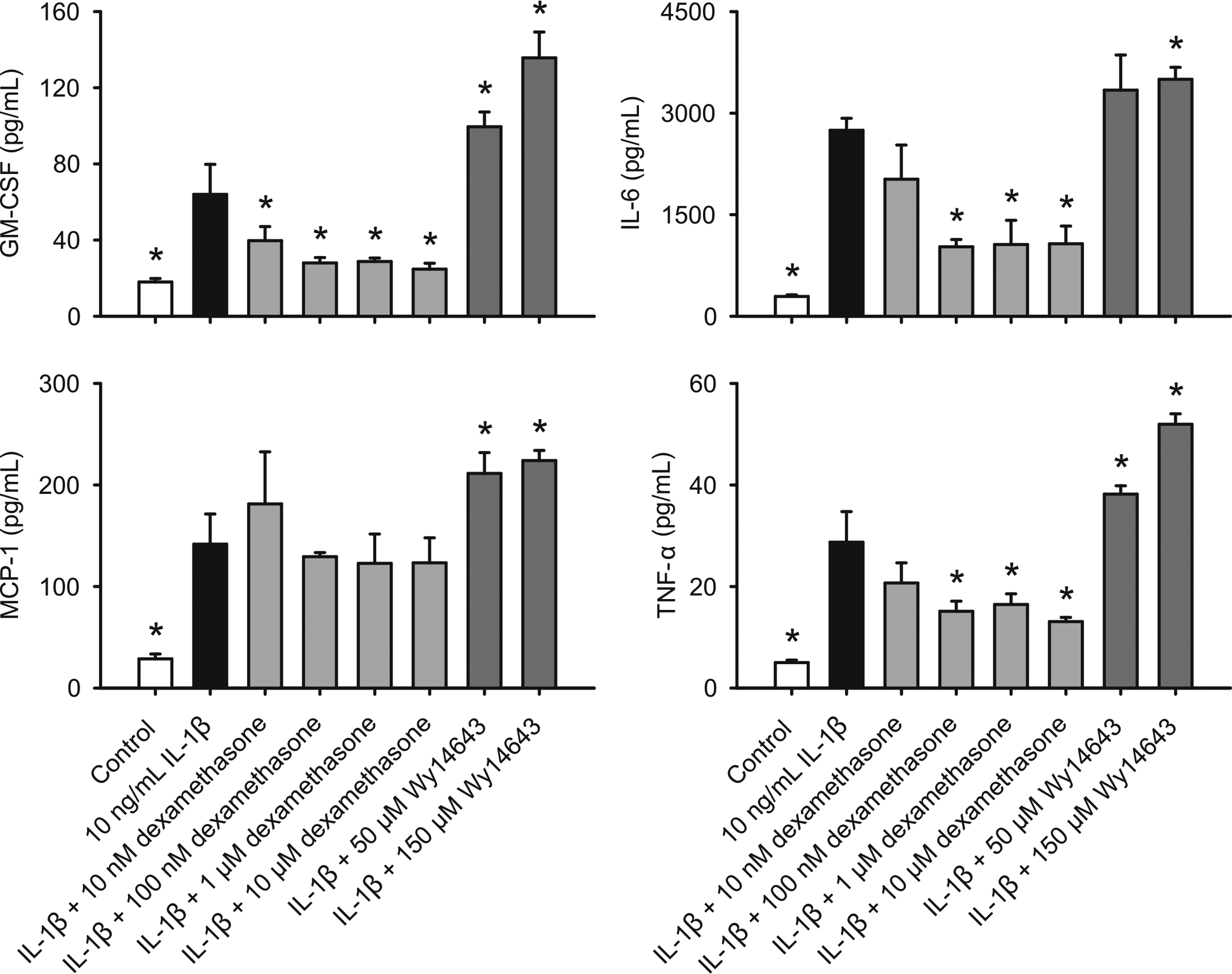
Effects of WY-14 643 and dexamethasone (DEX) on IL-1β-induced cytokine release in HCEpiCs. Cells were pretreated with WY-14 643 or dexamethasone for 2 hours, and then further treated with vehicle (0.1% DMSO), IL-1β, WY-14 643, dexamethasone, or their combinations in the basic medium for 18 hours. Cytokine content in the conditioned media was determined using Luminex technology. Data are means ± standard deviation (SD), n = 3. *P ≤ .05 versus IL-1β. IL indicates interleukin; HCEpiCs, human corneal epithelial cells; DMSO, dimethyl sulfoxide; TNF-α, tumor necrosis factor α; MCP-1, monocyte chemotactic protein-1; GM-CSF, granulocyte macrophage colony-stimulating factor.
In human keratocytes, 9 of 18 cytokines tested were detected (Eotaxin, G-CSF, GM-CSF, IL-6, IL-8, IP-10, MCP-1, RANTES, and VEGF), and 8 of 9 cytokines detected were induced by 1 ng/mL IL-1β (all except VEGF). Dexamethasone significantly inhibited IL-1β-induced G-CSF, GM-CSF, IL-6, and IL-8 release at 10 µmol/L whereas G-CSF, GM-CSF, IL-6, and IL-8 release was significantly increased by WY-14 643 at 100 µmol/L (Figure 2). In HRECs, dexamethasone significantly inhibited IL-1β-induced IL-1α and TNF-α release at all doses tested (10 nmol/L, 100 nmol/L, 1 µmol/L, and 10 µmol/L). However, WY-14 643 had no effects on IL-1α release, and significantly increased TNF-α release at 150 µmol/L (Figure 3). No statistically significant effects of dexamethasone or WY-14 643 on other cytokines were observed in human keratocytes and HRECs.
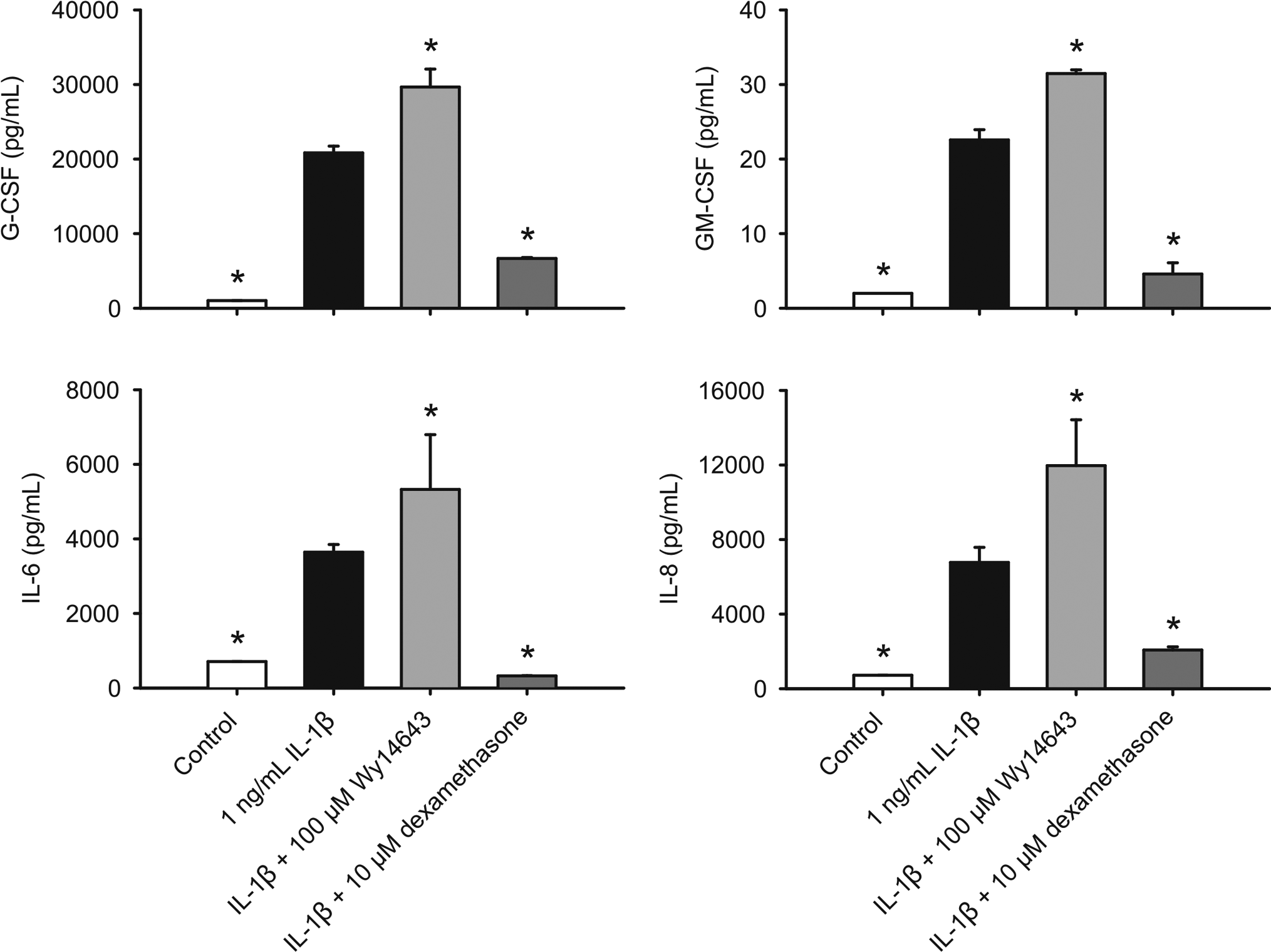
Effects of WY-14 643 and dexamethasone (DEX) on IL-1β-induced cytokine release in human keratocytes. Cells were pretreated with WY-14 643 or dexamethasone for 2 hours, and then further treated with vehicle (0.1% DMSO), IL-1β, WY-14 643, dexamethasone, or their combinations in the basic medium for 18 houors. Cytokine content in the conditioned media was determined using Luminex technology. Data are means ± standard deviation (SD), n = 3. *P ≤ .05 versus IL-1β. IL indicates interleukin; DMSO, dimethyl sulfoxide; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte macrophage colony-stimulating factor.
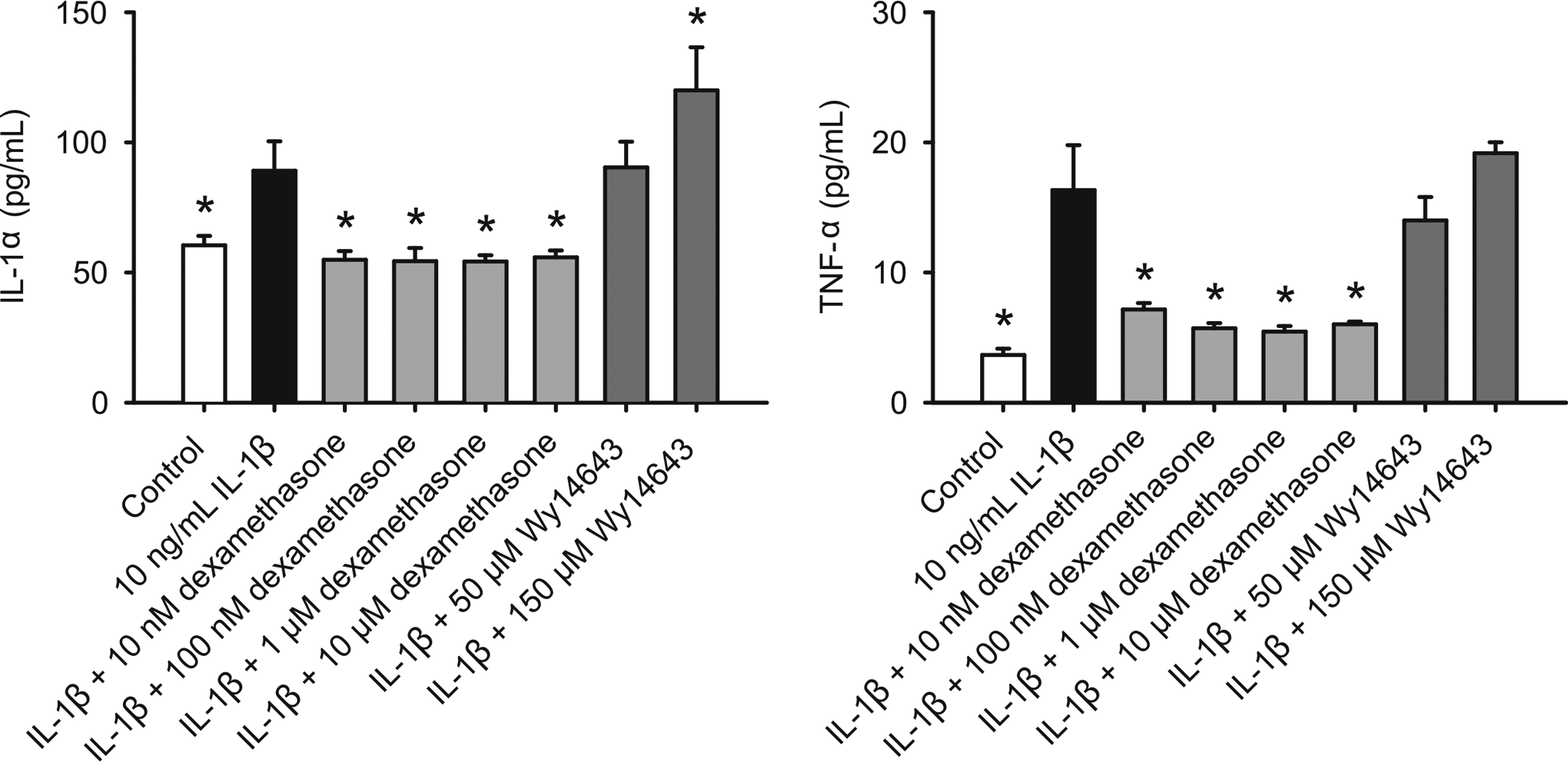
Effects of WY-14 643 and dexamethasone (DEX) on IL-1β-induced cytokine production in HRECs. Cells were pretreated with WY-14 643 or dexamethasone for 2 hours, and then further treated with vehicle (0.1% DMSO), IL-1β, WY-14 643, dexamethasone, or their combinations in the basic medium for 18 hours. Cytokine content in the conditioned media was determined using Luminex technology. Data are means ± standard deviation (SD), n = 3. *P ≤ .05 versus IL-1β.IL indicates interleukin; HRECs, human retinal endothelial cells; DMSO, dimethyl sulfoxide; TNF-α, tumor necrosis factor α.
Effects of WY-14 643 and Dexamethasone on IL-1β-Induced Cytokine Release in HASMCs
It has been reported that WY-14 643 inhibits IL-1β-induced IL-6 expression in HASMC. 14 To validate our assays, primary HASMC cells were treated with IL-1β, dexamethasone, WY-14 643, or their combinations as described above for the 3 ocular cell types. As shown in Figure 4, dexamethasone and WY-14 643 both significantly inhibited IL-6, IL-8, and MCP-1 release in HASMC. Furthermore, WY-14 643 significantly stimulated TNF-α release whereas dexamethasone inhibited its release (Figure 4).
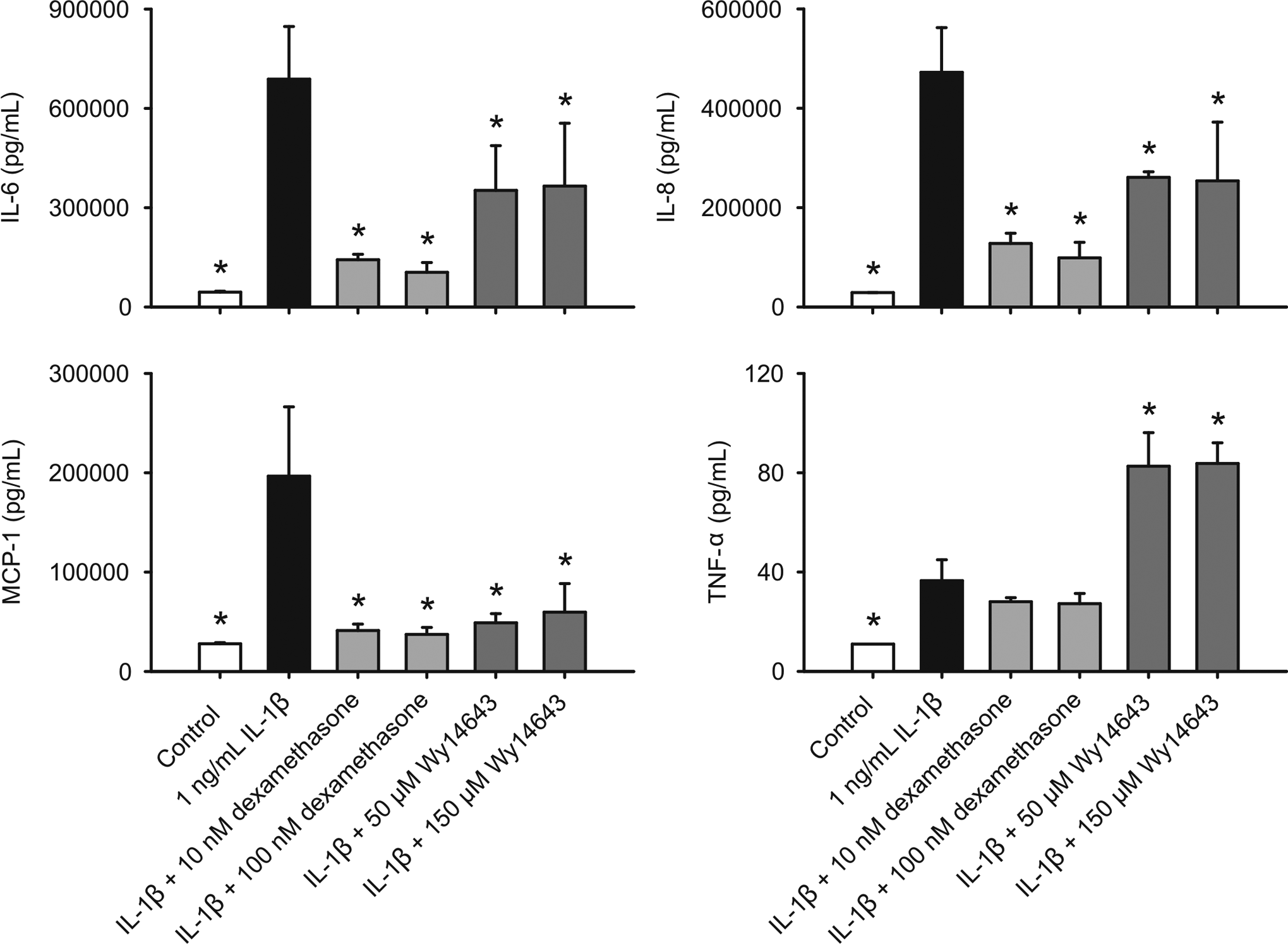
Effects of WY-14 643 and dexamethasone (DEX) on IL-1β-induced cytokine release in HASMCs. Cells were pretreated with WY-14 643 or dexamethasone for 2 hours, and then further treated with vehicle (0.1% DMSO), IL-1β, WY-14 643, dexamethasone, or their combinations in the basic medium for 18 hours. Cytokine content in the conditioned media was determined using Luminex technology. Data are means ± standard deviation (SD), n = 3. *P ≤ .05 versus IL-1β. IL indicates interleukin; HASMCs, human aortic smooth muscle cells; DMSO, dimethyl sulfoxide; TNF-α, tumor necrosis factor α; MCP-1, monocyte chemotactic protein-1.
Effects of WY-14 643 and Dexamethasone on VEGF Production in HCEpiCs and Human Keratocytes
As anticipated, IL-1β had no significantly stimulatory effect on VEGF production in HCEpiC and human keratocytes. However, WY-14 643 significantly increased the release of this proangiogenic molecule in HCEpiC at 50 and 150 µmol/L and in human keratocytes at 150 µmol/L whereas dexamethasone exhibited some inhibitory effect (Figure 5).
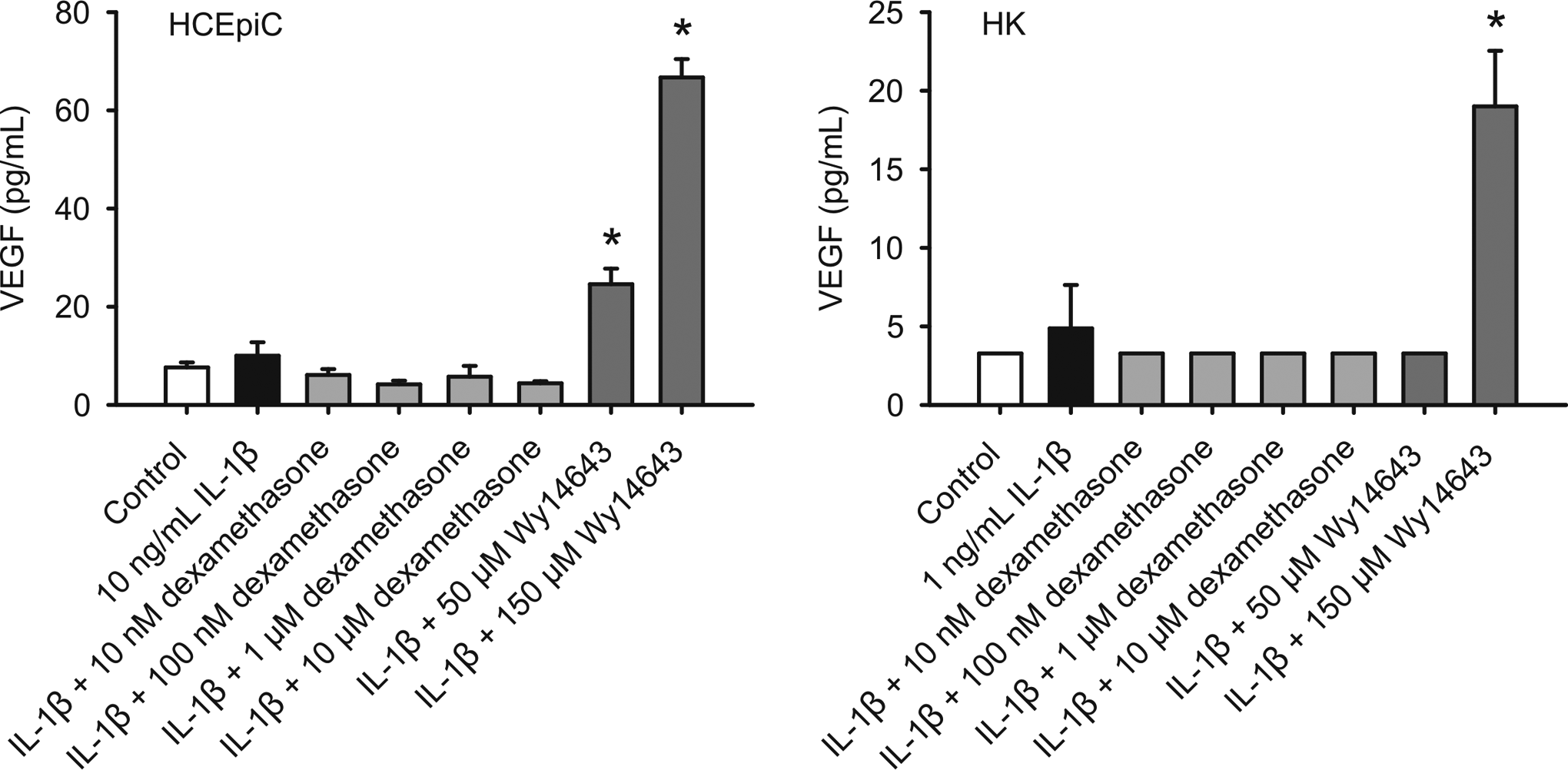
Effects of WY-14 643 and dexamethasone (DEX) on VEGF release in HCEpiCs and human keratocytes (HK). Cells were pretreated with WY-14 643 or dexamethasone for 2 hours, and then further treated with vehicle (0.1% DMSO), IL-1β, WY-14 643, dexamethasone, or their combinations in the basic medium for 18 hours. VEGF content in the conditioned media was determined using Luminex technology. Data are means ± standard deviation (SD), n = 3. *P ≤ .05 versus control. HCEpiCs indicates human corneal epithelial cells; HK, human keratocytes; DMSO, dimethyl sulfoxide; VEGF, vascular endothelial growth factor.
Expression of PPARα in HCEpiC, HK, HREC, and HASMC
The pharmacological target of interest, PPARα, was determined to be present in all cell types studied in this study by real-time qPCR using human PPARα-specific sequence as the primer (Figure 6). Expression of PPARα in corneal epithelial cells, fibroblasts, and endothelial cells has been previously demonstrated by PCR, Northern blot, or Western blot. 5,15 –19
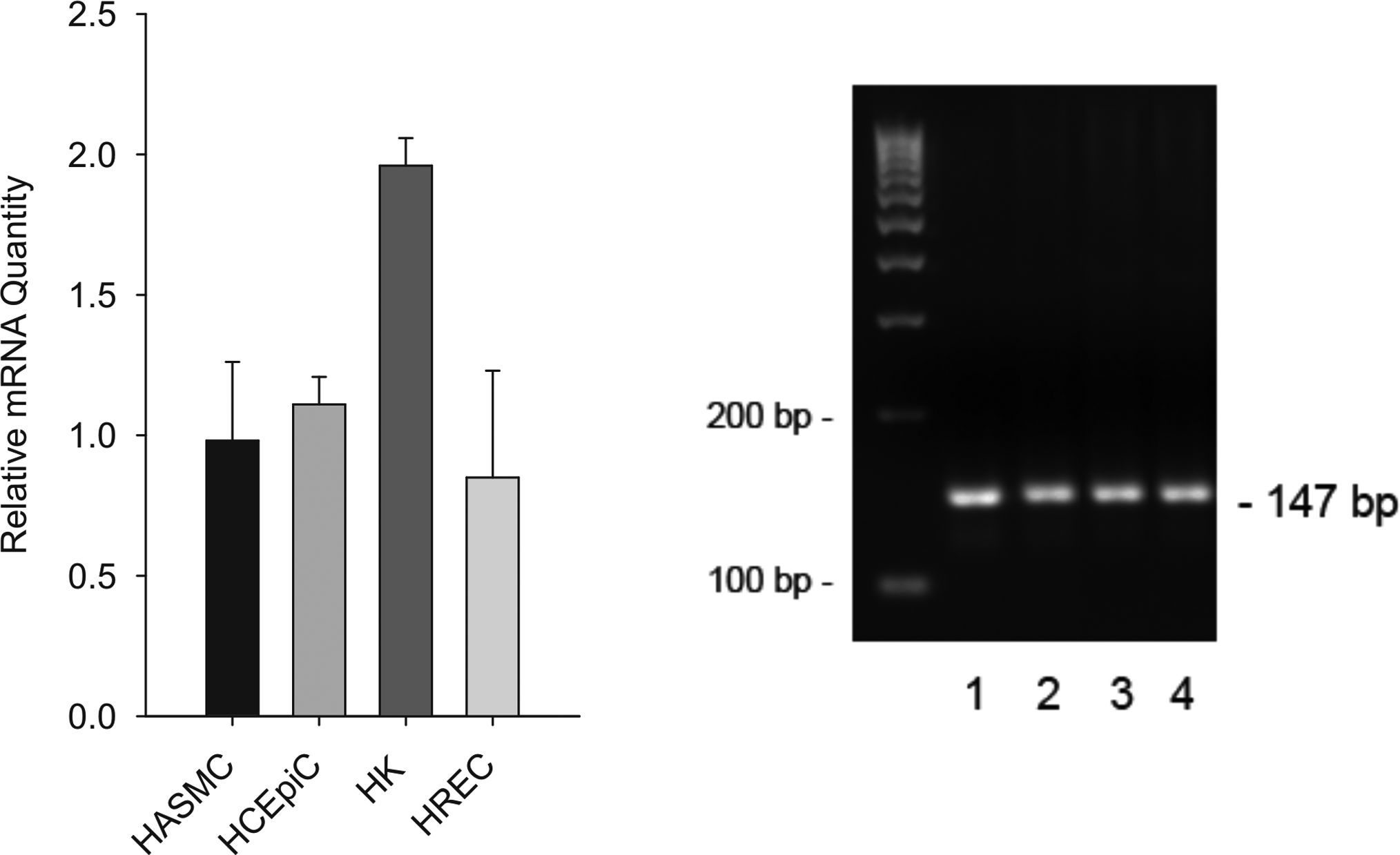
Relative levels of PPARα mRNA in HASMCs, HCEpiCs, human keratocytes (HK), and HRECs. Total RNA was prepared using RNeasy plus Mini kit and quantified using Quant-iT RNA Assay kit. First-strand cDNA was prepared using Affinity Script QPCR cDNA Synthesis kit. Real-time quantitative PCR was performed using Brilliant II qPCR Master Mix and inventoried Taqman human PPARα primers and probes. Relative quantities of PPARα mRNA levels in different cell types were determined in comparison to HASMCs, whose expression level was set equal to 1 (left panel). PCR products (147 bp) of PPARα for each cell type were also determined by agarose gel electrophoresis (right panel). Bio-Rad EZ Load 100 bp DNA ladder was used. 1, HASMC; 2, HCEpiC; 3, HK; 4, HREC. HCEpiCs indicates human corneal epithelial cells; PPARα, Peroxisome proliferator-activated receptor α; HASMCs, human aortic smooth muscle cells; HRECs, human retinal endothelial cells; HK, human keratocytes; cDNA, complementary DNA; PCR, polymerase chain reaction; mRNA, messenger RNA; qPCR, quantitative PCR.
Discussion
The results from the current series of studies strongly indicate that in ocular tissues, WY-14 643, a low potent but selective PPARα agonist, is not associated with cellular responses consistent with anti-inflammatory activity. In fact, the current data are consistent with a proinflammatory effect of WY-14 643 in the eye. Peroxisome proliferator-activated receptor agonists demonstrate marked anti-inflammatory effects in a variety of tissues. 20 In endothelium and vascular smooth muscle, agonism of PPARα is associated with the inhibition of cytokines and adhesion molecules. 21 –24 These in vitro results have been borne out experimentally in vivo; PPARα-deficient mice are prone to proinflammatory responses and exacerbation of various inflammatory diseases, and PPARα agonists decrease nitric oxide (NO) production in experimental autoimmune encephalomyelitis, decrease TNF-α and IL-1α and inflammation in allergic dermatitis, decrease damage in ischemia−reperfusion injury, and demonstrate neuroprotective effects in animal models of Alzheimer. 20,25 Altogether, these observations support the rationale for the anti-inflammatory potential of PPAR agonism.
Signaling pathways involving nuclear hormone receptors, including PPARs, are inherently complex, and are likely to involve both ligand-specific and cell type-specific effects. 5 For PPARs, despite the clear evidence from multiple investigations demonstrating the potential for anti-inflammatory efficacy, there have also been some suggestions to the contrary. For instance, activation of PPARα in human aortic endothelial cells has been associated with the upregulation of the proinflammatory cytokines IL-8 and MCP-1. 26 Furthermore, PPARα agonist treatment in lipopolysacharide (LPS)-stimulated mice resulted in increased TNF-α plasma levels. 27 Although the mechanisms by which such potentially proinflammatory effects occur is not understood, such data point to the need for careful evaluation of the effects of PPARα agonists in relevant cell types.
In ocular cells, potentially proinflammatory effects of PPARα agonism have also been reported. In corneal epithelial cells, PPARα agonists have been shown to induce expression of cyclooxygenase 2 (COX-2) and to increase the conversion of arachidonic acid to 12-hydroxy-5,8,11,14-eicosatetraenoic acid (12-HETE) and 12-hydroxy-5,8,14-eicosatrienoic acid (12-HETrE). 19,28 Additionally, the PPARα agonist fenofibrate has been shown to induce VEGF expression in HREC in an adenosine monophosphate (AMP)-activated protein kinase (AMPK)-dependent manner. 29 Again, although the molecular mechanisms for these effects are not well understood, their potential proinflammatory implications are important.
WY-14 643 is a selective and low-potent PPARα agonist, which has been extensively used to study the therapeutic effects of PPARα-mediated pathways in vivo and in vitro at micromole levels. 16,19,21,26,30 Our preliminary studies showed no effect of WY-14 643 on cell viability in cultured primary ocular cells up to 300 µmol/L. The data from the current study demonstrate for the first time the broad upregulation of proinflammatory cytokines in ocular cells, including HCEpiC, corneal fibroblasts (keratocytes), and retinal endothelial cells. Consistent with the previous studies in the literature, the current findings clearly demonstrate cell-specific effects of WY-14 643. It should be pointed out that although PPAR agonists such as fibrates and thiazoladinediones (TZDs) have been used for their hypolipidemic and anti-inflammatory effects, they do not selectively act on PPARs. Many reported PPAR activities may occur through PPAR-independent pathways. For instance, increasing evidence suggests that fibrates activate other nuclear receptors and also stimulate pathways that do not depend on PPARα. 31 –34
In addition to the effects of WY-14 643 on ILs and related molecules, the current study demonstrates an upregulation of VEGF expression in 2 ocular cell types. To date, studies on VEGF modulation by PPAR agonists have primarily focused on PPARγ agonists, especially the TZDs. In clinical practice, there have been a number of case reports of vision loss and profound macular edema associated with the use of such PPAR agonists in diabetic patients. 35,36 Recently, a biochemical mechanism has been postulated for these findings; PPARγ agonism has been demonstrated to produce tissue-specific upregulation of VEGF expression in retina and adipose tissue. 37 Previously, it was believed that such effects were associated primarily with agonism of PPARγ, and not PPARα. In fact, PPARα agonism has been shown to inhibit experimental corneal neovascularization in mice. FIELD study (Effect of fenofibrate on the need for laser treatment for diabetic retinopathy) suggests that treatment with fenofibrate in individuals with type 2 diabetes reduces the need for laser treatment of diabetic retinopathy. 38 However, more recent studies have demonstrated that fenofibrate upregulates VEGF expression in retinal endothelium, 29 as well as VEGF-dependent corneal neovascularization in mice. 15
In conclusion, although the majority of available data from cell-based and animal studies indicate an anti-inflammatory property of PPARα agonists, there is also clear evidence suggesting that these anti-inflammatory effects may be cell- or tissue-type specific. 20,27 The data from the current study are consistent with the emerging apparent role of PPARα agonism as a proinflammatory and proangiogenic stimulus in ocular tissue. Furthermore, these observations demonstrate that these effects are not limited to a single ocular cell type or cytokine, but form a consistent pattern indicating a role of PPARα agonism in ocular inflammation. However, additional studies are needed to further explore this issue, including using other selective PPARα agonists.
Footnotes
Notes
The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) received no financial support for the research and/or authorship of this article.
Acknowledgments
The authors thank Jessica Ackerman, Charu Agarwal, Megan Cavet, Yong-Qing Lin, Karl VanDerMeid, and Stepan Volhejn for technical assistance.