
Abstract
The synthetic retinoic acid analog, 9-cis-UAB30 [(2E,4E,6Z,8E)-8-(3′,4′-dihydro-1′(2′H)-naphthalen-1′-ylidene)-3,7-dimethyl-2,4,6-octatrienoic acid], is a specific ligand for the retinoid X receptor. Murine oncogenicity and pharmacokinetics studies were performed as part of the preclinical development of 9-cis-UAB30 for breast cancer chemoprevention. In the oncogenicity study, TSG-p53(+/-) (p53 knockout) mice (25 per sex per group) received daily gavage exposure to 9-cis-UAB30 doses of 0 (control), 30, 100, or 300 mg/kg/d for 6 months. Positive controls received p-cresidine (400 mg/kg/d) for 6 months. 9-cis-UAB30 had no biologically significant effects on survival, body weight, body weight gain, clinical signs, hematology, or clinical chemistry but induced dose-related hepatomegaly in both sexes and decreased thymus weights in high-dose females. Gross and microscopic pathology provided no evidence of 9-cis-UAB30 toxicity or oncogenicity; by contrast, p-cresidine induced urinary bladder neoplasms in more than 60% of male and female mice. It was concluded that 9-cis-UAB30 is not oncogenic in p53(+/-) mice. In the pharmacokinetics study, C57BL/6 mice received daily gavage exposure to 9-cis-UAB30 (100 or 300 mg/kg/d) for 1 or 7 days. Pharmacokinetic parameters were similar after 1 and 7 days of dosing. Dose-related peak plasma levels of 9-cis-UAB30 were seen between 0.25 and 3 hours; volume of distribution was comparable at both dose levels. Increases in area under the curve were less than proportional to dose and were associated with an increased rate of apparent clearance and decreased elimination half-life. These results suggest decreased absorption and/or possible induction of clearance mechanisms. Enzyme induction may underlie the hepatomegaly seen in mice treated with 9-cis-UAB30 for 6 months in the oncogenicity study.
Several natural and synthetic analogs of vitamin A (retinoids) have significant activity as cancer chemopreventive agents in the breast and other organs in laboratory animals. 1 Unfortunately, however, the results of clinical intervention trials with retinoids have generally failed to demonstrate broadly based chemopreventive activity against breast cancer. 2 In the largest such trial, N-(4-hydroxyphenyl)retinamide (fenretinide) achieved significant reductions in breast cancer risk in only a subset of high-risk patients. 3 As a result, efforts continue to identify novel retinoids with increased efficacy in breast cancer chemoprevention. Increased efficacy could be achieved by molecules with greater intrinsic anticarcinogenic activity and/or reduced systemic toxicity. Reductions in toxicity could permit drug administration at higher doses, thereby increasing desirable pharmacologic activity.
It is well-established that modifying the β-ionine ring and/or the polyene side chain of the retinoic acid (RA) molecule has substantial effects on both chemopreventive efficacy and toxicity. These effects appear to result at least in part from the relative ability of individual retinoic acid analogs to act as ligands for the retinoic acid receptor (RAR) and the retinoid X receptor (RXR), specific nuclear receptors with substantially different downstream effects. 4,5 Specific RAR agonists (eg, all-trans-RA and 13-cis-RA), specific RXR agonists (eg, bexarotene), and RAR/RXR pan-agonists (eg, 9-cis-RA) all demonstrate significant antitumor activity in rodent models for breast cancer. 6-8 Importantly, however, 9-cis-RA and bexarotene, retinoids whose chemopreventive activities appear to be at least partially mediated by RXR binding, have toxicity profiles that do not include classical “vitamin A-like” toxicities such as weight loss, bone fractures, and alopecia in either rodents 9,10 or humans. 11
9-cis-UAB30 ([2E,4E,6Z,8E]-8-[3′,4′-dihydro-1′(2′H)-naphthlen-1′-ylidene]-3,7-dimethyl-2,4,6-octatrienoic acid; Figure 1 ) is a conformationally constrained synthetic retinoic acid analog that binds specifically to RXR. 12 On the basis of its activity as a chemopreventive agent in a rat model for breast cancer 13-14 and the relatively limited toxicity of RXR agonists such as bexarotene in both experimental animals 10 and humans, 15,11 9-cis-UAB30 is being developed as a chemopreventive agent for human breast cancer. The compound is currently in phase 1 clinical trials.
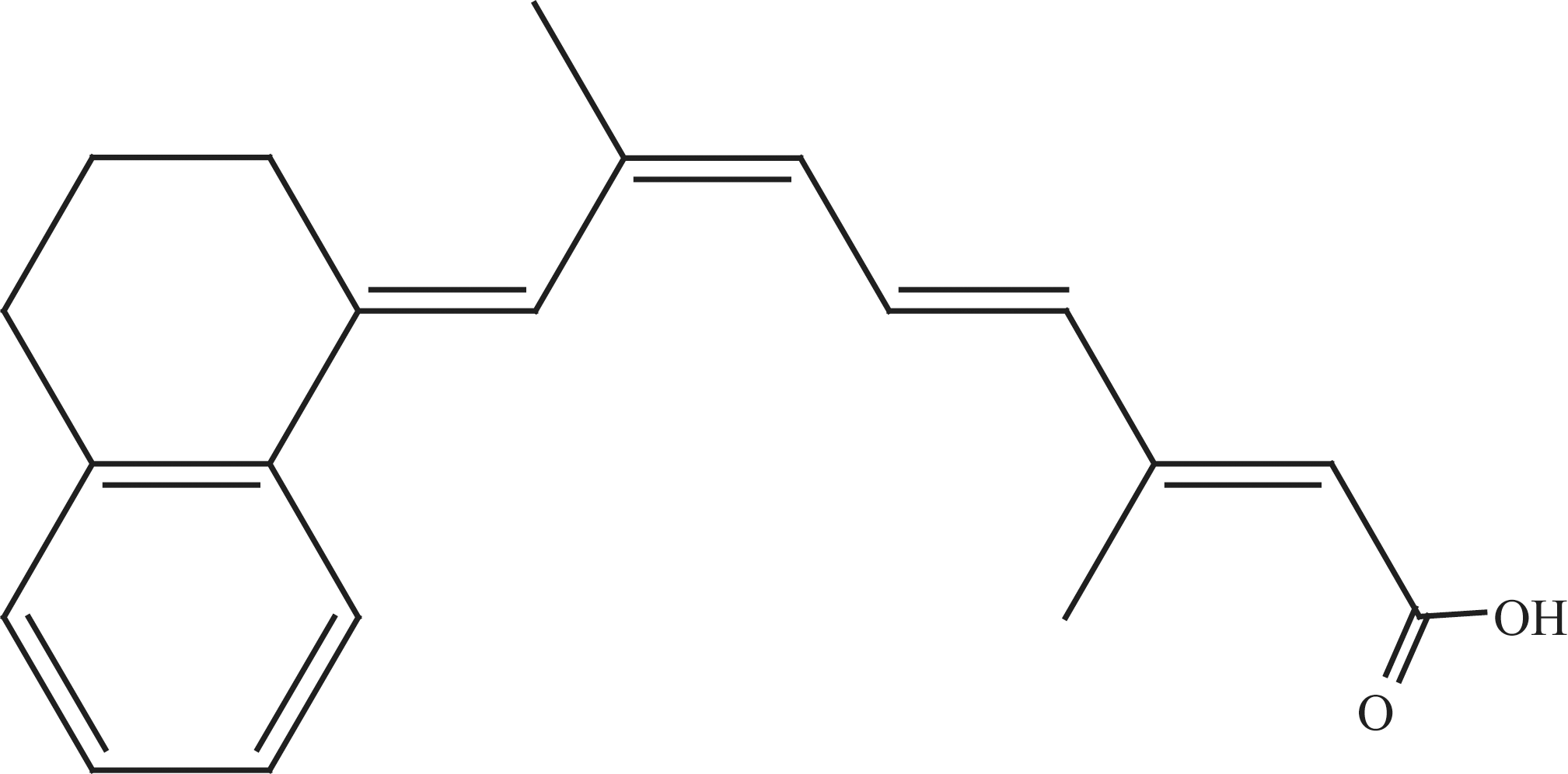
Chemical structure of 9-cis-UAB30.
In genotoxicity evaluations, 9-cis-UAB30 was negative in bacterial mutagenesis tests (Ames tests), an in vivo mouse micronucleus assay, and an in vivo mutagenicity evaluation performed using the B6C3F1 Big Blue lacI/cII transgenic mouse model (unpublished). However, the results of chromosomal aberration studies in Chinese hamster ovary (CHO) cells were inconclusive. In view of this equivocal result, the present studies were performed (a) to evaluate the oncogenicity of 9-cis-UAB30 in the TSG-p53(+/-) mouse (p53 knockout mouse) and (b) to characterize its plasma pharmacokinetics in mice following oral administration.
The p53 knockout mouse is broadly accepted by regulatory agencies as an alternative animal model for oncogenicity bioassays 16 and demonstrates a more than 85% concordance with the results of chronic rodent oncogenicity bioassays. 17 At the time of assay conduct, the position of the US Food and Drug Administration (FDA) was to “accept studies in the p53 model for drugs that are equivocally or clearly positive for genotoxic activity.” 18 As such, these studies addressed a key regulatory requirement for the entry of 9-cis-UAB30 into clinical efficacy trials for breast cancer prevention and provided critical laboratory data relevant to its possible utility as a cancer preventive agent in humans.
Materials and Methods
Animal Welfare
Prior to the initiation of experimentation, study protocols were reviewed and approved by the IIT Research Institute Animal Care and Use Committee. All work involving experimental animals was performed in full compliance with US National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Animals and Animal Husbandry
C57BL/6 and TSG-p53(+/-) (heterozygous p53 knockout) mice were purchased from Taconic, Germantown, New York. The p53 knockout mouse is a genetically engineered animal strain that is accepted by the US FDA and the European Committee for Proprietary Medicinal Products as an alternative model for oncogenicity bioassays. 16 The C57BL/6 mouse is the nontransgenic parental strain of the TSG-p53(+/-) mouse and is commonly used for range-finding and pharmacokinetics studies conducted in association with oncogenicity studies performed in the TSG-p53(+/-) mouse model system.
Mice were received at 6 to 8 weeks of age and were quarantined for a minimum of 1 week prior to study initiation. Mice were housed individually in suspended stainless steel cages in a windowless, climate-controlled room that was maintained on a 12-hour light/12-hour dark cycle. At all times during the quarantine and dosing periods, animals were permitted free access to certified Purina Laboratory Chow 5002 (PMI Nutrition International, Brentwood, Mo) and coarse-filtered City of Chicago water (delivered via an automatic watering system). After assignment to experimental groups, mice were identified by tail tattoo.
Test and Control Articles
9-cis-UAB30 [(2E,4E,6Z,8E)-8-(3′,4′-dihydro-1′(2′H)-naphthalen-1′-ylidene)-3,7-dimethyl-2,4,6-octatrienoic acid] (C20H22O2; lot number AC5003NC, purity = 99.1%) was manufactured by Alchem Laboratories (Alachua, Fla) and obtained from the Chemopreventive Agent Repository maintained by the Division of Cancer Prevention, National Cancer Institute. 9-cis-UAB30 was administered in a vehicle of 0.5% (wt/vol) aqueous methylcellulose (Sigma-Aldrich Chemical, St. Louis, Mo). Mice in the vehicle control group received daily gavage administration of vehicle only. Mice in the positive control group in the oncogenicity study received daily oral (gavage) exposure to p-cresidine (3-amino-4-methoxytoluene, 2-methoxy-5-methylaniline; C8H11NO; purity = 98.5%; obtained from Sigma-Aldrich). p-Cresidine induces urinary bladder cancers in TSG-p53(+/-) mice and has been used previously as a positive control article for oncogenicity bioassays in this animal model. 17,19
Oncogenicity Study Design
The oncogenicity study in p53 knockout mice was conducted in full compliance with FDA Good Laboratory Practice Regulations (Title 21, Code of Federal Regulations, part 58). Groups of 25 TSG-53(+/-) mice per sex received daily oral (gavage) exposure to 9-cis-UAB30 at doses of 30, 100, or 300 mg/kg of body weight per day for 6 months. 9-cis-UAB30 was administered in a vehicle of 0.5% (wt/vol) aqueous carboxymethylcellulose; dosing volume was 10 mL/kg. Mice in the vehicle control group (25 per sex) received daily gavage exposure to vehicle only (10 mL/kg body weight). Mice in the positive control group (25 per sex) received daily gavage exposure to p-cresidine (400 mg/kg of body weight per day) for the same period.
Dose levels of 9-cis-UAB30 were selected on the basis of the results of a 28-day range-finding study in C57BL/6 mice. In this study, a no observed adverse effect level (NOAEL) was identified as 30 mg/kg; doses above 30 mg/kg induced dose-related hepatomegaly in female mice (data not shown). Doses for the murine range-finding study were selected on the basis of data from subchronic (28-day) oral toxicity studies in rats and dogs. In the rat and dog studies, the NOAEL was identified as 3 mg/kg/d in rats and more than 100 mg/kg/d in dogs (unpublished data). The starting dose for phase 1 clinical trials is commonly selected on the basis of the NOAEL in the most sensitive preclinical test species; the rat is clearly the most sensitive species for the toxicity of 9-cis-UAB30. Interspecies scaling of the rat NOAEL (3 mg/kg/d) to mice using the mg/m2 approach yields an equivalent mouse dose of 6 mg/kg/d. Applying a 50× safety factor to this dose resulted in the selection of 300 mg/kg/d as the high dose used in the range-finding study in mice; because this dose did not induce limiting toxicity in the mouse range-finding study, it was deemed to be appropriate for use as the high dose in the oncogenicity study.
Mice were observed twice daily throughout the quarantine and dosing periods for mortality or evidence of toxicity. Body weights and food consumption were measured individually for each animal once per week throughout the dosing period, and each animal underwent a detailed, hand-held clinical and physical observation each week. Blood samples for clinical pathology evaluations were collected immediately prior to the terminal necropsy and were analyzed using automated instruments (clinical chemistry assays, Synchron LX-20, Beckman Coulter, Fullerton, Calif; hematology assays, Advia 120, Bayer Healthcare, Tarrytown, NY).
All mice underwent a complete necropsy with tissue collection. Intercurrent deaths were necropsied immediately after their discovery. At the end of the 6-month dosing period, surviving animals were euthanized in random order by CO2 asphyxiation; necropsy was initiated within 5 minutes of euthanasia. At the terminal necropsy, weights of the adrenals, brain, heart, kidneys, liver, spleen, testes/ovaries, and thymus were collected individually for each animal. Histopathologic evaluations on approximately 45 tissues per animal were performed on all mice in the vehicle control group and the group receiving the high dose of 9-cis-UAB30; the tissue list used in the study was consistent with that defined in section IV.C.6. (Carcinogenicity Studies With Rodents) of the FDA Redbook 2000. Histopathologic evaluations in groups receiving the low and middle doses of 9-cis-UAB30 were limited to identified target tissues only. Histopathologic evaluation of tissues from animals in the positive control group was limited to the kidney and urinary bladder, known target tissues for the oncogenicity of p-cresidine. 17
Pharmacokinetics Study Design
To characterize the plasma pharmacokinetics of 9-cis-UAB30, 220 C57BL/6 mice (110 per sex) received daily oral (gavage) doses of 100 or 300 mg 9-cis-UAB30 per kilogram of body weight for either 1 or 7 days. The 7-day period of repeat-dose administration was selected as sufficient to demonstrate possible enzyme induction, as is seen with a series of prototypic P450 inducers. 20 Five animals per sex per time point were euthanized for blood collection at predose and at 0.25, 0.5, 1.0, 1.5, 2, 3, 4, 6, 8, and 12 hours after dosing on day 1 and day 7. Plasma was collected and stored at –80°C prior to analysis.
To quantitate plasma levels of 9-cis-UAB30, 0.4 mL of 0.1 M phosphoric acid and 2 mL of a mixture of HPLC grade hexane isomers were added to 100 μL of sample. Samples were shaken for 15 minutes and centrifuged for 10 minutes. The organic layer was removed, evaporated to dryness under nitrogen, and reconstituted in methanol. Reconstituted samples were analyzed by HPLC, using a Waters Spherisorb ODS-2, 150 × 4.6 mm, 3-µ column held at 30°C, and a mobile phase of 0.1% formic acid in methanol/water (85:15, vol/vol). Absorbance was measured at 330 nm. At a flow rate of 1.0 mL/min, elution time of 9-cis-UAB30 was approximately 6.6 minutes. The 9-cis-UAB30 concentration in each sample was quantified by comparison to a standard curve prepared on each day of analysis.
Pharmacokinetic (PK) analyses were performed in WinNonlin (professional edition 4.1; Pharsight, Mountain View, Calif), using the noncompartmental model for extravascular input; all PK analyses were performed using mean plasma concentrations of 9-cis-UAB30 in 5 animals per group per time point. Area under the plasma concentration-time curve (AUC) from time zero to the last measured concentration was estimated using the linear trapezoidal rule up to Cmax (maximum observed plasma concentration), followed by the log trapezoidal rule for the remainder of the curve. The area under the plasma concentration-time curve extrapolated to infinity is defined as AUC0-∞ = AUC0-t + Ct/λz, where λz is the disposition rate constant estimated using log-linear regression during the terminal elimination phase, and Ct is the last measureable plasma concentration. Mean AUC values between treatment groups, genders, and day of dosing were compared using Bailer’s z test, 21 as modified by Nedelman et al 22 for hyperbolic curve-fitting. The type I error level was 5% (α = .05).
Statistical Analysis
Statistical analysis of continuous data (body weight, food consumption, clinical pathology, organ weights) was performed using analysis of variance, with post hoc comparisons made using Dunnett’s test. Incidence data (survival, clinical observations, histopathology) were compared using χ2 analysis. A minimum significance level of P < .05 was used for all comparisons.
Results
Oncogenicity Evaluation of 9-cis-UAB30 in p53(+/−) Mice
Daily gavage administration of 9-cis-UAB30 at doses of up to 300 mg/kg/d for 6 months induced no evidence of limiting toxicity that was identifiable through evaluations of animal survival, mean body weight, clinical signs, or the results of clinical pathology studies.
No pattern of gross toxicity attributable to 9-cis-UAB30 exposure was identified during detailed clinical observations that were performed weekly throughout the study. Mortality patterns also provided no evidence of 9-cis-UAB30 toxicity (Table 1). At study termination, 100% survival (25/25) was seen in male mice in vehicle control group and in groups exposed to the high and low (300 and 30 mg/kg/d) doses of 9-cis-UAB30. Survival in female mice in the vehicle control group was 96% (24/25) at study termination, compared with 100% survival (25/25) in female mice receiving the high and low doses of 9-cis-UAB30. In both sexes, the only deaths in mice exposed to 9-cis-UAB30 were seen in the middle-dose group (1/25 males, 3/25 females; P > .05 for both comparisons). In the positive control group, survival in both sexes of mice receiving p-cresidine was 88% (22/25).
Survival and Body Weight in p53(+/-) (p53 Knockout) Mice Receiving Chronic Oral Exposure to 9-cis-UAB30 or p-Cresidine
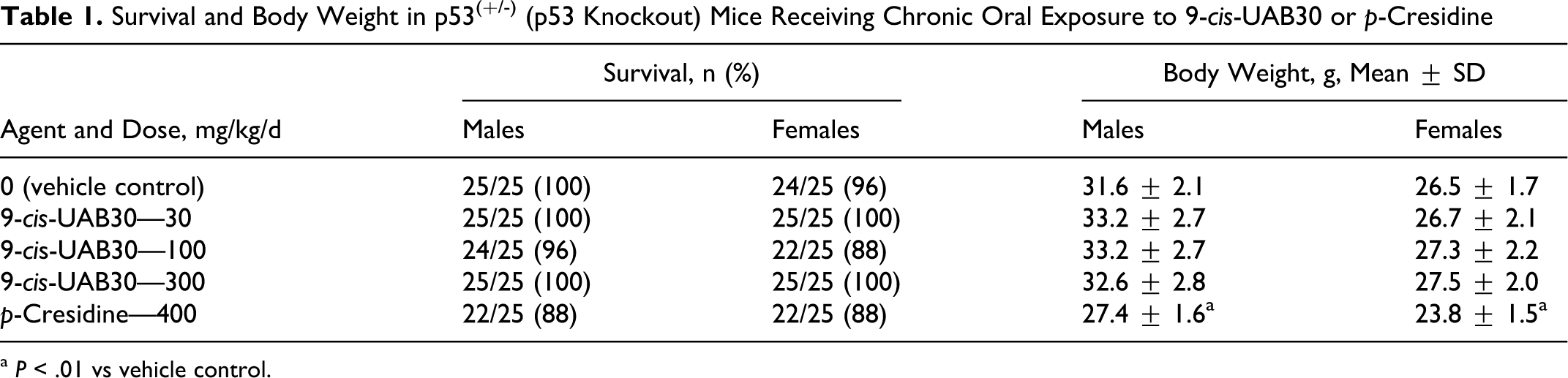
a P < .01 vs vehicle control.
Comparisons of group mean body weight also failed to identify any limiting toxicity associated with administration of 9-cis-UAB30; body weight curves in all groups exposed to 9-cis-UAB30 were comparable to those of sex-matched controls throughout the study (data not shown). At study termination at 26 weeks, mean body weights in groups of male mice exposed to the low, middle, or high doses of 9-cis-UAB30 ranged from 103.2% to 104.7% of male mice in the vehicle control group (Table 1; P > .05 for all comparisons). Similarly, group mean body weights in female mice exposed to 9-cis-UAB30 ranged from 100.8% to 103.8% of body weights in female vehicle controls (Table 1; P > .05 for all comparisons). By contrast, statistically significant reductions in group mean body weights in the positive control group treated with p-cresidine were seen in male mice beginning at week 7 and in female mice beginning at week 6. At study termination, mean body weights in the positive control group were 86.7% of control in male mice and 89.8% of control in female mice (P < .01 for both comparisons; Table 1).
Hematology evaluations performed on samples collected at the terminal necropsy provided no evidence of 9-cis-UAB30 toxicity (data not shown). Although mice treated with 9-cis-UAB30 demonstrated occasional differences from the vehicle control group, no dose-related changes in any hematology parameter were seen within a sex, nor were any consistent patterns of alterations in hematology parameters seen in both sexes. By contrast, male and female mice in the positive control group demonstrated statistically significant decreases in red blood cell count, hematocrit, and hemoglobin as well as changes in the differential white blood cell count. Significant increases in platelet counts in were seen in the positive control group in male mice only.
With the exception of plasma triglycerides in male mice, the results of clinical chemistry evaluations also failed to identify any evidence of 9-cis-UAB30 toxicity (data not shown). Plasma triglyceride levels were modestly (~25%) but significantly increased in male mice receiving the high dose of 9-cis-UAB30; smaller, nonsignificant increases in plasma triglycerides were seen in male mice in groups receiving the low and middle doses of 9-cis-UAB30. By contrast, triglyceride levels in all groups of female mice receiving 9-cis-UAB30 were 5% to 10% less than the mean triglyceride levels in female mice in the vehicle control group (P > .05 for all comparisons). Whereas 9-cis-UAB30 had minimal impact on clinical chemistry parameters, numerous statistically significant alterations in clinical chemistry values were seen in mice in the positive control group receiving p-cresidine. Male mice in the positive control group demonstrated statistically significant increases in plasma levels of Na+, Ca2+, alkaline phosphatase aspartate aminotransferase (AST), total bilirubin, and blood urea nitrogen (BUN) and in the BUN/creatinine ratio. Female mice in the positive control group demonstrated statistically significant increases in cholesterol, triglycerides, alanine aminotransferase, and AST.
Organ weight data collected at the terminal necropsy demonstrated statistically significant, dose-related increases in absolute and relative liver weights in both sexes of mice exposed to 9-cis-UAB30 (Table 2). Statistically significant increases in absolute liver weight were present at all dose levels in male mice; percentage increases in liver weight were 9.6%, 11.6%, and 23.6% in the low, middle, and high doses, respectively (P < .05 for all comparisons). Female mice demonstrated percentage liver weight increases of 1.8%, 9.2%, and 15.9% in the low-, middle-, and high-dose groups, respectively; liver weights in female mice in the middle- and high-dose groups were significantly different from vehicle control (P < .05).
Absolute Organ Weights (Grams) in p53(+/-) (p53 knockout) Mice Receiving Chronic Oral Exposure to 9-cis-UAB30
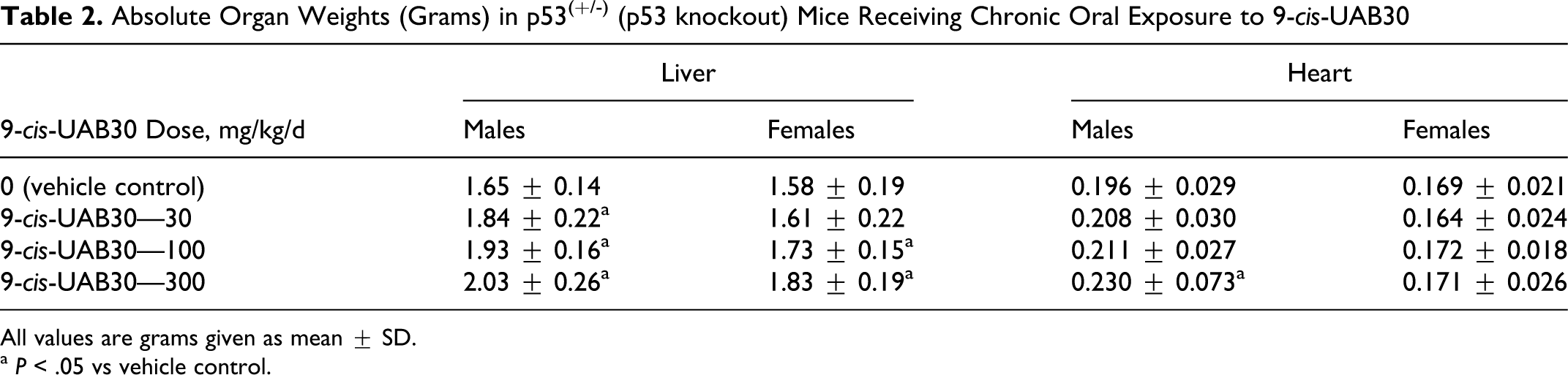
All values are grams given as mean ± SD.
a P < .05 vs vehicle control.
Absolute heart weights also demonstrated dose-related increases in male mice but not in female mice (Table 2); although mean heart weights in male mice were increased in all dose groups, only the 17.3% increase in mean heart weight in male mice receiving the high dose of 9-cis-UAB30 was statistically significant.
Histopathologic evaluation of tissues collected after 6 months of administration provided no evidence of oncogenicity of 9-cis-UAB30 in p53(+/-) mice and essentially no evidence of agent toxicity as demonstrated by the presence of nonneoplastic lesions. No malignancies were identified in any mouse exposed to 9-cis-UAB30 or in the vehicle control group (Table 3). Similarly, no exposure-related nonmalignant lesions were identified in groups exposed to 9-cis-UAB30. Chronic inflammatory changes were commonly observed in several organs in mice exposed to 9-cis-UAB30, and low incidences of several other nonneoplastic lesions were identified. However, lesion incidences in mice treated with 9-cis-UAB30 did not differ from those in sex-matched vehicle controls and are consistent with the background incidence of such lesions in p53(+/-) mice. As such, these nonneoplastic changes are considered to be incidental lesions and are interpreted as being unrelated to exposure to 9-cis-UAB30.
Number and Incidence of Exposure-Related Neoplastic and Nonneoplastic Lesions in p53(+/-) (p53 Knockout) Mice Receiving Chronic Oral Exposure to 9-cis-UAB30 or p-Cresidine
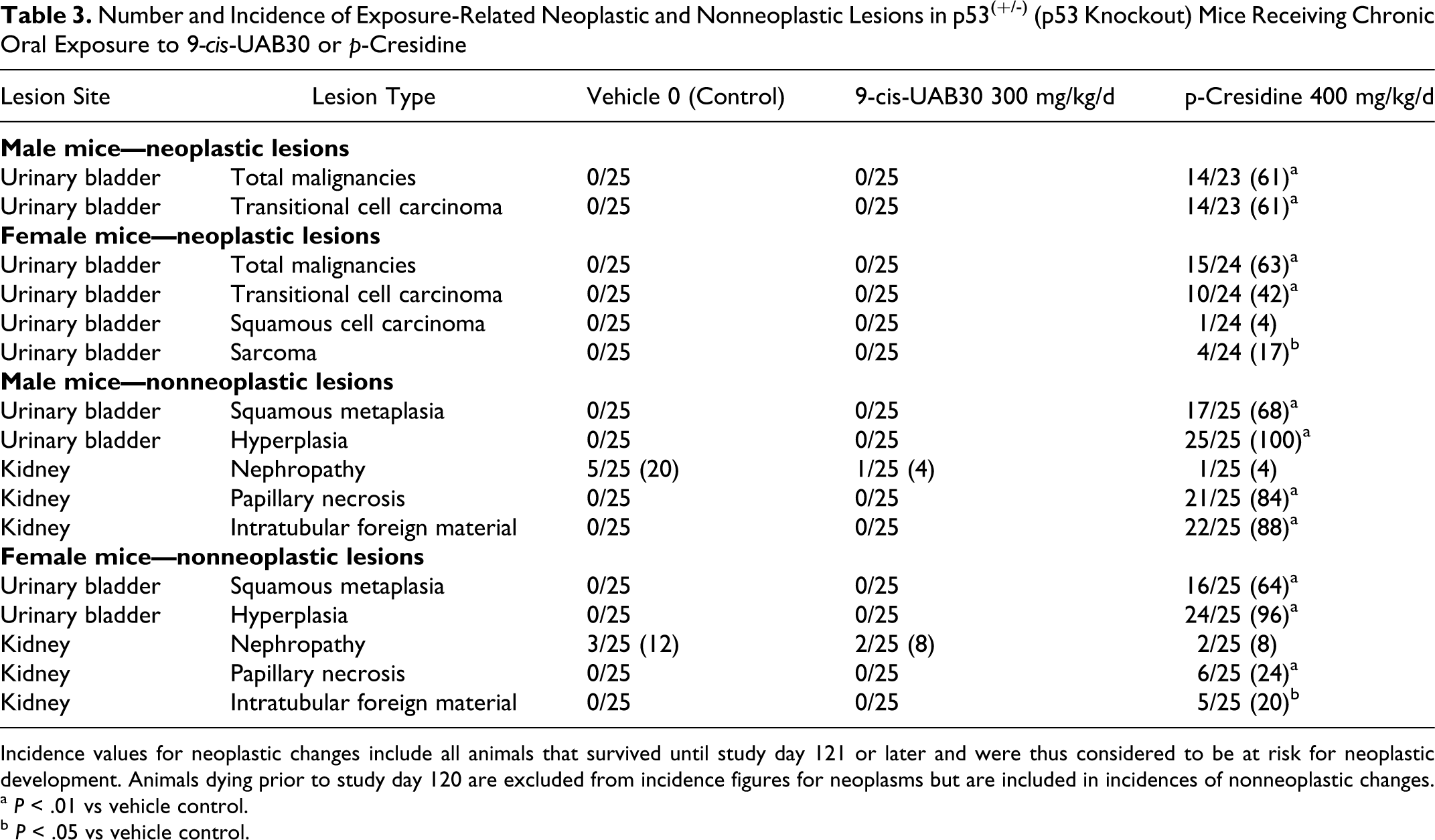
Incidence values for neoplastic changes include all animals that survived until study day 121 or later and were thus considered to be at risk for neoplastic development. Animals dying prior to study day 120 are excluded from incidence figures for neoplasms but are included in incidences of nonneoplastic changes.
a P < .01 vs vehicle control.
b P < .05 vs vehicle control.
By contrast to the lack of oncogenicity of 9-cis-UAB30, histopathologic evaluation of the urinary bladder from mice in the positive control group provided clear evidence of the oncogenicity of p-cresidine in both sexes (Table 3). In male mice, 14 of 23 positive controls (61%) exposed to p-cresidine demonstrated transitional cell carcinomas of the urinary bladder. In addition to these invasive malignancies, urothelial hyperplasia and squamous metaplasia, 2 precancerous lesions, were identified in 100% and 68% of male mice in the positive control group. Similarly, 15 of 24 female mice (63%) in the positive control group demonstrated invasive urinary bladder malignancies; malignant lesions identified in positive control females included transitional cell carcinomas (10/24; 42%), squamous cell carcinoma (1/24; 4%), and sarcomas (4/24; 17%). Urothelial hyperplasia was also a nearly universal finding (24/25) in females in the positive control group, and 16 of 25 (64%) of female mice in the positive control group demonstrated squamous metaplasia. Other nonneoplastic lesions identified in the positive control group included renal papillary necrosis and a low incidence of nephropathy in both sexes.
Pharmacokinetic Characterization of 9-cis-UAB30 in C57BL/6 Mice
Plasma drug concentration-time profiles in male and female mice receiving either 1 or 7 daily gavage doses of 9-cis-UAB30 are presented in Figure 2 (100 mg/kg/d) and Figure 3 (300 mg/kg/d), respectively. Comparisons of pharmacokinetic parameters in mice receiving 9-cis-UAB30 at 100 and 300 mg/kg demonstrated that increases in plasma drug levels were less than proportionate to increases in dose: as indicated in Tables 4 and 5 , both sexes demonstrated a shorter elimination half-life, a smaller AUC, and greater apparent clearance as the dose of 9-cis-UAB30 was increased from 100 mg/kg to 300 mg/kg.
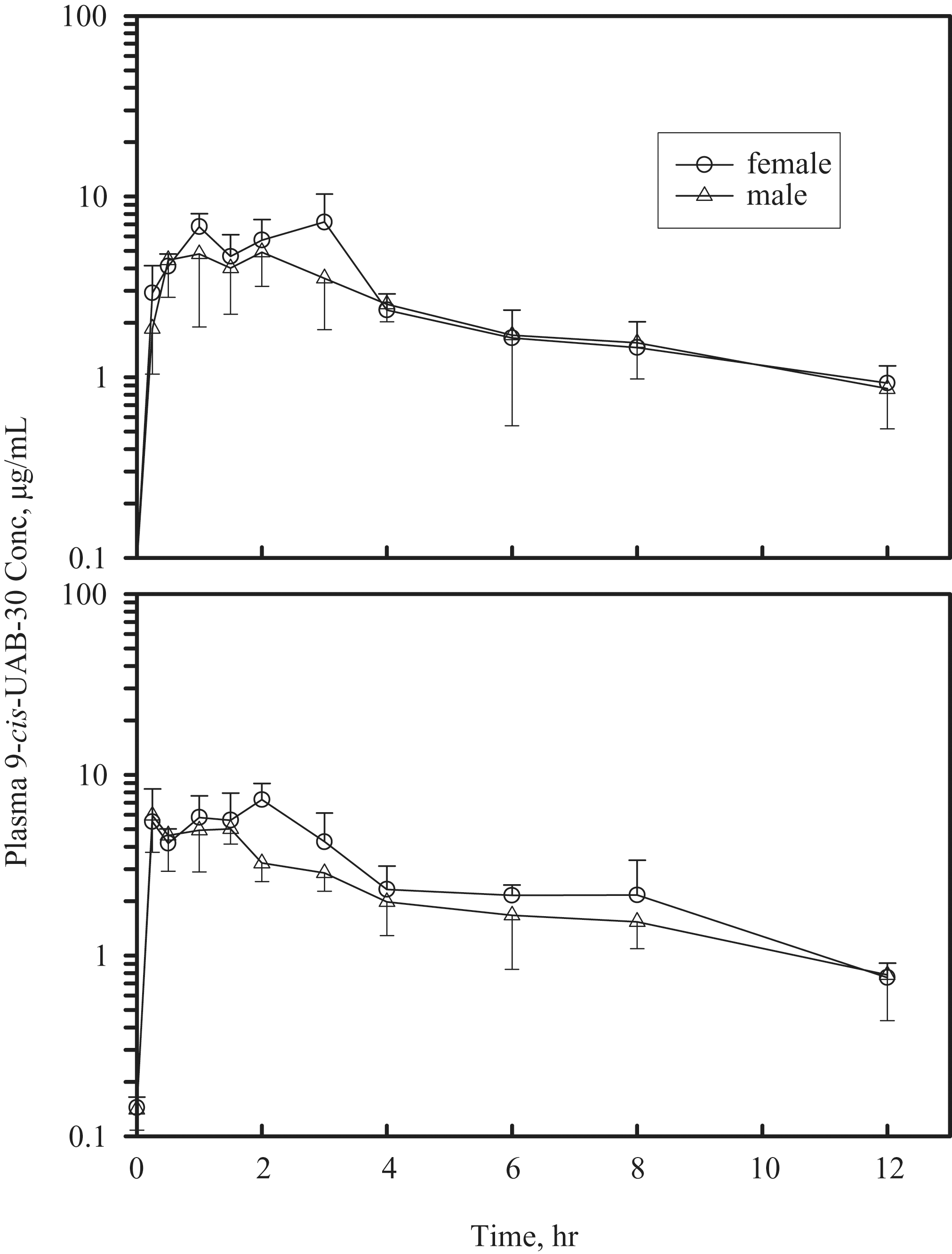
Plasma concentration-time profiles in mice treated with a single dose (top panel) or 7 daily doses (bottom panel) of 9-cis-UAB30 (100 mg/kg).
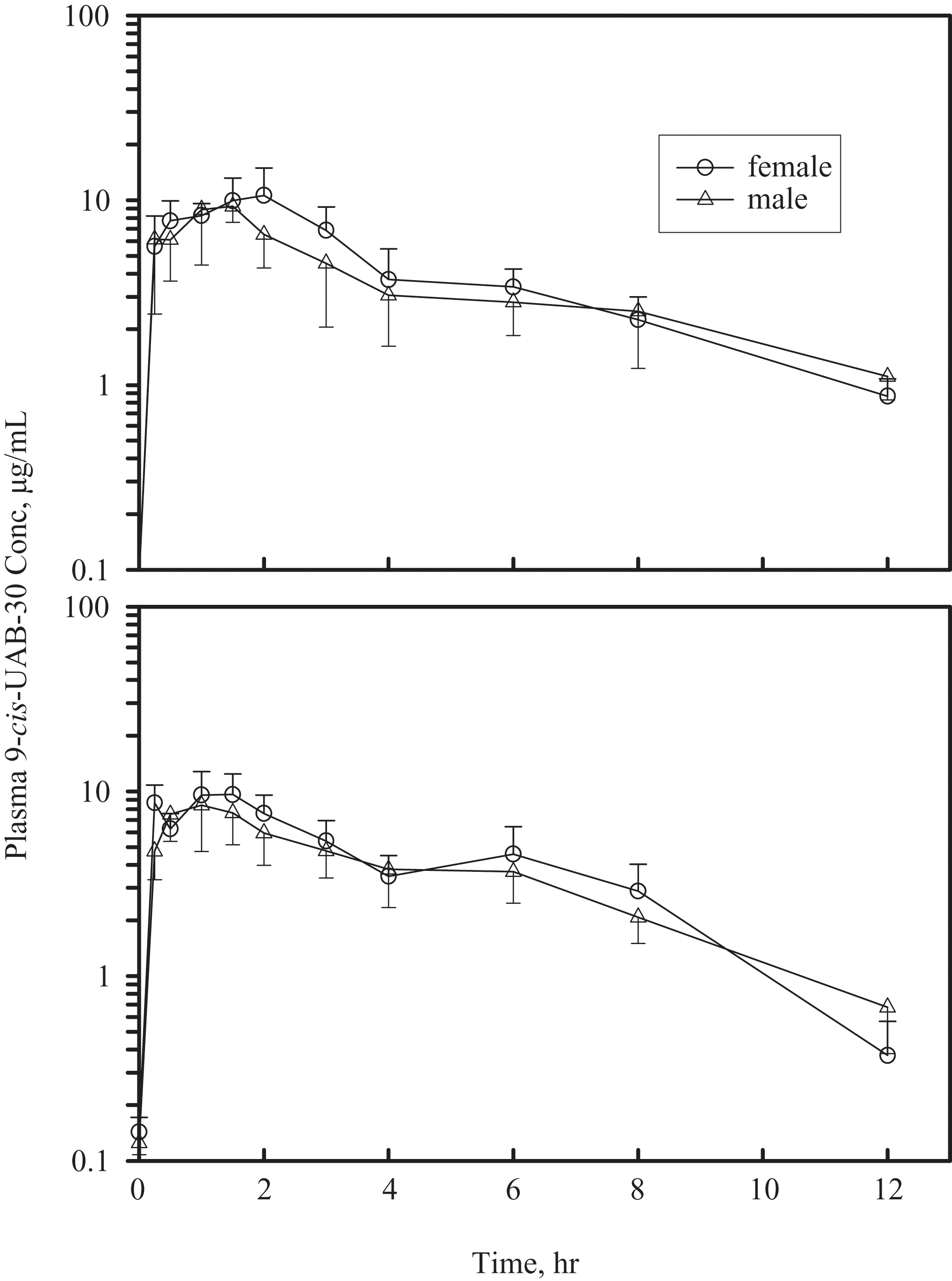
Plasma concentration-time profiles in mice treated with a single dose (top panel) or 7 daily doses (bottom panel) of 9-cis-UAB30 (300 mg/kg).
Pharmacokinetic Parameters of 9-cis-UAB30 in C57BL/6 Mice After 1 Day of Dosing
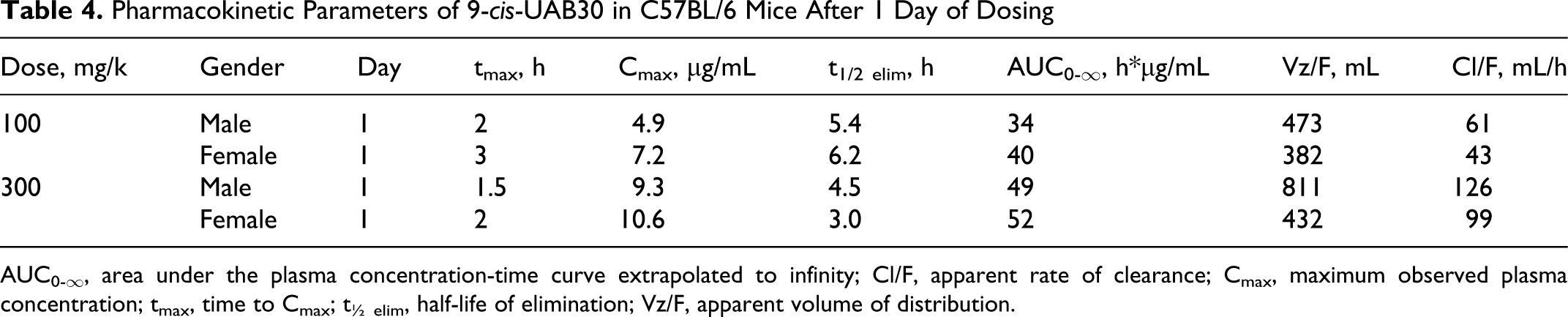
AUC0-∞, area under the plasma concentration-time curve extrapolated to infinity; Cl/F, apparent rate of clearance; Cmax, maximum observed plasma concentration; tmax, time to Cmax; t½ elim, half-life of elimination; Vz/F, apparent volume of distribution.
Pharmacokinetic Parameters of 9-cis-UAB30 in C57BL/6 Mice After 7 Days of Dosing
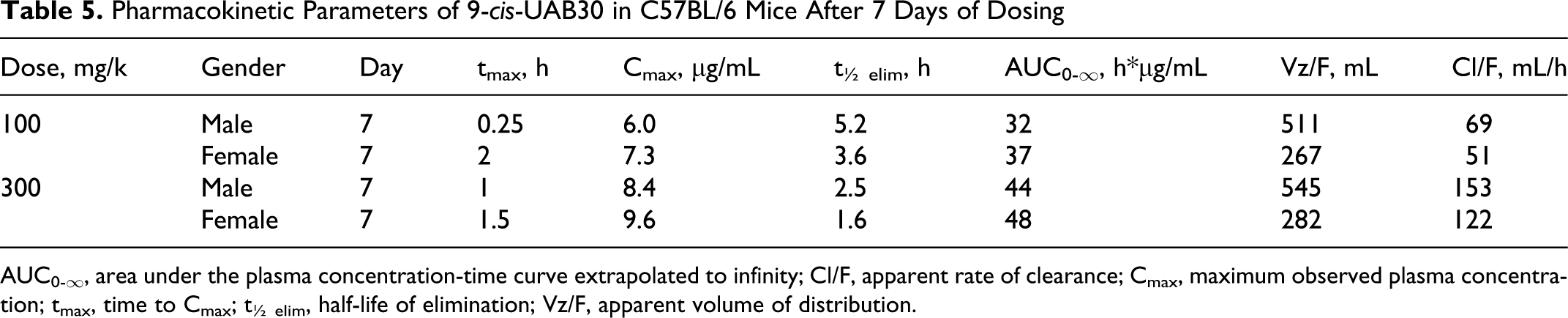
AUC0-∞, area under the plasma concentration-time curve extrapolated to infinity; Cl/F, apparent rate of clearance; Cmax, maximum observed plasma concentration; tmax, time to Cmax; t½ elim, half-life of elimination; Vz/F, apparent volume of distribution.
In general, female mice exhibited a longer elimination half-life, greater AUC, and lower apparent clearance for 9-cis-UAB30 (Table 4) than did males administered the same dose. Values for apparent clearance tended to be greater after 7 daily doses versus those seen following administration of a single oral dose of drug.
In addition to the parent drug, an unidentified metabolite was found eluting just prior to 9-cis-UAB30 in LC chromatograms. Using an analytical approach similar to that used to quantitate 9-cis-UAB30, the concentration of the metabolite was found to be similar to that of the parent (data not shown).
Discussion
Because cancer chemopreventive agents are likely to require chronic administration in order to sustain anticarcinogenic efficacy in humans, an essential element of the preclinical development of any chemopreventive agent is an assessment of its possible oncogenicity. The present results demonstrate that 9-cis-UAB30 is not oncogenic in TSG-p53(+/-) (p53 knockout) mice. Because the 6-month oncogenicity study in the p53 knockout mouse is accepted by the US FDA and other regulatory agencies as a suitable evaluation of agent carcinogenicity, the lack of oncogenicity of 9-cis-UAB30 in the p53 knockout mouse model is generally accepted as providing adequate evidence of lack of carcinogenicity in a murine model.
The lack of oncogenicity of 9-cis-UAB30, and its very limited toxicity, are not a function of poor oral bioavailability. Peak plasma levels of 9-cis-UAB30 ranged from 4.9 to 10.6 μg/mL (17-36 nM) in both sexes of mice receiving oral (gavage) exposure for either 1 or 7 days (Tables 4 and 5). These levels are within the range of 9-cis-UAB30 concentrations demonstrated to inhibit the oncogene-mediated transformation of epithelial cells in vitro 23 and are well above the plasma drug levels measured in rats in a chemoprevention program in which 9-cis-UAB30 was demonstrated to inhibit rat mammary carcinogenesis in vivo. 14
Plasma drug levels (Cmax and AUC) were higher in female mice than in male mice receiving the same dose of 9-cis-UAB30 (Tables 4 and 5); longer elimination half-lives and reduced clearance rates were a consistent finding in female animals when compared with males in the same dose group. Gender differences in hepatic drug metabolizing enzymes are well known, particularly in rodents, and have been proposed to be result from differences in plasma levels of growth hormone and resulting effects on transcription factors. 24
Although both Cmax and AUC were clearly dose-related in both sexes in the present study, increases in plasma drug levels were less than proportional to increases in dose. After both 1 and 7 days of dosing, the apparent half-life of elimination (t1/2 elim) in mice receiving the high dose (300 mg/kg/d) of 9-cis-UAB30 was shorter than in mice receiving the drug at 100 mg/kg/d; the reduced t1/2 elim of 9-cis-UAB30 in the high-dose group, and the associated increased apparent rate of clearance, appear to underlie the lack of proportionality between administered dose and plasma drug levels.
Within a sex and dose group, Cmax and AUC were generally comparable after both 1 and 7 days of dosing. The apparent rate of clearance of 9-cis-UAB30 after 7 days of administration was greater than after a single dose; this may reflect increased metabolism of the parent drug. In studies with human, rat, and dog microsomes, Gorman and colleagues 25 found that 9-cis-UAB30 is metabolized into 6 to 9 phase 1 metabolites (depending on species) and 1 glucuronide conjugate; these investigators also reported that phase 1 biotransformation of 9-cis-UAB30 is mediated primarily by CYP2C isozymes (CYP2C8, CYP2C9, and CYP2C19). Induction of CYP2Cs has been demonstrated in response to RXR heterodimers 26 ; through this mechanism, exposure to an RXR agonist may result in induction of CYP2C, with subsequent effects on metabolism and clearance of the inducing drug.
In this regard, the only clearly drug-related finding in the oncogenicity study was dose-related hepatomegaly seen in both sexes of mice receiving chronic oral exposure to 9-cis-UAB30. The observed increases in liver weight are consistent with possible induction of hepatic metabolizing enzyme systems by 9-cis-UAB30. Hepatomegaly was also seen in a subchronic oral toxicity study of 9-cis-UAB30 in rats (National Cancer Institute, unpublished data).
The results of the present studies provide no evidence that 9-cis-UAB30 is oncogenic in mice lacking 1 allele of the p53 tumor suppressor gene. The drug is orally bioavailable and was very well tolerated at all dose levels used in the study. Toxicity associated with chronic exposure to 9-cis-UAB30 was limited to dose-related hepatomegaly in both sexes; the observed increases in liver weights may be secondary to hepatic enzyme induction. Importantly, no evidence of vitamin-A-like toxicities such as weight loss, bone fractures, or epidermal changes were identified.
In consideration of the lack of toxicity of 9-cis-UAB30, its demonstrated chemopreventive activity in several experimental models for breast cancer, and the chemopreventive activity of other RXR ligands (such as bexarotene) in animal models for cancer in several sites, 9-cis-UAB30 merits consideration for further evaluation in human cancer chemoprevention trials.
Footnotes
Acknowledgements
We thank Leigh Ann Senoussi for assistance in preparing the manuscript.