
Abstract
The protection against 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD; 5 µg/kg body weight) toxicity by the antioxidant 4b,5,9b,10-tetrahydroindeno[1,2-b]indole (THII) was examined in female C57BL/6J mice. TCDD produced increases in the levels of hepatic lipid-derived aldehydes, rates of mitochondrial production of hydrogen peroxide and superoxide, and the oxidation state of cytosolic GSH. In contrast, mitochondrial GSH increased in reduction state, correlating with an increase in mitochondrial membrane potential. Systemically, TCDD lowered body weight gain, percentage body fat, and hepatic ATP levels, parameters prevented by concomitant administration of 100 µM THII in drinking water. However, TCDD-induced increases in mitochondrial respiration and decreased mitochondrial membrane fluidity were not prevented by THII. These results suggest that TCDD-mediated oxidative stress was not responsible for changes in mitochondrial respiration or membrane fluidity. Furthermore, although TCDD produced a large increase in mitochondrial oxygen consumption, this was not associated with the poor gain in weight produced by TCDD.
The ubiquitous environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is the prototypic polynuclear aryl hydrocarbon that can cause cancer, cardiovascular disease, birth defects, and metabolic diseases such as wasting syndrome and diabetes. 1 TCDD and TCDD-like compounds are hydrophobic and metabolically recalcitrant, resulting in persistent activation of aromatic hydrocarbon receptor (AHR)–mediated pathways. The AHR, a cytosolic ligand-activated transcription factor, modulates the expression of hundreds of diverse genes 2-5 and is responsible for most of the adverse effects of TCDD. 6-8 In addition to transcriptional changes, TCDD may also act through nontranscriptional pathways, although these still appear to involve the AHR. 9
One important pathway for the biological activity of TCDD, both genomic and nongenomic in nature, is a robust oxidative stress response. 10 TCDD increases the cellular production of reactive oxygen in both the mitochondrial 11,12 and cytosolic 13,14 compartments. TCDD-mediated oxidative stress 8,10 is characterized by increases in such biomarkers as liver heme oxygenase and metallothionein, 15 increases in lipid peroxidation and DNA damage, 11 and oxidative perturbations in glutathione homeostasis. 11 Most cells have an elaborate antioxidant defense system that includes chemical antioxidants and inducible enzyme scavengers to reduce toxic radicals and electrophiles. However, chronic excessive production of reactive oxygen, such as following TCDD exposure, may produce cellular damage. In such cases, dietary antioxidants may reduce toxicity. This study examines the ability of 4b,5,9b,10-tetrahydroindeno[1,2-b]indole (THII), a potent hydrophobic antioxidant, to reduce TCDD-induced biochemical toxicity. 16 THII and its congeners protect animals and cells in culture against toxicity from a variety of chemicals and appear to act directly as chemical antioxidants, with the parent compound THII being about 10-fold more efficacious than vitamin E. 17-20 THII alters neither enzyme activities nor endogenous antioxidant levels associated with drug metabolism or oxidative stress, and it does not activate the AHR. 18 Therefore, THII may be used to separate those effects evoked by TCDD that are associated with oxidative stress from those events not so associated. In a 40-week study in mice, THII was effective in reducing benzo[a]pyrene-induced skin tumorigenesis when delivered via drinking water, suggesting that via this route of delivery, THII has a favorable pharmacokinetic profile to distribute in a pharmacologically active form throughout the body. 19
Materials and Methods
Chemicals
TCDD was purchased from Accustandard (New Haven, CT). All other chemicals and reagents were obtained from Sigma-Aldrich Chemical Company (St Louis, MO) as the highest available grades.
Animals and Treatment
Experiments involving mice were performed according to the National Institutes of Health standards for care and use of experimental animals and the University of Cincinnati Institutional Animal Care and Use Committee. Animals were group housed, maintained on a 12-hour light-dark cycle, and had access to standard rodent chow and water ad libitum. Female C57BL/6J mice (10 weeks of age) were purchased from The Jackson Laboratories (Bar Harbor, ME).
Mice were administered a single dose of TCDD (5 µg/kg body weight) in corn oil by intraperitoneal injection; vehicle controls were given equivalent volumes of corn oil. Beginning with TCDD treatment and continuing until sacrifice, mice were administered 100 µM THII in drinking water ad libitum. The consumption of water remained constant at ~3.5 mL/d, such that the dosage was about 3.5 mg THII/kg body weight/d.
At 28 days following treatment, the mice were killed by carbon dioxide asphyxiation. Immediately, 0.1 g of liver from each animal was frozen in liquid nitrogen for tissue ATP determination. The rest of the liver was excised and washed in ice-cold 0.9% NaCl. A 10% whole homogenate was prepared in 250 mM sucrose, 1 mM EDTA, 1 mM EGTA, 0.1% defatted and recrystallized bovine serum albumin, 10 mM HEPES, pH 7.2. A mitochondrial fraction was prepared as described previously, 21 and suspended in a potassium chloride respiratory buffer (KCl-RB), consisting of 140 mM KCl, 0.1 mM EDTA, 2.5 mM KH2PO4, 2.5 mM MgCl2, and 0.1% bovine serum albumin, in 5 mM HEPES (pH 7.4).
Assays for Products of Lipid Peroxidation and Reactive Oxygen
The hepatic content of malondialdehyde and 4-hydroxyalkenals, products of lipid peroxidation, were estimated using the colorimetric probe 1-methyl-2-phenylindole (BIOXYTECH LPO-586; OXIS Health Products, Inc, Portland, OR), following the procedure described by the manufacturer. H2O2 was monitored in freshly prepared mitochondria as 5 µM luminol (5-amino-2,3-dihydro-1,4-phthalazinedione) chemiluminescence that was inhibited by 500 U/mL catalase. 14,21 The reaction mixture consisted of 5 µM luminol, 2.5 U/mL horseradish peroxidase, 6 mM sodium succinate, 50 µg mitochondrial protein and KCl-RB, in a final volume of 1.0 mL maintained at 37°C. Superoxide production was monitored as 20 µM lucigenin (bis-N-methylacridinium) chemiluminescence that was inhibited by 5 µM of the superoxide dismutase mimetic Mn(III)tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride. 21
GSH, GSSG, and Redox Potential
GSH and GSSG were determined fluorometrically using the o-phthalaldehyde procedure. 22 To calculate the molar mitochondrial concentrations of GSH and GSSG, we estimated mitochondrial volume at 1 µL/mg protein. 23 For the GSSG/2GSH half-reaction, GSSG + 2H+ → 2GSH, the reduction potential at pH 7 (ΔE′) is the standard reduction potential (ΔE0′) adjusted for the mass action ratio of the reactants and products. 24 The ΔE value was calculated as ΔE = {ΔE0′ + (RT/nF) × (actual pH – standard pH)} – {RT/nF ln ([GSH]2/[GSSG])}, where n is the number of electrons transferred, R is the universal gas constant (8.31 J K–1 mol–1), T is the temperature in Kelvin, and F is the Faraday constant (9.65 Coulombs × 104 mol–1).
At 37°C, ΔE = {−240 mV + (−61.5 mV/2e−) × (actual pH − 7.0)} − {(61.5 mV/2e−) × log ([GSH]2/[GSSG])]}. For calculating reduction potential, the cytosol was assumed to be pH = 7.2, and the mitochondria pH 7.8.
Mitochondrial Respiration, Membrane Potential, and Membrane Fluidity
Mitochondrial oxygen consumption was measured polarographically with a Clark-type oxygen electrode (Hansatech Instruments, Norfolk, UK). 21 The rate of state 4 respiration (ADP limited) was determined in the presence of 0.5 mL KCl-RB, 50 µg mitochondrial protein, and 6 mM succinate at 37°C with stirring. Following addition of 0.4 mM ADP, the rate of state 3 respiration was measured. The respiratory control ratio was calculated as the ratio of state 3 to state 4 respiration.
The mitochondrial inner-membrane potential was quantified using the cationic lipophilic dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide). 25 To 10 µg mitochondrial protein in 1.0 mL KCl-RB was added 0.05 nmol JC-1, and fluorescence ratios were determined at 37°C. Membrane potential was estimated as the ratio of red (Ex = 488 nm; Em = 595 minus 620 nm) to green fluorescence (Ex = 488 nm; Em = 535 minus 510 nm). 11
Fluorescence probes were used to probe mitochondrial membrane fluidity at the interior (1,6-diphenyl-1,3,5-hexatriene; DPH) and surface (1-(4-trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene p-toluenesulfonate; TMA-DPH), as described. 12 Briefly, 1 µM final probe concentration was added to 1 mg mitochondrial protein/mL KCl-RB, and fluorescence intensity measurements were determined at 25°C with excitation (357 nm) and emission (428 nm) polarization filters in the parallel and perpendicular orientations. Fluorescence polarization anisotropy was calculated as an r value, 26 where lower values correspond to greater membrane fluidity.
Body Weight and Composition
Body weights and food and water consumption were measured twice weekly. Body composition was assessed in live, unanesthetized mice by nuclear magnetic resonance (EchoMRI; EchoMedical Systems, Houston TX, www.echomri.com), providing a valid estimate of total fat tissue. 27
Other Assays
ATP levels in liver were quantified using luciferin-luciferase bioluminescence as described. 21 Protein was measured by the bicinchoninic acid method (Pierce Chemical Co, Rockford, IL), according to details provided by the manufacturer.
Statistics
Statistical significance of the differences between group sample mean values was determined by 1-way analysis of variance, followed by the Student-Newman-Keuls test for pairwise comparison of means. Statistics were performed using SigmaStat Statistical Analysis software (SPSS Inc, Chicago, IL).
Biohazard Precaution
TCDD is highly toxic and a possible human carcinogen. All personnel were instructed as to safe handling procedures. Lab coats, gloves, and masks were worn at all times, and contaminated materials were collected separately for disposal by the Environmental Health & Safety Office of the University of Cincinnati. TCDD-treated mice were housed separately, and their carcasses were treated as contaminated biological materials.
Results
THII Protects Against TCDD-Mediated Oxidative Stress
A single exposure to 5 µg TCDD/kg body weight resulted on day 28 in a 4- to 5-fold increase in the hepatic content of products of lipid peroxidation, malondialdehyde, and 4-hydroxyalkenals (Figure 1 ). THII treatment reduced these elevated levels of aldehydes by 90% to 95%. Correlating with this was the finding that the THII abolished the TCDD-induced increases in the succinate-dependent mitochondrial production of reactive oxygen species, hydrogen peroxide, and superoxide.
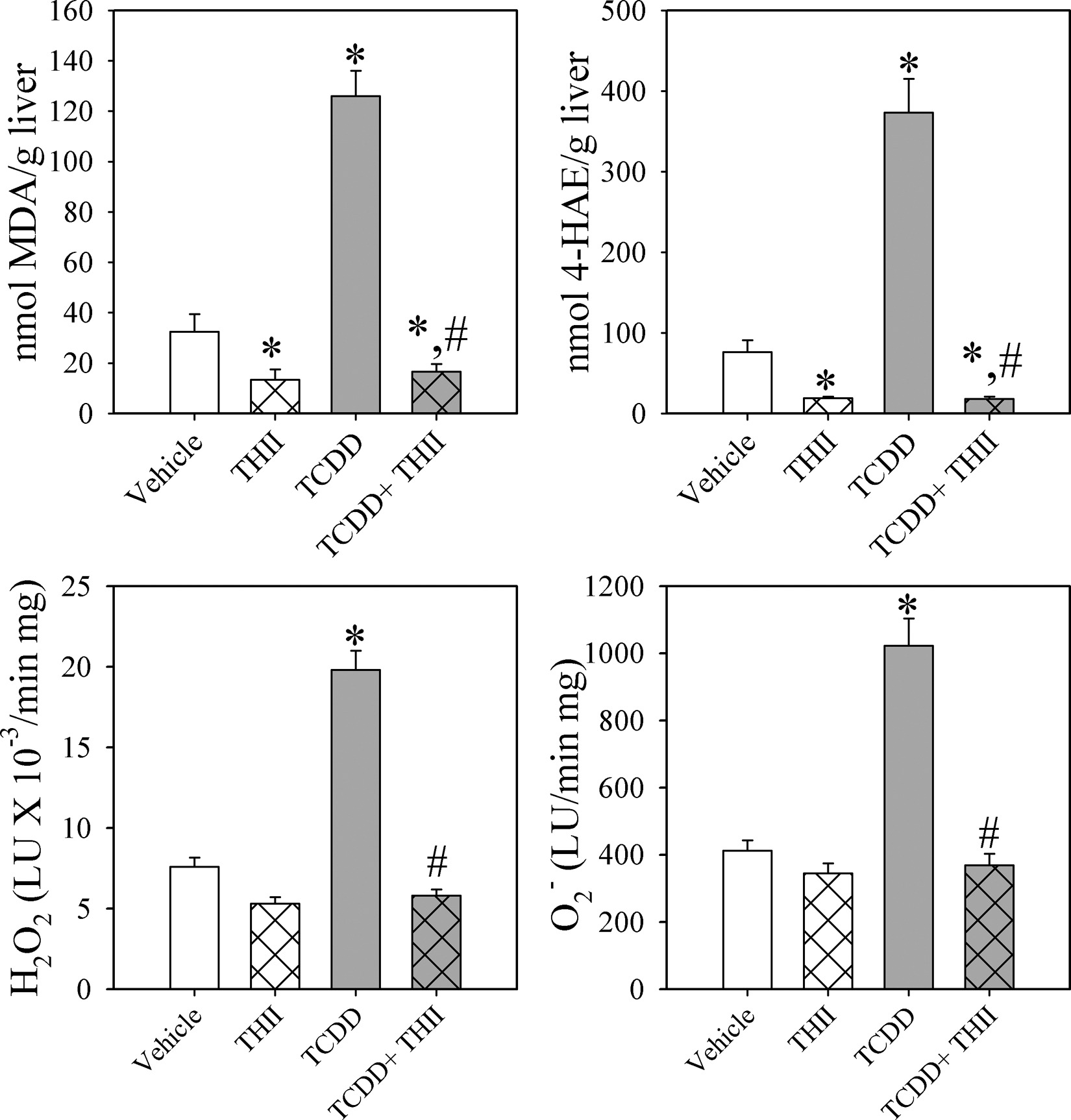
4b,5,9b,10-tetrahydroindeno[1,2-b]indole (THII) inhibits 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)–induced oxidative stress. Livers were harvested following 28 days of treatment with corn oil (vehicle) or a single dose of TCDD (5 µg/kg body weight), with or without continuous THII in drinking water. Tissue was homogenized and content of malondialdehyde (MDA; top left) and 4-hydroxyalkenals (4-HAE; top right) determined. Mitochondria were prepared, and succinate-dependent H2O2 and superoxide production were estimated using the luminescence probes luminol (bottom left) and lucigenin (bottom right), respectively. Reactive oxygen data are presented as luminescence units (LU)/min/mg protein. All data are shown as mean values ± SE (N = 4). *Statistically different (P < .05) from the mean values for vehicle-treated mice. #Mean values for mice treated with TCDD + THII statistically different (P < .05) from the mean values for mice treated with TCDD only.
THII Protects Against the Changes in TCDD-Induced Cytosolic and Mitochondrial Glutathione Redox States
Concomitant with the TCDD-induced oxidative stress response were changes in both cytosolic and mitochondrial redox state of glutathione, as previously reported (Figure 2 11 ). In the cytosol, TCDD increased the concentration of GSSG to a greater extent than GSH, resulting in a 17 mV less negative redox potential (more oxidized state) for the GSSG/2GSH redox couple. In mitochondria, however, TCDD caused GSH levels to nearly double, while GSSG increased only slightly, resulting in a 29 mV more negative redox potential (more reduced state) for the GSSG/2GSH redox couple. The more reduced glutathione redox state in mitochondria from TCDD-treated mice is due to the ability of mitochondria to strongly accumulate GSH and export excess GSSG. 11 All of the TCDD-mediated changes in GSH and GSSG in both cytosolic and mitochondrial compartments are prevented by administration of THII (Figure 2).
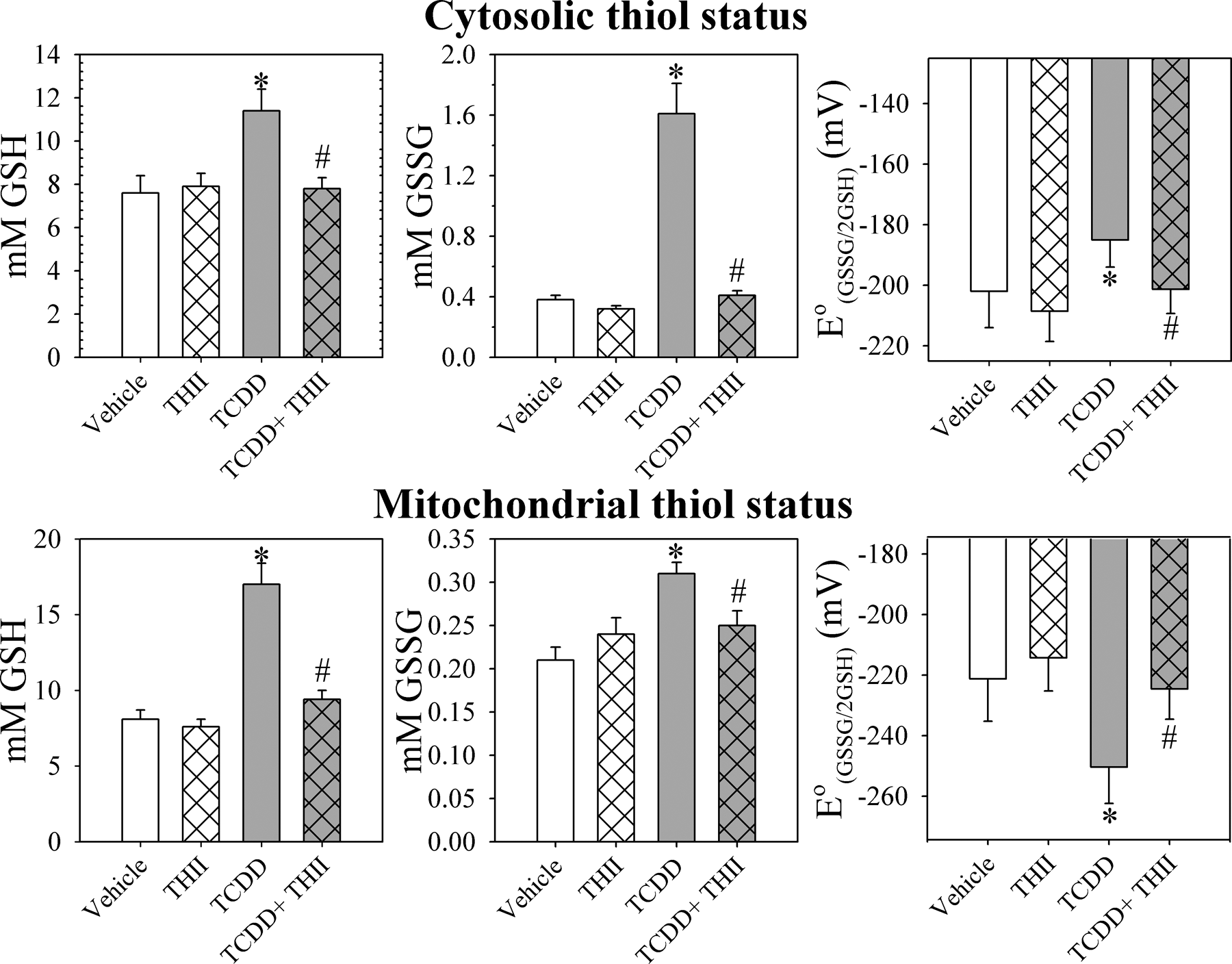
4b,5,9b,10-tetrahydroindeno[1,2-b]indole (THII) prevents changes in glutathione redox status in cytosol (top) and mitochondria (bottom). Mice were treated and cell fractions prepared as described in Figure 1 and the “Materials and Methods” section. Glutathione-related parameters are shown for cytosol (top) and for mitochondria (bottom). Data shown are GSH (left), GSSG (center) concentrations, and the GSSG/2GSH half-cell redox potential (right). Data are presented as the mean values ± SE (N = 4). *Statistically different (P < .05) from the mean values for vehicle-treated mice. #Mean values for mice treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) + THII statistically different (P < .05) from the mean values for mice treated with TCDD only.
THII Protects Against TCDD-Mediated Changes in Membrane Potential and Fluidity but not Parameters of Respiration in Isolated Mitochondria
We next examined various parameters of mitochondrial function that could be related to oxidative stress and glutathione redox state. Confirming our prior findings (Figure 3 , top), in liver mitochondria from TCDD-treated mice, we found an increase in mitochondrial membrane potential indicated by the succinate-dependent increase in the JC-1 fluorescence ratio 11 and increases in membrane fluidity (decreases in r value) using a lipid bilayer probe (DPH) and a surface probe (TMA-DPH). 12 We also confirmed the increases in mitochondrial respiration in phosphorylating state 3 and ADP-limited state 4, with no change in the respiratory control ratio (Figure 3, bottom 21 ). THII treatment prevented TCDD-induced mitochondrial membrane hyperpolarization; however, THII affected neither the TCDD-induced increases in membrane fluidity nor the increases in states 3 or 4 respiration.
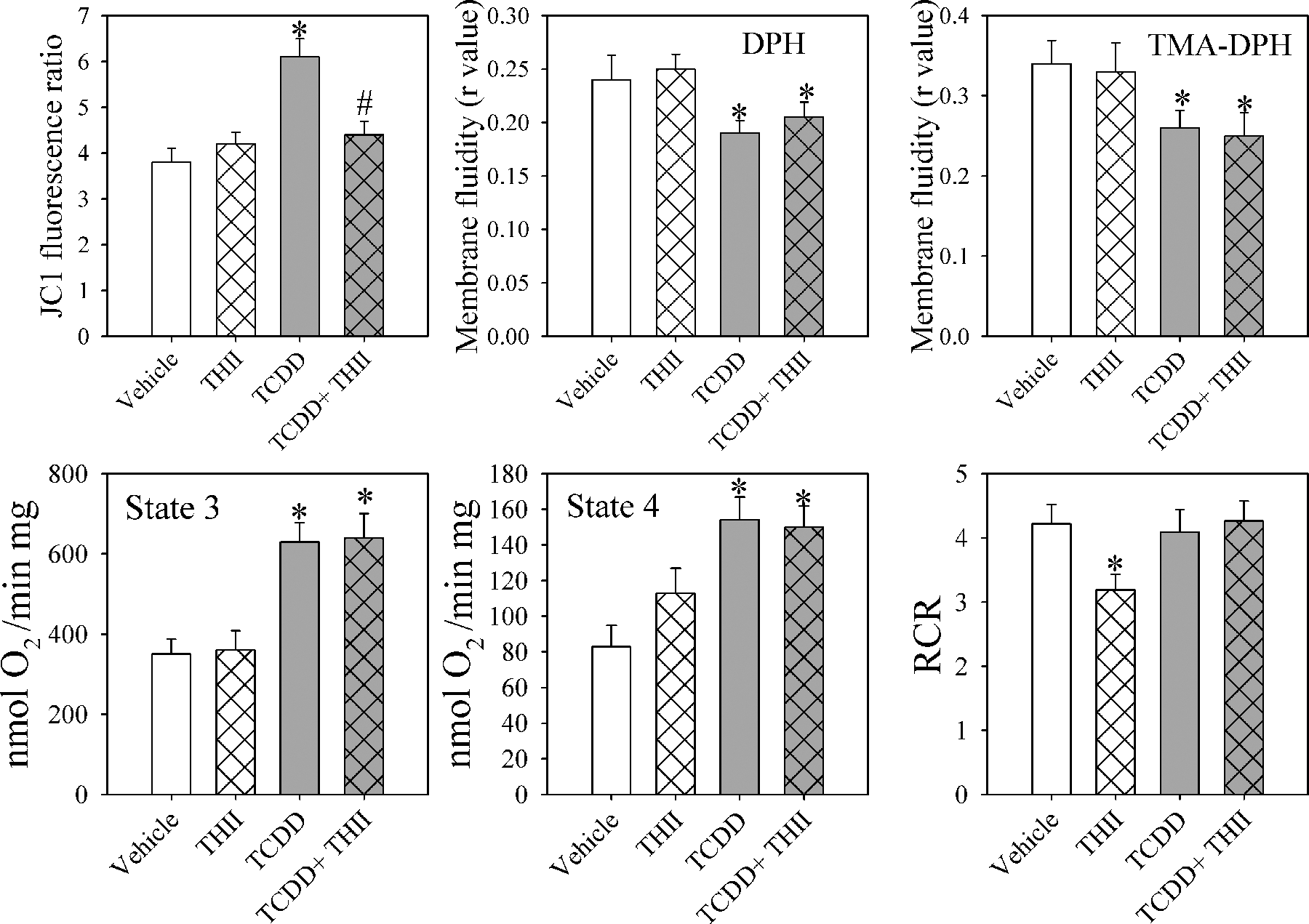
In mitochondria, 4b,5,9b,10-tetrahydroindeno[1,2-b]indole (THII) prevents 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)–mediated changes in membrane potential but not membrane fluidity or parameters of respiration. Mice were treated and mitochondria were prepared as described in Figure 1 and the “Materials and Methods” section. Data shown are membrane potential determined as JC-1 fluorescence ratios (top left) and membrane fluidity (polarization anisotropy, calculated as an r value) using the deep membrane probe diphenylhexatriene (DPH; top center) or the surface probe trimethylammonium diphenylhexatriene (TMA-DPH; top right). Rates for state 3 respiration (with ADP, bottom left), state 4 respiration (ADP limited, bottom center), and respiratory control ratio (state 3/state 4, bottom right) are shown. For standardizing JC-1 fluorescence ratios to membrane potential, known concentration gradients of potassium were applied across the membrane, in the presence of 2 µg valinomycin/mL. Using the Nernst equation, a calculated transmembrane potassium diffusion potential of about –50 mV could be attained. 11 If we assume linearity, a fluorescence ratio of 2 is equivalent to a membrane potential of about 100 mV. Data are presented as the mean values ± SE (N = 4).*Statistically different (P < .05) from the mean values for vehicle-treated mice. #Mean values for mice treated with TCDD + THII statistically different (P < .05) from the mean values for mice treated with TCDD only.
THII Prevents Changes in Energy Homeostasis Associated With TCDD Exposure
TCDD is hepatotoxic in part because of severe depletion of tissue ATP levels, and in addition, it causes loss of body weight and adipose tissue mass (Figure 4 ). These parameters of liver and system toxicity by TCDD are prevented by THII.
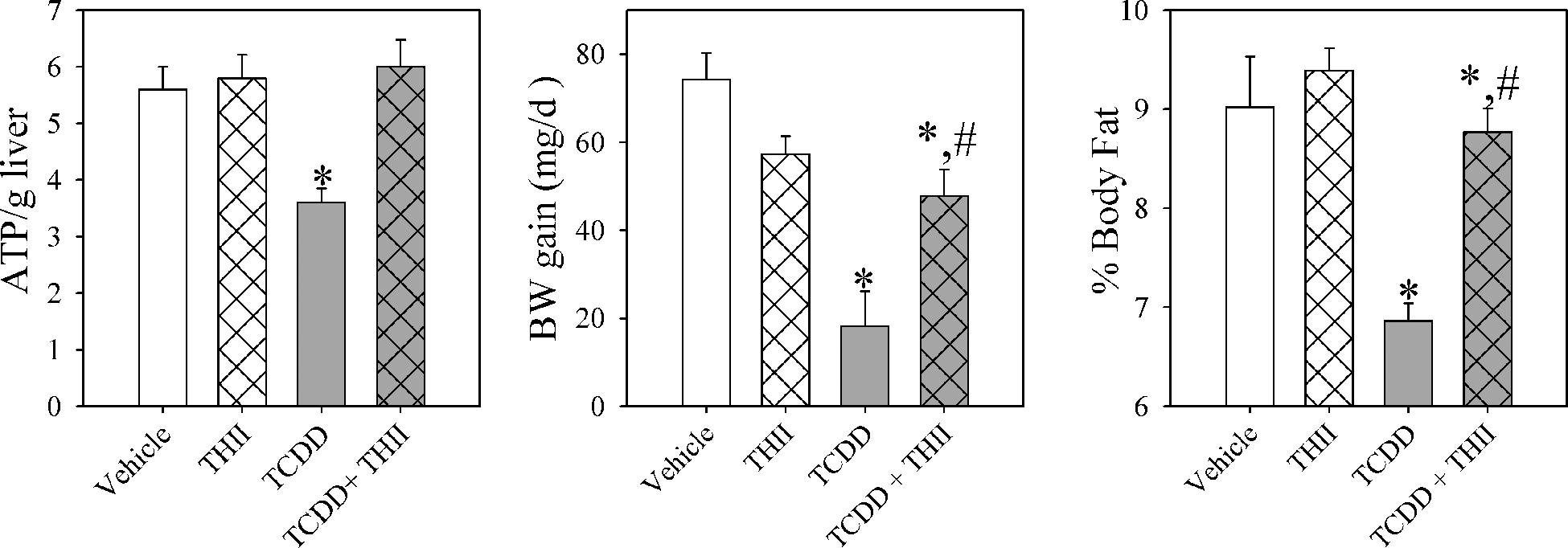
4b,5,9b,10-tetrahydroindeno[1,2-b]indole (THII) prevents 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)–mediated depletion of hepatic ATP and decreases body fat content and rate of body weight (BW_ gain. Mice were treated and liver homogenates prepared as described in Figure 1 and the “Materials and Methods” section. Body fat content was estimated using nuclear magnetic resonance with restrained mice. Liver ATP levels (left), rate of BW gain (center), and percentage body fat (g fat/g BW × 100; right) are shown. Data are presented as the mean values ± SE (N = 4). *Statistically different (P < .05) from the mean values for vehicle-treated mice. #Mean values for mice treated with TCDD + THII statistically different (P < .05) from the mean values for mice treated with TCDD only.
Discussion
TCDD exposure elicits a variety of serious health issues, including cancer, cardiovascular disease, birth defects, and diabetes. 1 The etiology of these diverse diseases is complex, involving both genomic 5 and nongenomic 9 pathways. In either case, the ligand-activated AHR is involved, and the progression of the various disease states involves oxidative stress. 8 However, at the biochemical level, it has not been clear which effects of TCDD are associated with oxidative stress and how these biochemical events are associated with the poor rate of weight gain, or actual loss in weight, a hallmark of TCDD exposure. Although intervention studies regarding TCDD toxicity would help to clarify these associations, there have been few studies in this regard. A recent study showed that resveratrol, a phenolic component of red grapes and peanuts, was able to prevent TCDD-induced hepatic steatosis, oxidative stress, and loss in body weight in C57BL/6J mice. 28
We had previously hypothesized that an increase in the reduction state of mitochondrial glutathione would result in inner mitochondrial membrane hyperpolarization and increased reactive oxygen production by redox cycling in complex 3. 11,12 Furthermore, futile cycling of ATP synthesis and hydrolysis in complex 5 (ATP synthase), facilitated by increased inner membrane fluidity, would increase mitochondrial respiration. However, the pattern of inhibition of TCDD-mediated mitochondrial parameters by THII (Figure 3) suggests a different pattern of events. While THII prevented the TCDD-mediated increase in inner membrane hyperpolarization, it did not prevent the increases in state 3 and state 4 oxygen consumption. Moreover, THII abolished succinate-dependent reactive oxygen production but did not inhibit the TCDD-mediated increase in membrane fluidity. This finding suggests that the TCDD-induced increase in membrane fluidity is not mediated by membrane fatty acid oxidation. A more likely explanation is that TCDD, being both biologically persistent and extremely hydrophobic, modifies mitochondrial membrane fluidity by direct interaction with both the lipophilic interior (probed with DPH) and the less lipophilic surface (probed with TMA-DPH). These considerations are supported by the report that TCDD, when added directly to mitochondria, increases state 3 and state 4 respiration. 12
It is difficult to ascribe a specific chemoprotective mechanism of action to inhibitors such as antioxidants, which can frequently alter a number of biochemical and genetic pathways. For example, in Jurkat T cells, THII inhibits apoptosis induced by chemical agents and ultraviolet radiation. 20 The inhibition is downstream of mitochondrial membrane depolarization and involves the inhibition of caspase 3. This effect appeared to be due to the ability of THII both to inhibit mitochondria-derived reactive oxygen and to generally increase the redox state of the cell. In the present study, THII most likely prevents TCDD toxicity by more than one mechanism, just as TCDD toxicity itself involves multiple mechanisms. However, in each study, the primary mechanism of action for protection by THII appears to be the quenching of radicals and reactive oxygen.
The role of GSH in TCDD toxicity remains unclear. Cells maintain at least 2 distinct regulated pools of GSH: cytosolic and mitochondrial. 29 It is certainly important to maintain the physiological levels of mitochondrial GSH. Depletion of mitochondrial GSH by electrophilic chemicals, by inhibiting GSH synthesis, or by inhibiting mitochondrial GSH transport leads to hepatic steatosis and cirrhosis, and respiratory toxicity, with a decrease in ATP production, as well as increased production of reactive oxygen and an augmented oxidative stress response. 30-33 Cells, however, are able to maintain mitochondrial GSH levels, even when cytosolic GSH is substantially depleted, such as occurs in mice when the gene for the catalytic subunit for glutamate cysteine ligase is ablated. 34 It is not surprising, therefore, that while TCDD increases the oxidation state of the GSSG/2GSH redox couple in the cytosol, it increases the reduction state in mitochondria. Although it is unusual for chemical agents to increase the glutathione redox status in mitochondria, TCDD is not the only agent that does so. Schisandrin B, a dibenzocyclooctene component from the fruit of the Chinese herb, Schisandra chinensis, was found to increase the liver mitochondrial GSH/GSSG ratio by almost 3-fold, while protecting completely against hepatotoxicity due to oxidative stress caused by carbon tetrachloride. 35 TCDD may evoke this same protective response, in that the increase in mitochondrial GSH levels may convey protection, despite the generation of a moderate oxidative stress response. 11 The ability of THII to prevent TCDD-induced hyperpolarization of the mitochondrial inner membrane is likely to be secondary to THII’s preventing thiol redox changes to both the cytosolic and mitochondrial compartments.
The biochemical pathways delineated in this article are outlined in Figure 5 . Although poor weight gain appears to be associated with the TCDD-induced oxidative stress response, and not changes in mitochondrial membrane fluidity or respiration, the biochemical pathways leading to loss in body fat and body weight remain unclear.
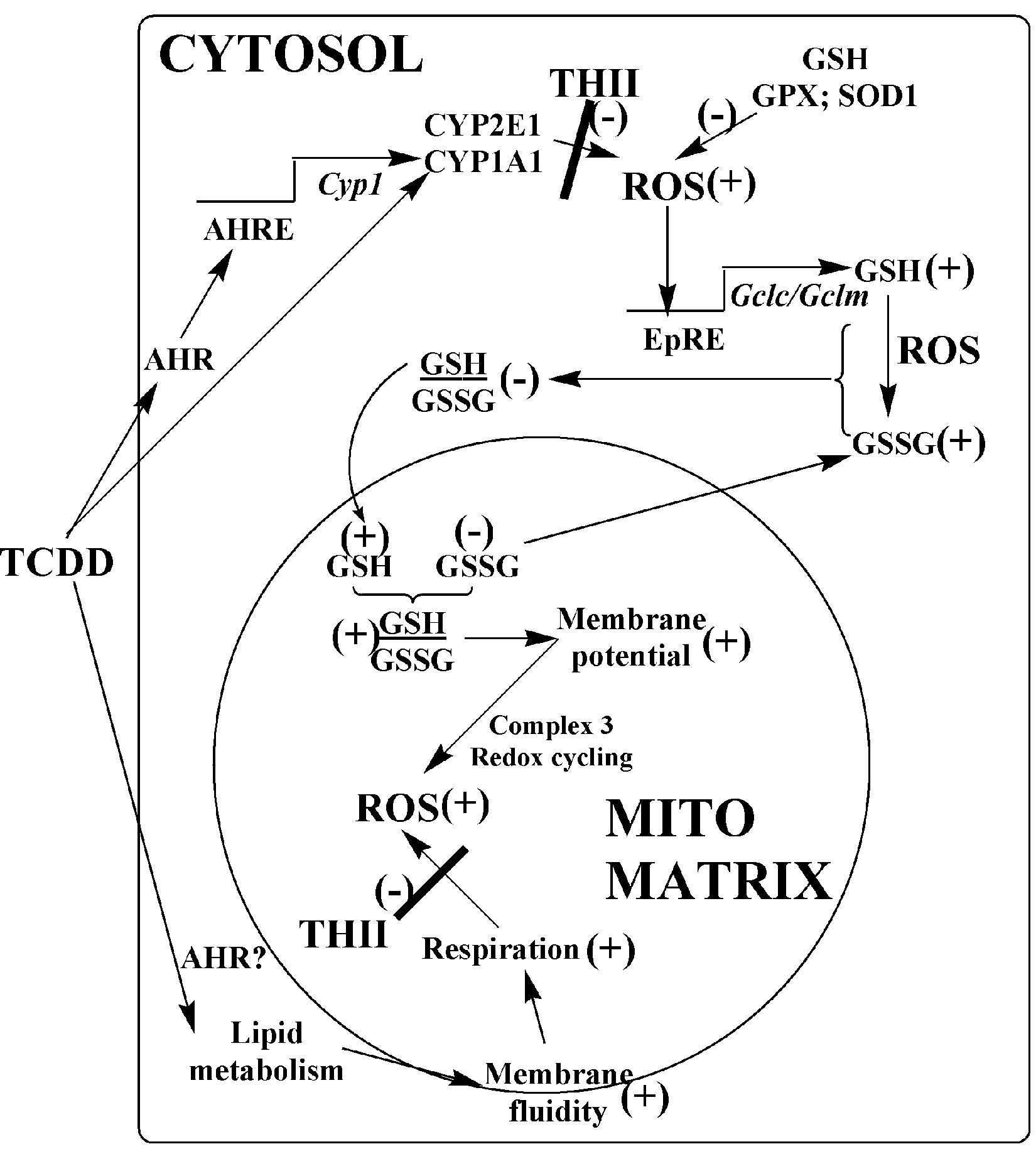
Proposed pathways for prevention of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) toxicity by 4b,5,9b,10-tetrahydroindeno[1,2-b]indole (THII). The toxicity of low dosages of TCDD occurs through the aromatic hydrocarbon receptor (AHR) and involves both transcriptional mechanisms (upper pathways of figure) and nontranscriptional mechanisms (lower pathways). There may be nontranscriptional pathways that do not involve the AHR. Regardless, both genomic and nongenomic pathways may elicit an oxidative stress response. THII blocks the production of reactive oxygen species (ROS) in both the cytosol and mitochondria. However, in mitochondria, TCDD-mediated changes in membrane potential, respiratory rates, and energy coupling are not dependent on ROS. AHRE indicates AHR response element; CYP, cytochrome P450; DPH, (1,6-diphenyl-1,3,5-hexatriene); Gclc/Gclm, glutamate cysteine ligase catalytic and modifier subunits; GPX, glutathione peroxidase; SOD, superoxide dismutase.
Conclusions
In summary, we have shown that THII completely inhibits the oxidative stress response caused by TCDD exposure. Associated with oxidative stress were glutathione redox states in the cytosolic and mitochondrial compartments, mitochondrial membrane potential, hepatic ATP levels, and poor weight gain and body fat percentage. Unassociated with TCDD-induced oxidative stress were mitochondrial respiration and membrane fluidity. There were 2 unexpected findings. First, reactive oxygen–initiated lipid peroxidation was neither responsible for changes in mitochondrial membrane fluidity nor mitochondrial respiration. Second, although TCDD produced a large increase in mitochondrial oxygen consumption, this did not contribute to the poor rate of weight gain observed in TCDD-treated mice.