
Abstract
Using time-resolved fluorescence resonance energy transfer (FRET), we have developed and validated the first high-throughput screening (HTS) method to discover compounds that modulate an intracellular Ca2+ channel, the ryanodine receptor (RyR), for therapeutic applications. Intracellular Ca2+ regulation is critical for striated muscle function, and RyR is a central player. At resting [Ca2+], an increased propensity of channel opening due to RyR dysregulation is associated with severe cardiac and skeletal myopathies, diabetes, and neurological disorders. This leaky state of the RyR is an attractive target for pharmacological agents to treat such pathologies. Our FRET-based HTS detects RyR binding of accessory proteins calmodulin (CaM) or FKBP12.6. Under conditions that mimic a pathological state, we carried out a screen of the 727-compound NIH Clinical Collection, which yielded six compounds that reproducibly changed FRET by >3 SD. Dose–response of FRET and [3H]ryanodine binding readouts reveal that five hits reproducibly alter RyR1 structure and activity. One compound increased FRET and inhibited RyR1, which was only significant at nM [Ca2+], and accentuated without CaM present. These properties characterize a compound that could mitigate RyR1 leak. An excellent Z′ factor and the tight correlation between structural and functional readouts validate this first HTS method to identify RyR modulators.
Introduction
Embedded in the sarcoplasmic reticulum (SR) membrane of striated muscle, ryanodine receptors (RyRs) are responsible for intracellular Ca2+ release that triggers muscle contraction. Dysregulation of skeletal (RyR1) and cardiac (RyR2) isoforms is implicated in the pathology of severe diseases, including malignant hyperthermia (MH), central core disease (CCD), muscular dystrophy, sarcopenia, heart failure (HF), diabetes, and Alzheimer’s disease (AD).1–5 In most of these cases, pathogenesis is, in part, attributed to excess SR Ca2+ “leak” via RyR under resting cellular conditions, and thus RyR is an intensely studied therapeutic target. Indeed, there are RyR small-molecule modulators that have been identified as possible therapeutic agents, including dantrolene, flecainide, tetracaine, K201, and S44121. However, most of these candidate drugs are known to also deleteriously interact with and functionally modulate other targets, and thus have side effects that render them unsuitable for chronic therapeutic use.6,7 Certainly, a large expansion in the list of RyR regulators would greatly benefit the advancement of RyR-targeted pharmaceutical approaches to treating several skeletal and cardiac myopathies.
The RyR is tightly regulated by endogenous proteins, such as calmodulin (CaM) and FK-506 binding proteins (FKBP) 12.0 and 12.6, that are associated with maintaining normal Ca2+ cycling. 8 Binding of CaM and FKBP to RyR has been shown to be sensitive to small-molecule RyR modulators (dantrolene, K201, and S44121) and RyR posttranslational modifications.9–12 Despite some controversy regarding the correlation between RyR phosphorylation and FKBP association, 8 the collective of studies indicate that CaM, and possibly also FKBP, binding to RyR may be used as a sensitive indicator of the RyR structural state associated with Ca2+ leak.11–15
Ideally, at the foundation of a target-directed high-throughput screen (HTS) is an effective method of monitoring the target’s activity that can be adapted for high-throughput measurement. As of yet, a true RyR-directed HTS method has not been reported, which may be attributed to the low-throughput nature of the methods that are currently used to directly assess RyR activity. Thus, a method that detects a signal that correlates with RyR function and is truly measurable in a high-throughput manner is highly desirable for identifying drug-like RyR modulators within libraries of small-molecule compounds. In this study, we aimed to validate an HTS-compatible method of seeking small-molecule modulators of RyR using time-resolved fluorescence resonance energy transfer (TR-FRET) as readout for a structural change in RyR1 or a shift in levels of CaM or FKBP binding. Our established molecular toolkit, as shown in Figure 1A , involves measuring FRET between an excited donor fluorophore attached to a single-Cys FKBP12.6 (D-FKBP) and an acceptor fluorophore attached to a single-Cys CaM (A-CaM), when both of these fluorescently labeled proteins are bound to RyR in native SR membranes.16,17 When A-CaM is bound to RyR in the proximity of D-FKBP, FRET is measured as a decrease in donor fluorophore fluorescence intensity 16 or fluorescence lifetime (FLT; calculated from the TR-fluorescence decay waveforms; Fig. 1B ). Changes in FRET due to an RyR modulator reflect changes in FKBP or CaM binding and/or in RyR structure, as shown in Figure 1C . We integrated this FRET-based molecular toolkit that targets RyRs with the use of an FLT plate reader (FLT-PR)18,19 to undertake a duplicate screen of the 727-compound NIH Clinical Collection (NCC). Using this technology, a high-precision (S/N > 100) TR-fluorescence decay, averaged from 200 fluorescence waveforms, is read in 200 ms, and a whole 384-well plate is scanned within 3 min. In this screen, we identified six compounds that reproducibly altered FRET in duplicate runs. Five of these compounds also reproducibly altered FRET in dose–response assays, and all these compounds altered RyR1 activity, as measured by [3H]ryanodine binding, in a fashion that typically inversely correlated with the shift in FRET. Therefore, we present an HTS assay to detect both activators and inhibitors of the RyR, and thus validated an HTS-capable method that will be used to identify compounds of high therapeutic potential.
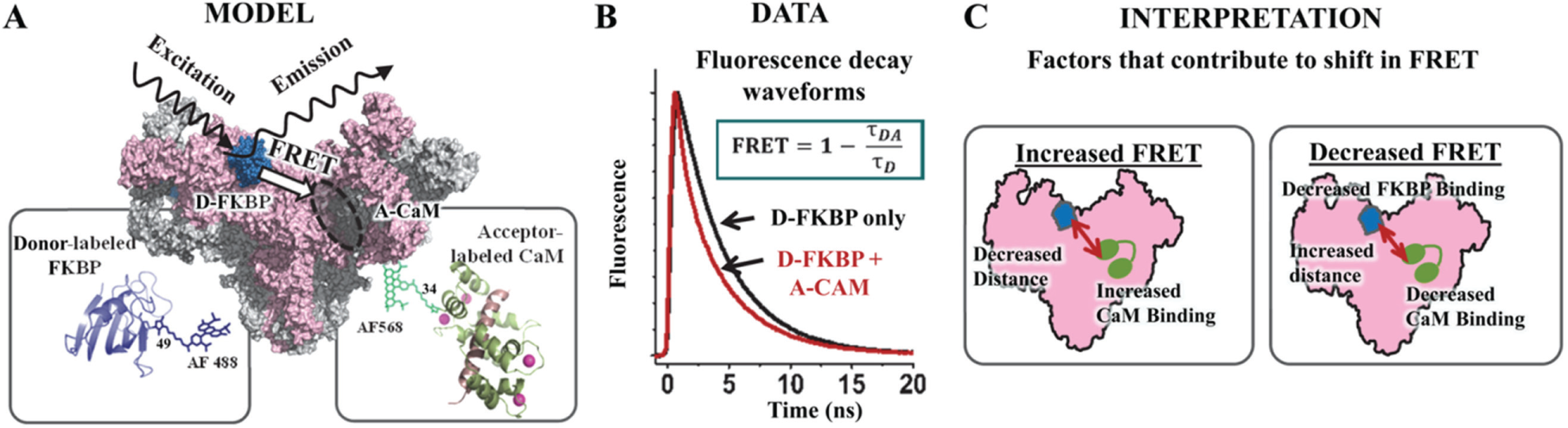
FRET-based HTS-compatible system to monitor RyR interaction with CaM and FKBP. (
Materials and Methods
The 727-compound NCC was purchased from Evotec (Hamburg, Germany) as 10 mM solutions in DMSO. The pig skeletal muscle (longissimus dorsi) was processed and purchased from Pel-Freez Biologicals (Rogers, AR). Fluorophores Alexa Fluor 488 C5-maleimide (AF488) and Alexa Fluor 568 C5-maleimide (AF568) were purchased from Life Technologies (Eugene, OR). Individual NCC compounds of interest (hits) were purchased separately, as follows: disulfiram and ebselen from Thermo Fisher Scientific (Rockford, IL); chloroxine, diazoxide, and fluphenazine from Sigma-Aldrich (St. Louis, MO); cefatrizine propylene glycol (PG), cefixime trihydrate, and tacrolimus from Sequoia Research Products Ltd. (Pangbourne, UK); and pravastatin and lofepramine from Santa Cruz Biotechnology (Santa Cruz, CA).
Compound Handling and Preparation of 384-Well Assay Plates
The NCC compounds were received in 96-well plates and reformatted into 384-well polypropylene intermediate plates (Greiner Bio-One, Kremsmunster, Austria) using a multichannel liquid handler, BioMek FX (Beckman Coulter, Miami, FL), and then transferred to 384-well Echo Qualified source plates (Labcyte, Inc., Sunnyvale, CA). Assay plates were prepared by transferring 50 nL of the 10 mM compound stocks or DMSO from the source plates to 384-well black polypropylene plates (Greiner), using an Echo 550 acoustic dispenser (Labcyte). NCC compounds were formatted in three plates, with the first two and last two columns loaded with 50 nL of DMSO and used for drug-free controls. These assay plates were then heat-sealed using a PlateLoc Thermal Microplate Sealer (Agilent Technologies, Santa Clara, CA) and stored at −20 °C prior to usage.
Isolation of Sarcoplasmic Reticulum Vesicles
SR membrane vesicles were isolated from pig longissimus dorsi muscle by differential centrifugation of homogenized tissue, in accordance with Fruen et al. 20 Heavy SR (HSR) vesicles, which are enriched in RyR1, were isolated by fractionation of crude skeletal SR vesicles using a discontinuous sucrose gradient. 20 All vesicles were flash frozen and stored at −80 °C. Immediately prior to the fluorescence or [3H]ryanodine binding studies described below, the SR vesicles were stripped of residual endogenous CaM by incubation with a peptide derived from the CaM binding domain of myosin light chain kinase, followed by sedimentation, in accordance with Fruen et al. 21
Expression, Purification, and Labeling of FKBP and CaM
Single-cysteine mutants of FKBP12.6 (C22A/R49C/C76I) and CaM (T34C) were expressed in Escherichia coli BL21(DE3)pLysS (Agilent Technologies), purified, and respectively labeled with fluorescent probes AF488 and AF568 as described previously.17,22 Stoichiometric labeling of FKBP and CaM with fluorescent probes to ≥95% was indicated by comparing the molar concentration of bound dye (determined from its absorbance) to the protein concentration, which was determined via a bicinchoninic acid assay (Thermo Fisher Scientific) and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) densitometry. Essentially complete labeling was confirmed by electrospray ionization mass spectrometry. We have previously demonstrated that our system of donor labeling FKBP12.6 (D-FKBP) with AF488 and acceptor labeling CaM (A-CaM) with AF568 alters neither the endogenous binding nor modulatory activity of these RyR accessory proteins.16,17
Preparation of SR Vesicles for FRET Measurement
HSR (0.4 mg/mL) membranes were preincubated with 60 nM D-FKBP for 90 min at 37 °C in a solution containing 150 mM KCl, 5 mM glutathione (GSH), 0.1 mg/mL bovine serum albumin (BSA), 1 µg/mL aprotinin/leupeptin, 1 mM DTT, and 20 mM PIPES (pH 7.0). To remove unbound D-FKBP, the SR membranes were spun down at 110,000g for 20 min, and then resuspended to 1 mg/mL in binding buffer consisting of 150 mM KCl, 5 mM glutathione disulfide (GSSG), 0.1 mg/mL BSA, 1 µg/mL aprotinin/leupeptin, and 20 mM PIPES (pH 7.0). These samples were then incubated with 0.3 µM A-CaM for 30 min at 22 °C in binding buffer containing 0.065 mM CaCl2 to give 30 nM Ca2+ in the presence of 1 mM EGTA (calculated by MaxChelator). This labeled SR sample was applied to the NCC assay plates in 50 µL aliquots using a Multidrop Combi reagent dispenser (Thermo Scientific) with a standard tube cassette that loaded a single plate within 30 s. An additional set of the NCC assay plates were loaded with HSR labeled with only D-FKBP, not A-CaM.
Fluorescence Data Acquisition
FLT measurements were conducted in a prototype top-read FLT-PR designed and built by Fluorescence Innovations, which reads each 384-well plate in ~3 min. AF488 donor fluorescence was excited with a 473 nm microchip laser from Concepts Research Corporation (Belgium, WI), and emission was acquired with 490 nm long-pass and 520/17 nm band-pass filters (Semrock, Rochester, NY). This instrument uses a unique direct waveform recording technology that enables high-throughput FLT detection at high precision. 23 We have previously demonstrated the performance of this plate reader with known fluorescence standards, as well as with a FRET-based HTS method that targets SERCA.18,19
HTS Data Analysis
TR-fluorescence waveforms for each well were fitted based on a two-exponential decay function using least-squares minimization global analysis software (Fluorescence Innovations). As indicated, the FRET efficiency (E) was determined as the fractional decrease of donor FLT (τD), due to the presence of acceptor fluorophore (τDA), using the following equation:

Assay quality was determined based on controls (DMSO-only samples) on each plate, as indexed by the Z′ factor: 24
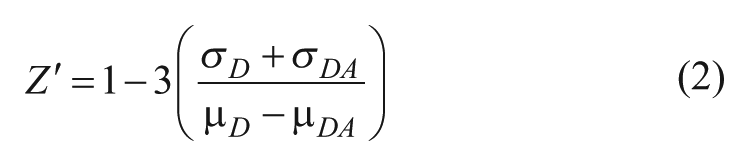
where σD and σDA are the standard deviations (SDs) of the controls τD and τDA, respectively, and µD and µDA are the means of the controls τD and τDA, respectively. The Z′ for the fluorescence peak data was calculated using eq 2 with σD and µD of control FD and σDA and µDA of FDA, based on controls present in each plate. A compound was considered a hit if it changed E by >3 SD relative to that of control samples (E0) that were exposed to 0.1% DMSO. The 3 SD hit selection threshold is typical for normally distributed HTS data, whereby 0.27% of the readings are expected to fall outside this limit. The threshold may be further adjusted to constrain the number of hits according to the resources available for evaluation via secondary (orthogonal) assays.
[3H]ryanodine Binding to SR Vesicles
Skeletal SR membranes (1 mg/mL) were preincubated with 0.02% DMSO or hit compound, with or without 800 nM CaM, for 30 min at 4 °C in a solution containing 150 mM KCl, 5 mM GSSG, 1 µg/mL aprotinin/leupeptin, 1 mM EGTA, 65 µM or 1.02 mM CaCl2 (as determined by MaxChelator to yield 30 nM or 30 µM Ca2+, respectively), 0.1 mg/mL of BSA, and 20 mM K-PIPES (pH 7.0). Nonspecific and maximal [3H]ryanodine binding to SR were separately assessed by addition of 20 µM nonradioactively labeled ryanodine or 5 mM adenylyl-imidodiphosphate, respectively. Binding of [3H]ryanodine (15 nM) was determined following a 3 h incubation at 37 °C and filtration through grade GF/B Glass Microfiber filters (Brandel, Inc., Gaithersburg, MD) using a Brandel Harvester. In 4 mL of Ecolite Scintillation cocktail (MP Biomedicals, Solon, OH), [3H] on filter was counted using a Beckman LS6000 scintillation counter (Fullerton, CA).
Results
HTS Performance
To validate the performance of our FRET-based HTS method for identification of novel RyR1 modulators that may have therapeutic benefit, we screened the NCC, which is a 727-compound library of small molecules that have a history of use in human clinical trials. This diverse collection of small molecules is ideal for a pilot screen, particularly given that some molecules are known RyR modulators, such as dantrolene, tacrolimus, and ebselen. The complete NCC was applied to columns 3–22 of three 384-well black-wall/black-bottom Greiner plates (drug-containing wells). Plate columns 1 and 2 and 23 and 24 were loaded with 50 nL of DMSO (and these wells were used for drug-free controls), using an Echo 550 acoustic dispenser (Labcyte). For each screen, one set of plates was loaded with D-FKBP-labeled SR membranes (donor-only sample), while the other set was loaded with the D-FKBP-labeled SR that was additionally incubated with 0.3 µM A-CaM (donor–acceptor sample) for 30 min prior to loading on the plates. This subsaturating concentration of A-CaM was chosen to provide a readout that is sensitive to library compounds that may increase or decrease CaM binding to RyR. Final assay conditions also included 30 nM Ca2+ to represent resting [Ca2+], and 5 mM oxidized glutathione (GSSG), which exaggerates the conditions associated with oxidative stress. 25 With these conditions, the objective was to promote a homogenous population of RyR1 that are associated with muscle pathologies. This is with a future goal of identifying RyR-targeted therapeutics for SR Ca2+ leak. These biological samples were loaded using a Multidrop Combi automated sample dispenser, as described in Materials and Methods.
TR-fluorescence decay waveforms were read in the FLT-PR over a time course of 5, 20, 40, 60, and 120 min after applying the SR sample to the assay plates. Although some compounds (chloroxine and ebselen) substantially altered FRET by the 20 min time point, the effect of small molecules that altered FRET was greatest or had plateaued by the 2 h time point (
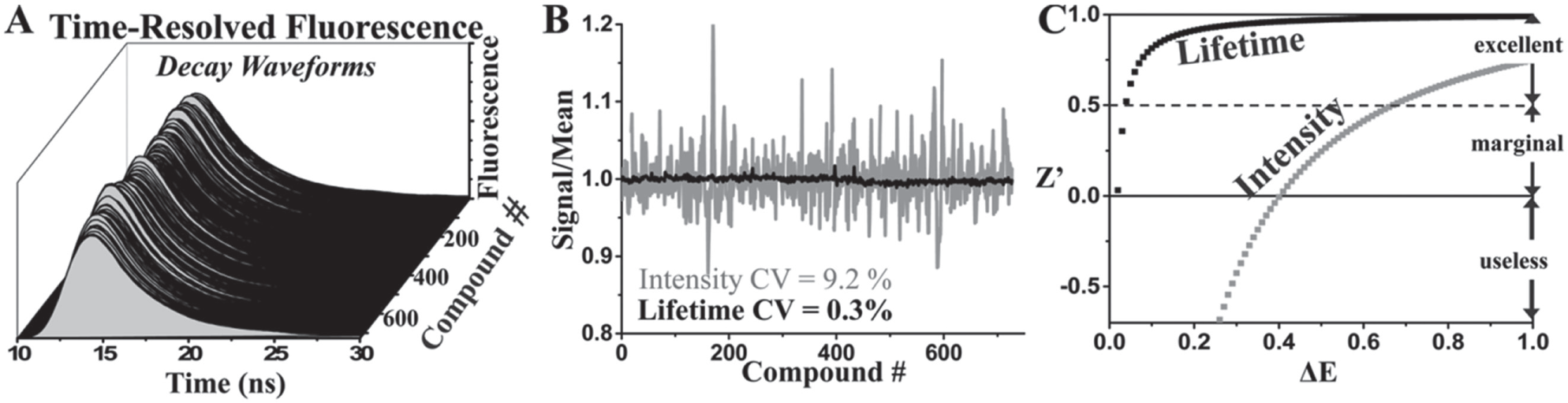
Performance of TR-fluorescence properties: lifetime vs. peak intensity. (
The overall effects on FRET, expressed as E/E0, are shown in
Figure 3A
, but with the readouts from fluorescent compounds excluded. Of 14 and 16 hits from each screen (based on the [+] or [–] 3 SD criterion), only 6 were reproducible, shown in
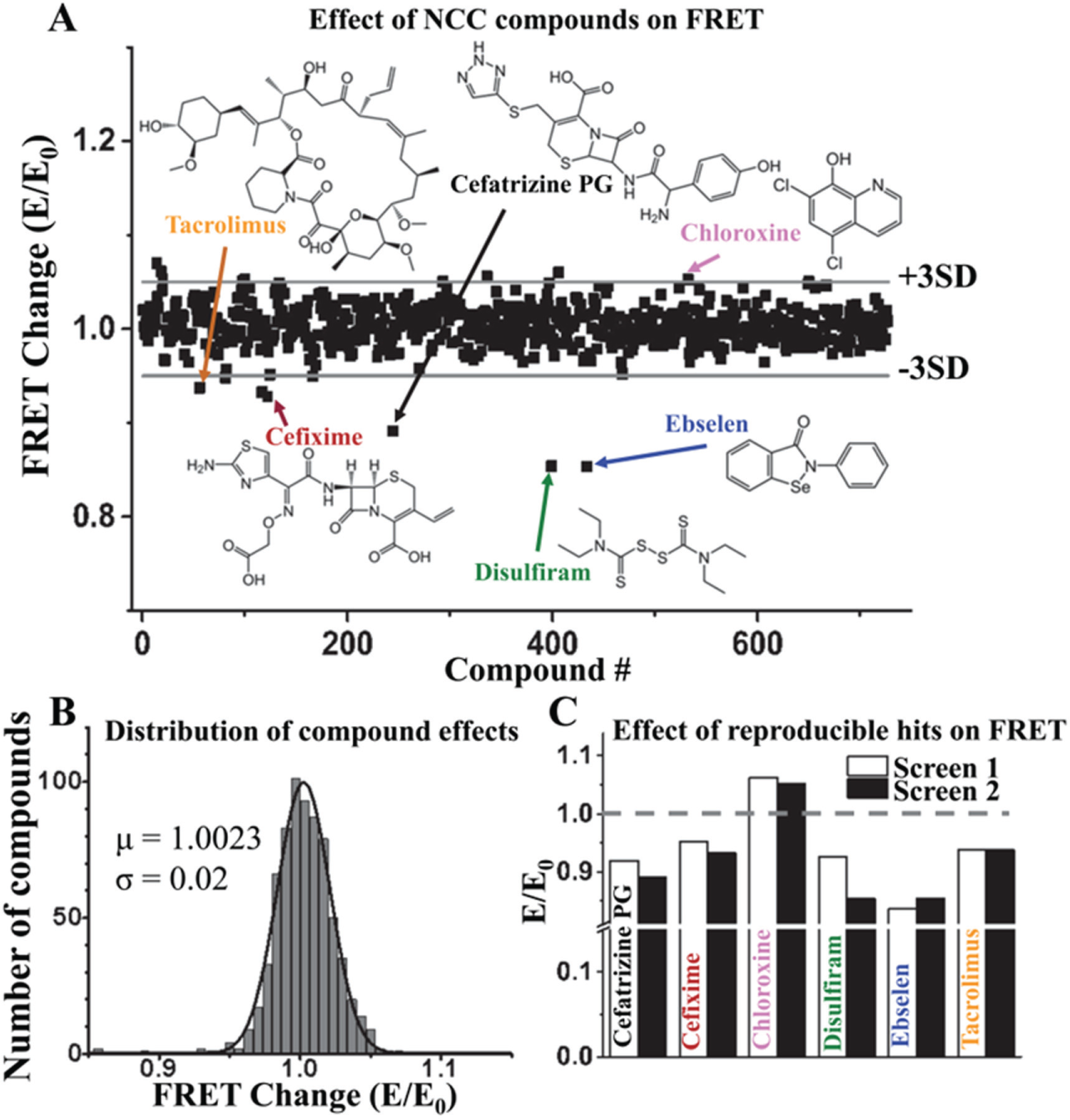
HTS results. A FRET screen of the NCC library (727 compounds, 10 µM) to detect modulators of the RyR/FKBP/CaM complex was undertaken in duplicate. The FRET of donor-labeled FKBP (D-FKBP) by acceptor-labeled CaM (A-CaM) was calculated from donor FLT. Then the FRET for each compound (E) was normalized to the DMSO control (E0). (
FRET Dose–Response Assays
Using the same assay condition as in the screens, we tested the dependence of FRET on the concentration of the reproducible hits (0.01–100 µM compound). We examined the effect of hits under Ca2+ conditions corresponding to muscle relaxation and contraction (30 nM or 30 µM free Ca2+, respectively). The effect of the compounds largely replicated the effect observed in the screens, with one exception, cefixime ( Fig. 4A ), which was not further examined in functional studies. As a minor variation, disulfiram and cefatrizine were both found to decrease FRET in the screen, but increased FRET at 10 µM in the follow-up dose–response assays. However, both disulfiram and cefatrizine biphasically affected FRET, with exposure to >20 µM compound decreasing FRET in a manner consistent with the HTS readout ( Fig. 4A ).
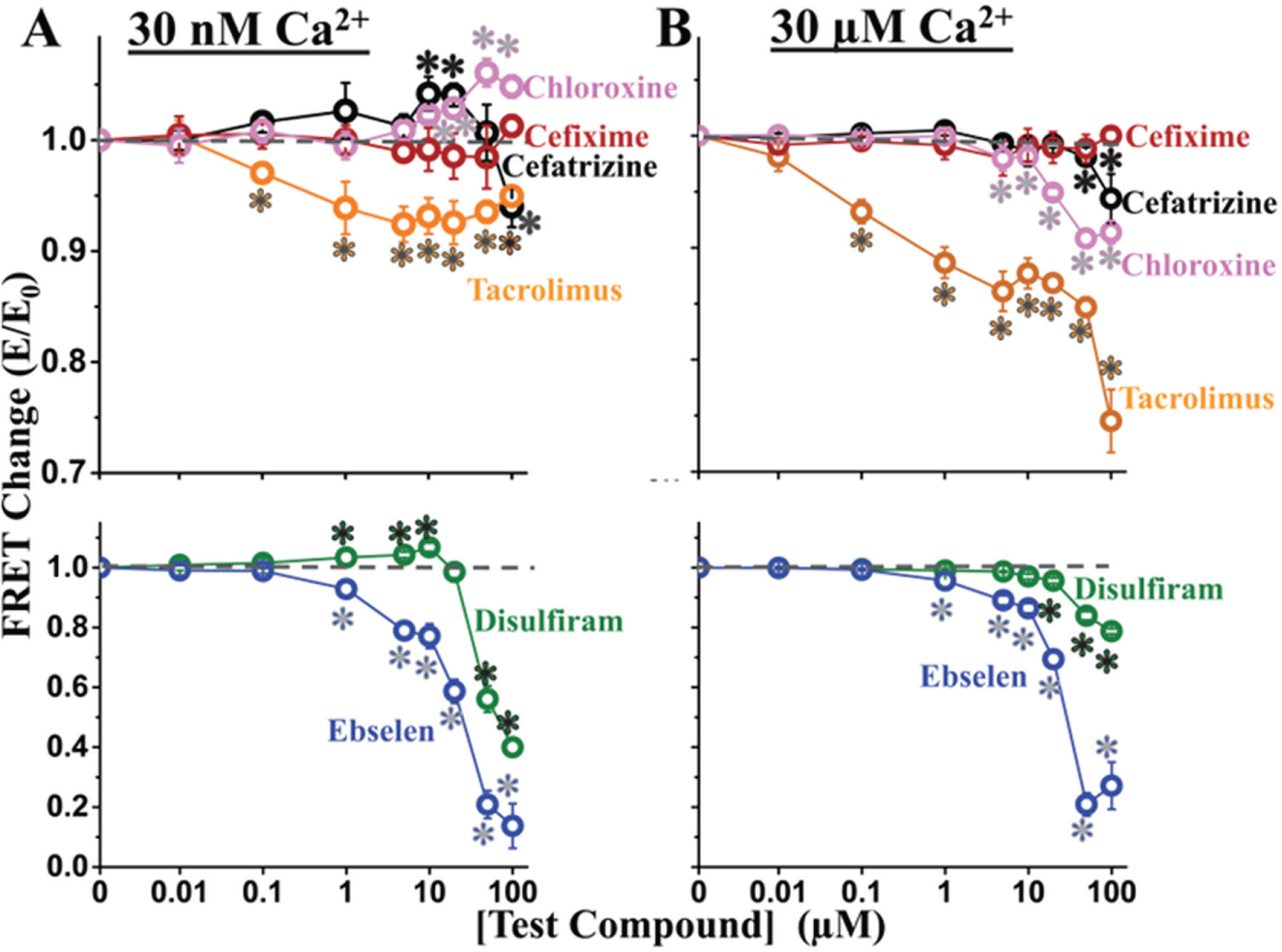
Dose–response of screen hits on FRET at 30 nM Ca2+ (
Given the role of tacrolimus (also known as FK506) in blocking both FKBP12 and FKBP12.6 from binding to RyRs, 28 we were pleased that our primary screen identified this compound as a significant inhibitor of FRET at both 30 nM and 30 µM Ca2+ ( Fig. 4 ). Similarly, ebselen is another known RyR modulator, which has been shown to affect RyR1 activity, in a biphasic manner, via oxidation of RyR1 free thiols. 29 However, ebselen has not been previously reported to perturb CaM or FKBP12.6 binding to RyR. Of the known RyR modulators present in NCC, we did not observe an effect of 10 µM dantrolene on FRET with skeletal SR under these particular experimental conditions. This is consistent with previous reports that dantrolene does not alter CaM binding affinity to RyR1 in porcine skeletal SR membranes. 30
Disulfiram has been shown to increase Ca2+ uptake by SR in saponin-permeabilized rat skeletal muscle fibers without modulating caffeine sensitivity of Ca2+ release, 31 thus suggesting that it did not act on the RyR. Yet, we observed an interesting biphasic effect of disulfiram on FRET at 30 nM Ca2+: it increased at 1–10 µM and decreased at 20–100 µM compound ( Fig. 4A ), although this effect is greatly attenuated at 30 µM Ca2+ ( Fig. 4B ).
Both cefatrizine and chloroxine are utilized therapeutically for their antibacterial properties, but neither drug has been reported, to date, to alter RyR activity or cellular Ca2+ homeostasis. As shown in Figure 4 , chloroxine slightly increased FRET (ΔE ≈ 0.05) at ≥10 µM at 30 nM Ca2+, but this effect is reversed to an inhibition of FRET in 30 µM Ca2+. Similar to the effects of disulfiram, a biphasic effect was observable with cefatrizine ( Fig. 4A ), which also decreased FRET >20 µM, consistent with the effect we observed in HTS mode. Furthermore, at 30 µM Ca2+, we observed only a small FRET decrease (rather than a biphasic effect) at >20 µM cefatrizine ( Fig. 4B ).
Overall, the dose–response data confirm that five of the six hits modulate FRET, even a compound (chloroxine) with a very small effect on FRET (ΔE ≈ 0.02). The reproducibility of these compounds at 10 µM is high, as evidenced by the low CV (ranging from 0.5% to 4.1%).
Effect of Hits on RyR1 Activity, as Determined Using [3H]ryanodine Binding to HSR
To evaluate the ability of our structure-based screen to identify functional modulators of RyR, we used [3H]ryanodine binding to SR. The level of [3H]ryanodine binding is a well-established index of the channel activity in an RyR population, as presented in native SR membranes. 22 Furthermore, this technique enables assay experimental conditions to be similar between RyR activity and FRET experiments. To determine whether a correlation exists between the FRET readout and RyR activity, we used the same overall experimental conditions in both types of assay.
Since CaM is a potent regulator of RyR activity and A-CaM was used in the FRET (structural) component of this study, we sought to determine whether CaM influences the functional effects of the hits. Thus, we carried out the [3H]ryanodine binding measurements in the absence and presence of CaM (800 nM), over the same range of hit concentrations as in the FRET dose–response studies above ( Fig. 4 ). In accord with the well-established functional effect of CaM on RyR1, addition of CaM increased activity ([3H]ryanodine binding) by ~4-fold in nM Ca2+, and inhibited activity by ~70% in µM Ca2+ ( Fig. 5A ). This response to a well-established RyR modulator indicates that our experimental conditions are adequate to detect functional effects by hits. Overall, relative to the matched DMSO controls, each compound modulated RyR1 activity in a Ca2+-dependent manner ( Fig. 5B–F ). All compounds had the strongest effect on RyR1 activity at 30 nM Ca2+ and in the absence of CaM, strongly indicating that CaM binding RyR1 is not required for these compounds to affect RyR1 function.
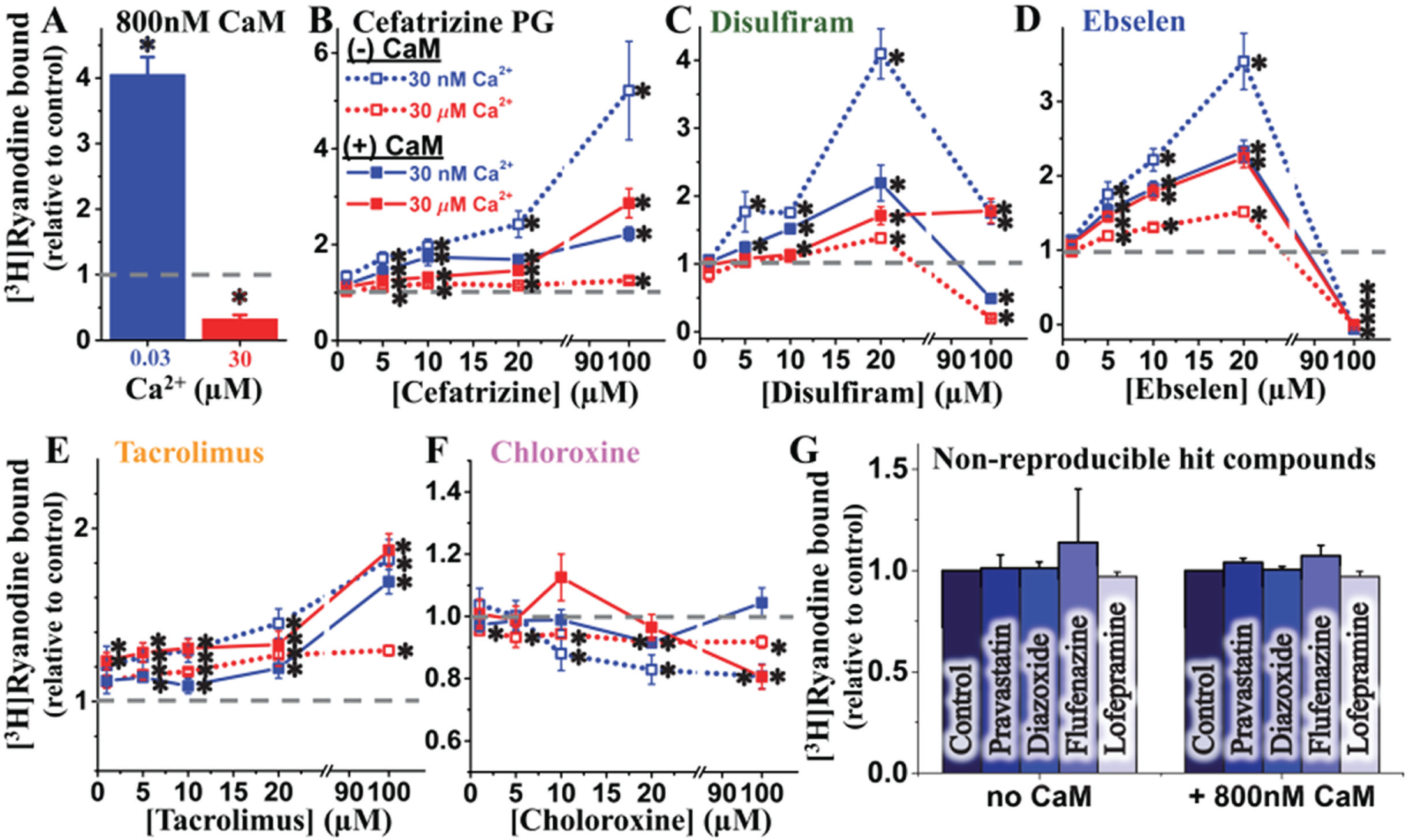
[3H]Ryanodine binding assays to determine the dose-dependent effect of hits on the RyR1 function. (
At 30 nM Ca2+, cefatrizine, disulfiram, ebselen, and tacrolimus significantly increased RyR1 activity, whereas chloroxine inhibited it. For all compounds, the amplitudes of these effects were significantly reduced when CaM (800 nM) was present in the sample.
At 30 µM Ca2+, the effects of all compounds were smaller and CaM influenced them in a less consistent manner than in 30 nM Ca2+. Namely, CaM increased the effects of cefatrizine and ebselen, did not significantly change those of disulfiram and tacrolimus, and changed that of chloroxine from slight inhibition to a biphasic profile peaking in a ~20% activation at 10 µM compound, and then turning into inhibition at the higher µM concentrations tested.
For the purpose of identifying RyR modulators, dantrolene is a false negative in our method’s readout. For a broader estimation of the rate of false negatives, we randomly selected a few nonreproducible hits (pravastatin, diazoxide, flufenazine, and lofepramine) and found that, at 10 µM, they have no significant effect on RyR1 activity, as indicated by [3H]ryanodine binding assays ( Fig. 5G ). This assessment also suggests that there is a moderate rate of false positives in one screen, but this is much lower in duplicate screens. Overall, all the compounds yielded by HTS as hits that were confirmed as FRET effectors in follow-up dose–response assays were found to functionally modulate RyR1. However, the presence of CaM in 30 nM Ca2+ strongly impacts the functional effect of hits.
Discussion
Excess SR Ca2+ leak under resting intracellular conditions is implicated in the pathology of several severe cardiac and skeletal myopathies, diabetes, and neurological disorders. The dysregulation of RyR that promotes this leaky state of the channel not only is associated with disease-causing mutations in RyR, but also altered posttranslational modifications that have been implicated in altering binding of RyR stabilizers, CaM, and FKBP12/12.6.1,3,4,6,10–12 Moreover, excessive posttranslational modifications are typically thought to be key players in late-stage pathology. Therefore, the pursuit of RyR-directed treatment agents is expected to produce clinically relevant outcomes—given the general appreciation in the field—based on compelling experimental evidence that targeting RyR-mediated Ca2+ leak is effective for treating animal and cellular models of sarcopenia, muscular dystrophy, or arrhythmias.4,32 Ultimately, identification or design of novel small molecules that inhibit RyRs under resting (nanomolar [Ca2+]) conditions is highly desirable in the pursuit of RyR-targeted therapeutics of myopathies. As of yet, however, advancement of RyR-targeted therapies, particularly pharmaceutical, is arrested by the lack of high-throughput methodologies to identify RyR modulators. We report on a key first step to address this important unmet clinical need.
To discover compounds that modulate RyR1, we adapted our well-established FRET system for rapid screen of a 727-compound library (NCC). By using FLT analysis of decay waveforms, we were able to identify even subtle changes of FRET, which could reflect a shift in the binding of CaM or FKBP to RyR, or a structural change in the RyR structure ( Fig. 1 ). With a threshold of 3 SD of the mean, modulators that altered the FRET efficiency by >0.03 were considered hits. From follow-up FRET dose–response assays of the individually purchased compounds, we found that five of the screen hit compounds significantly altered FRET, albeit >20 µM disulfiram and cefatrizine were necessary to produce the same reduction in FRET that was produced by 10 µM compound in the screen. The probable reason for these differences is variable compound degradation or their effective concentration in the source library. Based on this small screen, the apparent rate of false positives is 16%, which indicates that the 3 SD hit selection threshold is economically tenable.
The validity of our FRET readout as an indicator of RyR1 functional modulation is strongly supported by the observation that all five compounds that altered FRET in the dose–response assays also modulated RyR activity, as measured using [3H]ryanodine binding assays. Thus, we have found that most hits reproducibly identified via HTS—that is, using a structural readout—also have a functional effect on RyR1. Particularly encouraging is the finding that one reproducible HTS hit that increased FRET (chloroxine) inhibited RyR1 function (as indicated by [3H]ryanodine binding; Fig. 5F ), and this effect was largest under nM [Ca2+] (resting) in the absence of CaM. These are the properties that we expect—at the stage of initial secondary studies following HTS—to characterize a compound that is likely to mitigate RyR1 leak in situ.
We compared changes in FRET to changes in activity for four different concentrations of each hit (≤20 µM), under nanomolar and micromolar Ca2+ ( Fig. 6A ). As indicated by the slope of the linear fit ( Fig. 6B ), most of the hits affect FRET and RyR activity in an inverse manner. That is, an increase in FRET is correlated with inhibition of [3H]ryanodine binding (the effect we seek in a leak inhibitor), and a decrease in FRET correlates with increased [3H]ryanodine binding (an effect indicative of potentially deleterious side effects due to increased RyR leak). Of the five tested hits, three—ebselen, disulfiram, and chloroxine—display a strong correlation between alteration in FRET and RyR activity, as indicated by the correlation coefficient of the linear fit ( Fig. 6C ). The apparently weak correlation in the effects of cefatrizine and tacrolimus may be due to a complex compound–RyR interaction, as suggested by the biphasic FRET response. In particular, the subtle effect of a change in RyR conformation would not be directly comparable to the larger FRET effect from dissociation of fluorescently labeled proteins (D-FKBP and A-CaM).
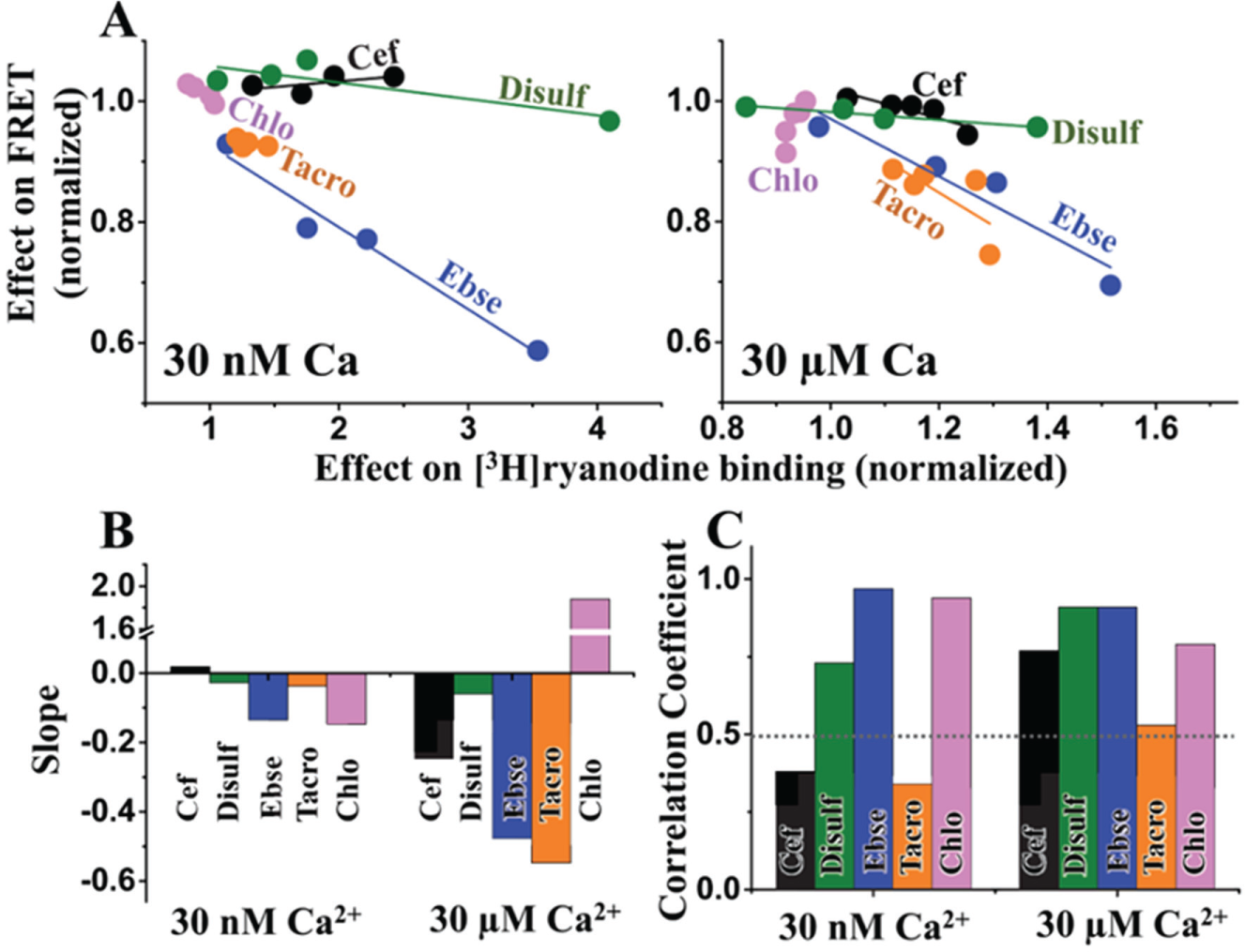
Correlation between hit effects on FRET and RyR1 activity. (
The detailed mechanism of hit action on the RyR complex is beyond the scope of this report. In this first pilot screen targeting RyR, the objective was simply to validate a method that can identify RyR modulators in HTS mode. Nevertheless, it is clear that (1) the hits affect [3H]ryanodine binding in the absence of CaM, and (2) these effects are influenced by the presence of CaM. Since CaM itself is a modulator of RyR1 function (activator at nM [Ca2+]; inhibitor at µM [Ca2+]), we expected that a hit (which was identified due to its disruption of CaM binding) would affect RyR1 function in a CaM-dependent manner. Moreover, we might expect that a hit would affect the CaM-mediated modulation of [3H]ryanodine binding by altering CaM-RyR1 binding. However, given the saturating [CaM] we used to test the impact of CaM on compound regulation of RyR1, this would only be apparent if a compound dramatically reduced CaM-RyR1 affinity—like disulfiram or ebselen—as indicated by FRET ( Fig. 4 ). In this instance, a shift in CaM binding might be reflected in a shift of RyR1 activity that is additive to the separate effect of the hit compound directly on the RyR. For example, if a hit directly activates RyR1 in low [Ca2+] and also inhibits CaM binding to RyR1, we expect the overall functional effect to be impacted by a combination of the direct activating effect of the hit (that can be observed in the absence of CaM) and a reduced CaM-mediated RyR1 activation. This would then become apparent in the functional results as the activating effect of a compound being attenuated by the presence of CaM in low [Ca2+]. This could easily be said for the effect of disulfiram (5–20 µM; Fig. 5C ) and ebselen (5–20 µM; Fig. 5D ) on RyR1 activity in the presence of CaM, whereby the putative drug-induced dissociation of CaM attenuates the activating effect of the drugs in 30 nM Ca2+ and enhances the effect of the drug in 30 µM Ca2+, due to a reduction in CaM-mediated activation and inhibition at the respective [Ca2+]. However, this model would not explain the CaM-dependent effects on other compounds that marginally alter FRET, given that this mode of action would likely be masked by the use of saturating [CaM] in [3H]ryanodine binding assays. In the case of marginally significant modulators of FRET, the more plausible explanation is that CaM binding to RyR1 allosterically alters compound binding or its effect. This may be the case for at least cefatrizine ( Fig. 5B ), tacrolimus ( Fig. 5E ), and chloroxine ( Fig. 5F ), but their actual mechanisms of action can only be determined through detailed studies beyond the scope of this report.
In summary, we have developed and validated an HTS platform designed to resolve small-molecule compounds that modulate RyR, laying the foundation for RyR-targeted HTS approaches to identify and develop agents that target RyR-mediated SR Ca2+ leak. In one screen of 727 compounds, we detected five modulators of RyR1 activity; two are known modulators, while three novel modulators were detected. To predict feasibility for identification of therapeutic agents that target RyR Ca2+ leak, one such compound may be a good candidate for further testing as a leak inhibitor in situ. With a screen of 20,000 compounds, as we previously carried out, 33 we anticipate detecting ~150 novel effectors of RyR, which would extensively expand our current knowledge of RyR1 pharmacomodulation. Of these, we would expect ~30 small inhibitors of RyR as viable candidates to be used in secondary screens to validate their therapeutic potential for myopathies or neurological disorders.
Footnotes
Acknowledgements
The NCC assay plates were prepared by Samantha Yuen and Ji Li. Kurt Peterson and Ji Li provided helpful discussions for undertaking the HTS, and Octavian Cornea helped prepare the manuscript for publication. HTS was performed using the facilities provided by Fluorescence Innovations, Inc. (Minneapolis, MN), with assistance from Kurt Peterson. Spectroscopy for dose–response assays was performed at the Biophysical Technology Center, Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: DDT and RLC hold equity in, and serve as executive officers for, Photonic Pharma LLC. These relationships have been reviewed and managed by the University of Minnesota. Photonic Pharma had no role in this study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by American Heart Association Grant-in-Aid 15GRNT25610022 (to RLC) and Postdoctoral Fellowship 14POST20310024 (to RTR), NIH grants R01HL092097 (to RLC and DMB) and R37AG026160 (to DDT), and STTR Phase II grant R42DA037622 (to GDG and DDT).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.