
Abstract
Facioscapulohumeral muscular dystrophy is a genetically dominant, currently untreatable muscular dystrophy. It is caused by mutations that enable expression of the normally silent DUX4 gene, which encodes a pathogenic transcription factor. A screen based on Tet-on DUX4-induced mouse myoblast death previously uncovered compounds from a 44,000-compound library that protect against DUX4 toxicity. Many of those compounds acted downstream of DUX4 in an oxidative stress pathway. Here, we extend this screen to an additional 160,000 compounds and, using greater stringency, identify a new set of DUX4-protective compounds. From 640 hits, we performed secondary screens, repurchased 46 of the most desirable, confirmed activity, and tested each for activity against other cell death–inducing insults. The majority of these compounds also protected against oxidative stress. Of the 100 repurchased compounds identified through both screens, only SHC40, 75, and 98 inhibited DUX4 target genes, but they also inhibited dox-mediated DUX4 expression. Using a target gene readout on the 640-compound hit set, we discovered three overlooked compounds, SHC351, 540, and 572, that inhibit DUX4 target gene upregulation without nonspecific effects on the Tet-on system. These novel inhibitors of DUX4 transcriptional activity may thus act on pathways or cofactors needed by DUX4 for transcriptional activation in these cells.
Keywords
Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is among the most prevalent of muscular dystrophies, affecting more than 25,000 people in the United States alone. The disease typically presents in the second decade and progresses variably and unpredictably, and often asymmetrically. 1 While virtually all muscles are affected to some degree, loss of function is usually most severe in facial muscles and upper arm and back muscles. In many patients, the hip girdle is severely affected, and eventual wheelchair use is common. There is no treatment for FSHD; therefore, there is a great need to discover new therapeutic options that may slow the advance or reverse the course of this disease.
FSHD is genetically dominant and usually caused by a reduction in copy number of a macrosatellite repeat at 4q35.2 called D4Z4.2,3 The D4Z4 repeat is subject to repeat-induced silencing, and the reduction in copy number results in epigenetic changes that disrupt silencing on the contracted allele. 4 FSHD can also be caused by mutations in the chromatin protein SMCHD1, 5 in which case all D4Z4 repeats become demethylated.6,7 Loss of silencing at D4Z4, combined with a specific allele of chr4 referred to as 4qA, 8 which provides a poly(A) signal, 9 leads to accumulation of an RNA encoding the transcription factor DUX4. 10 The DUX4 protein shows pathological effects on myoblasts: at high levels, it causes cell death,11,12 while at low levels, it interferes with myogenesis 13 and sensitizes cells to oxidative stress. 11 DUX4 is therefore the primary target for drug development in FSHD.
Mechanisms of DUX4 activity are currently actively being investigated. The N-terminus of DUX4 contains two homeodomains, which together bind to a sequence related to TAATCTAATCA.14,15 Several hundred DUX4 target loci have been identified by ChIP-seq.14,16 DUX4 acts as a pioneer factor at the majority of these loci, displacing nucleosomes to bind DNA in inaccessible chromatin, and its C-terminus recruits p300 and/or CBP, inducing acetylation of nearby histones, replacing repressive chromatin marks with markers of activation, leading to the expression of nearby genes. 16
We previously developed a high-throughput screening assay based on the loss of viability resulting from DUX4 expression and used it in a screen of 44,000 compounds. 17 This screen identified 183 compounds, with the majority clustering into 39 chemical series. Fifty-two compounds were confirmed by purchasing, performing mass spectrometry, and retesting. Interestingly, when tested in other assays, the majority of these 52 compounds also protected cells from another cell death–inducing insult, namely, oxidative stress. This revealed that the majority of these compounds were not acting directly on DUX4, but rather on downstream pathways, in particular, one specific downstream pathway (oxidative stress) that plays a major role in DUX4-mediated toxicity. In order to expand the repertoire of DUX4-protective compounds, and to further explore mechanisms of protection, we have screened an additional 160,000 compounds and evaluated these against various cell death–inducing insults. To identify those compounds that might have direct effects on mechanisms of DUX4 molecular activity rather than on pathways downstream of DUX4, we have also evaluated the newly identified compounds for effects on transcriptional changes induced by DUX4. We report a collection of new compounds that do not block DUX4-mediated transcription, but do inhibit toxicity, presumably by acting on downstream pathways. Some of these fall into chemical series previously identified, and some represent new chemical series. In addition, we describe three compounds that inhibit both toxicity and induction of DUX4 target gene expression.
Materials and Methods
Composition of the Library
We screened 160,000 compounds from the UT Southwestern Chemical Library that comprised three large compound collections acquired from ChemBridge (San Diego, CA), ComGenex (Budapest, Hungary), and ChemDiv (San Diego, CA). The compounds in the library were selected to avoid undesirable characteristics for medicinal chemistry and to satisfy a relaxed version of Lipinski’s rules for good predicted bioavailability. The set of 160,000 compounds screened was selected based on diverse representation of chemical structure across the library.
PAINS Identification
We used the Drugs3 web server (fafdrugs3.mti.univ-paris-diderot.fr) 18 to filter pan-assay interference compounds (PAINS).
Repurchased Compounds
Repurchased compounds were obtained from a variety of sources (ChemBridge, ChemDiv, Vitas M Labs, Enamine, Life Chemicals, and InterBioScreen).
Cell Culture
C2C12 derivative lines were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 20% fetal bovine serum (FBS; Atlanta Biologicals, Flowery Branch, GA),
Luciferase Assay
Cells were plated at 1000 cells per well in 384-well plates. Twenty-four hours later, luciferase was induced with doxycycline (dox) at 500 ng/mL. The Bright-Glo Luciferase Assay System (Promega, Madison, WI) was used to detect and quantify luminescence.
High-Throughput Screen
To minimize variation due to cell line genetic and epigenetic drift, we had previously generated a large frozen stock (80 vials) of cells from a single-cell subclone of iC2C12-DUX4. 17 Cells were expanded and all vials were frozen at the same time. For screening, a single vial was thawed, expanded over 6 days, and plated in 384-well plates at a density of 1000 cells per well. Compounds were added 24 h later at a concentration of 5 µM from a 500 µM stock in DMSO. Following compound addition, dox was added at 500 ng/mL. After 24 h of exposure to compounds and dox, cell viability was measured using the ATPlite system (PerkinElmer, Waltham, MA) according to the manufacturer’s directions.
Cell Death Inhibition Assays
C2C12 cells were plated in 384-well plates at 625 cells/well in 25 μL of medium using a Biomek FX robot (Beckman, Indianapolis, IN) and cultured for 24 h. Cell death–inducing reagents were then added at the following concentrations: tert-butyl-hydroperoxide (tBHP; 25 μM; Sigma), ionomycin (12.5 μM; Cayman, Ann Arbor, MI), staurosporine (0.0125 μM; Cayman), etoposide (12.5 μM; Cayman), ABT-263 (12.5 μM; Selleckchem, Houston, TX), and tunicamycin (2.5 μM; Cayman), using an Echo 550 robot (LabCyte, Sunnyvale, CA). Plates were cultured for a further 24 h, after which the plates were equilibrated to room temperature and medium was removed. To quantify ATP, CellTiter-Glo reagent (Promega) diluted (1:1) in PBS was added to each well and the plates were read on an Analyst AD 96/384 plate reader (LJL Biosystem, Sunnyvale, CA).
Dose–Response Analyses
iC2C12-DUX4 cells were exposed to compounds at various doses (7.44, 2.47, 0.81, 0.27, 0.09, 0.03, and 0.01 μM), followed by dox at 250 ng/mL. Cell viability was measured 24 h later using CellTiter-Glo.
High-Throughput qRTPCR
iC2C12-DUX4 cells were plated in 24-well plates at 4 × 104 cells/mL. Twenty-four hours later, compounds were treated at a concentration of 3 μM and then dox (50 ng/mL) was added for 4 h. Total RNA was extracted using the ZR-96 Quick-RNA extraction kit according to the manufacturer’s instructions (Zymo Research, Irvine, CA). Reverse transcription (RT) was performed using the Vero cDNA synthesis kit (Thermo Fisher Scientific, Waltham, MA), and quantitative PCRs (qPCRs) were conducted using Taqman probes (Myo1g, Mm00617991_m1; Gapdh, Mm99999915_g1; DUX4, Hs03037970_g1) (Applied Biosystems, Waltham, MA). mRNA expression was analyzed by the ΔCt method and values expressed relative to the expression of GAPDH.
Results
We previously developed a primary screen for inhibitors of DUX4 toxicity based on colorimetric detection of ATP levels in C2C12 myoblasts conditionally expressing DUX4 in response to dox. 17 When dox is added to these cells, they die in large numbers over a 24 h period and ATP content is markedly reduced. Using this same screening platform, we screened 160,000 new small molecules from the UT Southwestern Chemical Library. The compounds in this library represent structurally diverse druglike small molecules that have been selected to avoid medicinal chemically undesirable characteristics, and were screened at a concentration of 5 μM.
Selection of Hits and Secondary Screening
Using a stringent z score cutoff of 5, we selected the top 640 compounds. These were transferred from the primary library onto secondary screening plates, for further study. First, the primary screen was repeated at three concentrations: 1.67, 5, and 15 μM. As expected from the relatively high cutoff, the majority of compounds repeated at the screening concentration, and were active at the other concentrations, although a subset lost activity at the lowest concentration ( Fig. 1A ).
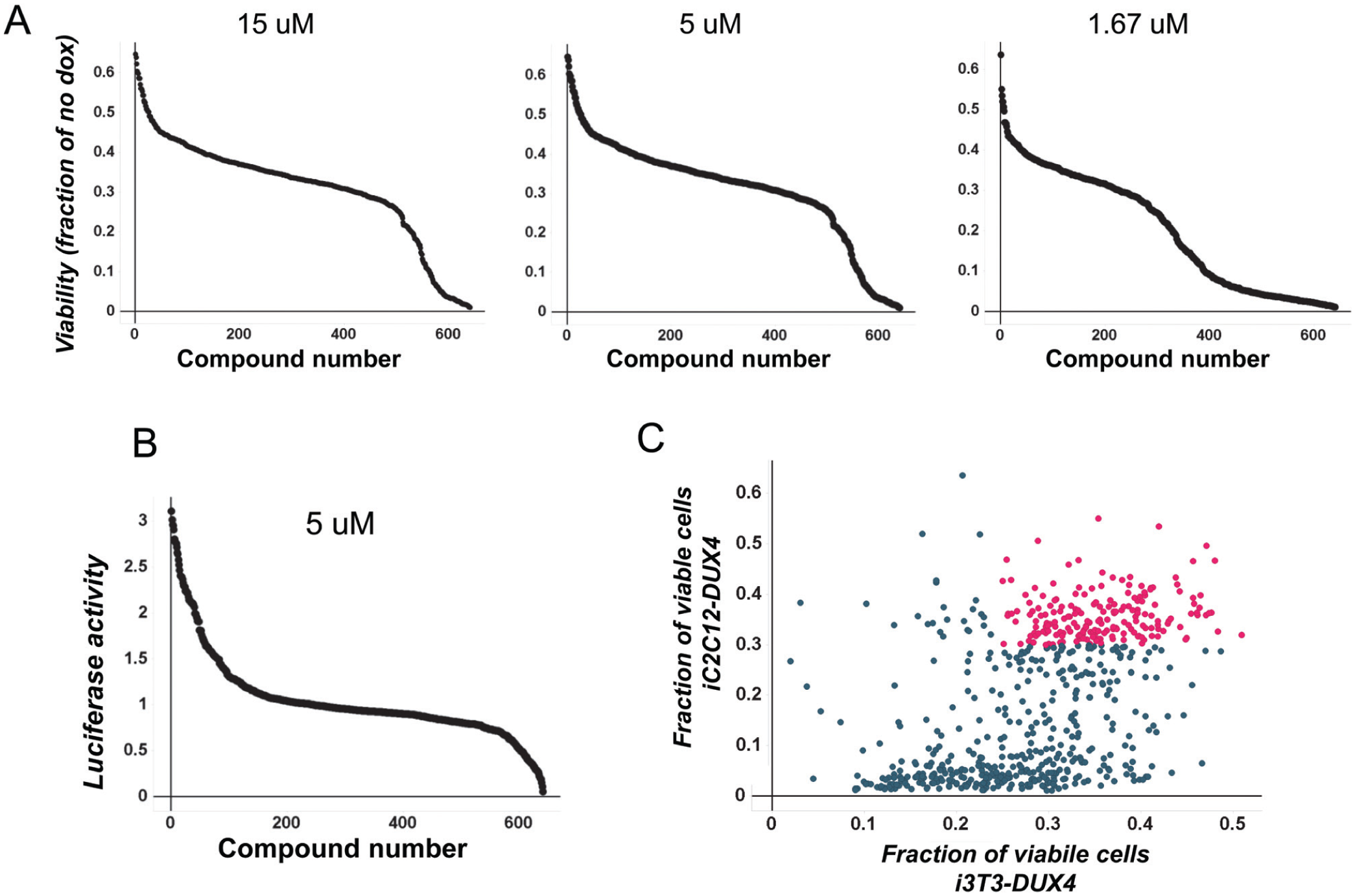
Secondary screens. (
Because we used the Tet-on system to express DUX4, some of our primary hits were expected to be indirectly protective through inhibition of the Tet-on system; that is, in their presence, DUX4 would not be expressed, or expressed at lower levels, resulting in less cytotoxicity. We therefore made use of a C2C12 cell line carrying a dox-inducible luciferase transgene. 11 A small number of compounds were found to inhibit the dox-mediated induction of luciferase by the Tet-on system in these cells, and these were eliminated from further consideration ( Fig. 1B ).
Finally, we tested each compound in 3T3 cells engineered to express DUX4 in response to dox ( Fig. 1C ). This revealed that approximately two-thirds of compounds that were active in C2C12 myoblasts were also active in 3T3 fibroblasts.
Identification of Chemical Series within the Set of Confirmed Hits
Evaluating the structures in the primary hit set, together with those of the previous screen, we identified 46 chemical series. To identify “actives” within this set, we filtered using the secondary screening data to identify compounds that were active at 1.67 μM, did not inhibit Tet-on luciferase, and were also active in both C2C12 and 3T3 cells. From the 640 hits, 193 compounds passed secondary screening. We sorted out the 193 compounds using the core definitions for these series, and selected at most three compounds from each active series. The three compounds selected were the best with respect to activity in iC2C12-DUX4 cells at 1.67 μM. No compounds were selected from core series that contained fewer than three representatives. In total, compounds from 22 of these core series were selected for follow-up from the two screens ( Fig. 2 ). Seven of these series were unique to the current screen, 10 series had representatives from both screens, and 5 only had representatives from the first phase of screening. 17
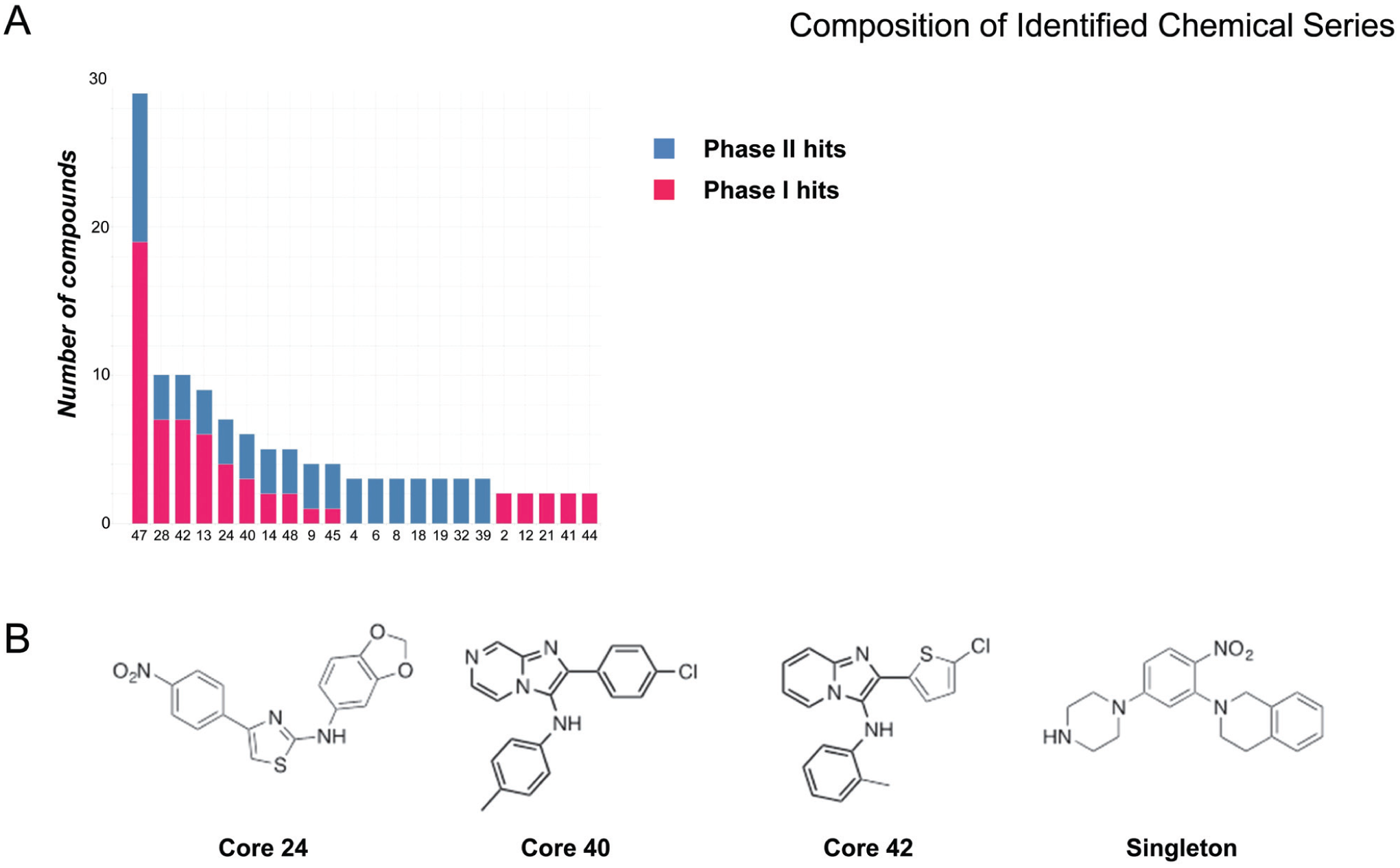
Chemical series selected for follow-up. (
We selected at most three compounds from each series (those compounds with the greatest activity, i.e., that gave the greatest percent viability) for purchase and retesting. In addition, we purchased six compounds from the singletons class for a total of 48 compounds (
Effects on Downstream Cell Death Pathways
In our previous application of this screen to a smaller library using a lower z-score threshold, we determined that a large subset of the hit compounds protected cells indirectly, by interfering with a cell death pathway involving oxidative stress. To determine to what extent compounds identified through this more stringent and much larger screen were acting at the level of downstream pathways versus acting at the level of DUX4 itself, we tested each purchased compound for activity against six different cell death–inducing insults. No compounds were protective against the Bcl2 inhibitor, ABT-263 ( Fig. 3A ); the broad kinase inhibitor, staurosporine ( Fig. 3B ); the Ca permeabilizer, ionomycin ( Fig. 3D ); or the endoplasmic reticulum stress inducer, tunicamycin ( Fig. 3F ). As seen in the previous screen, the great majority (83%) of identified compounds were protective against the oxidative stress–inducing agent tBHP ( Fig. 3E ). Interestingly, the more stringent cutoff (z ≥ 5 vs z ≥ 3) seems to have increased the likelihood of identifying inhibitors in this pathway (in the previous screen, 60% of compounds acted in the oxidative stress pathway). The current screen has also identified two compounds (SHC23 and 38) that protect against etoposide, a DNA double-strand break-inducing compound ( Fig. 3C ). Interestingly, these two compounds are also protective against oxidative stress, although not against other cell death pathways.
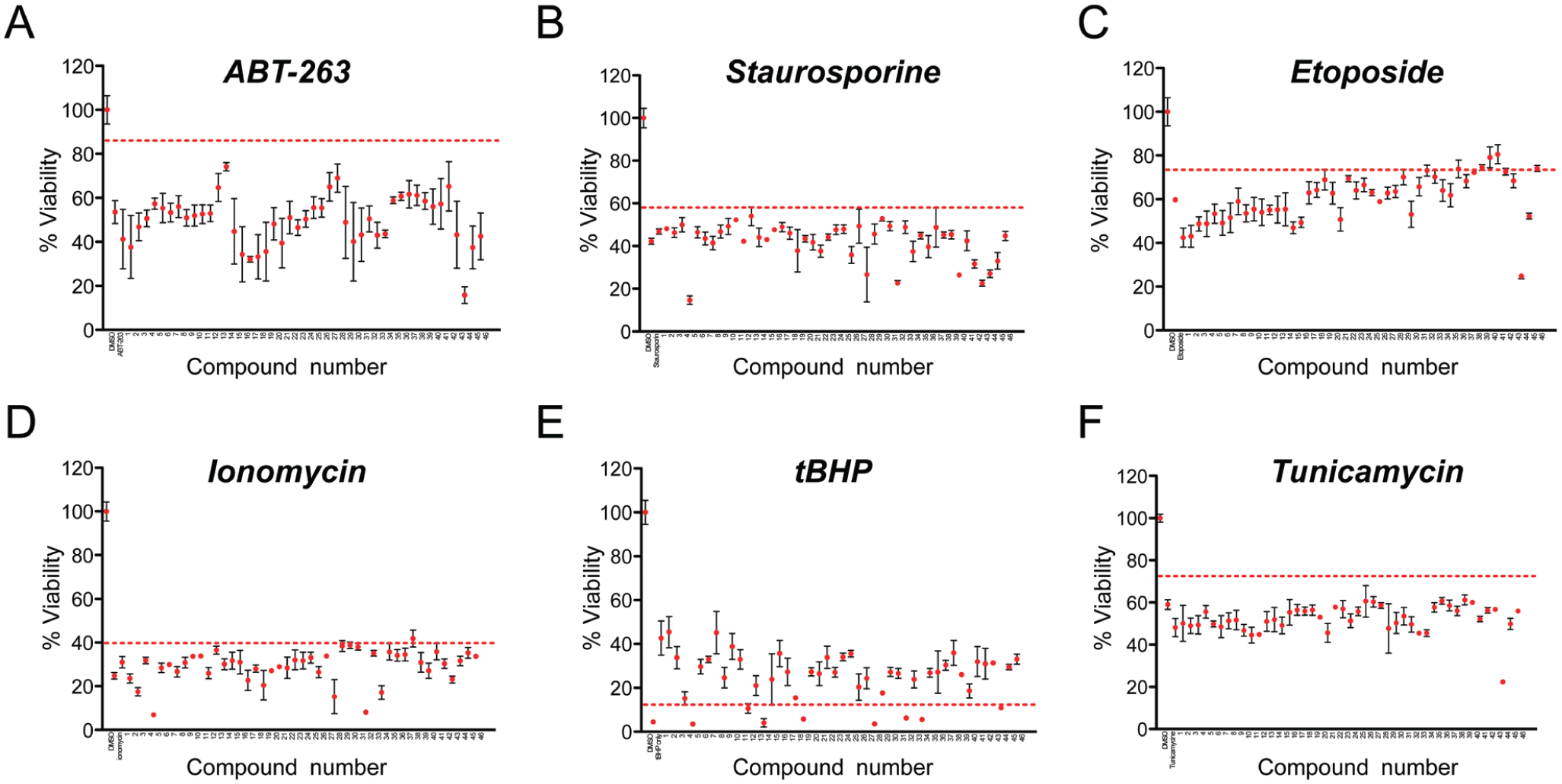
Protection from other cell death–inducing pathways. WT C2C12 cells were exposed to cell death–inducing drugs acting on various pathways in the presence of 5 µM purchased compounds. Viability (ATP content) is shown on the y axis. The first two points in each series represent controls: cells not treated with toxic agent, followed by cells treated with toxic agent plus DMSO alone. Compounds tested for protection are indicated from 1 to 46 on the x axis, error bars = SEM. A cutoff of three standard deviations above control (toxic agent + DMSO alone) is shown as a dotted red line. (
Effects on Transcription of DUX4 Targets
Compounds that directly inhibit DUX4 would not be expected to protect against other cell death–inducing toxins. Because both screens produced a set of compounds that did not have activity in these cell death–inducing insults, we wished to determine whether compounds in this subset were acting directly at the level of DUX4. A more direct readout of DUX4 activity is transcriptional changes in DUX4 target genes. We previously identified Myo1g as one of the most rapidly and potently upregulated mouse targets. 11 We therefore tested each compound for its ability to inhibit Myo1g upregulation by DUX4. For this work, we included the compounds previously identified from the 44,000-compound library screen, 17 which are here referred to as compounds 47–100. Three compounds were potent inhibitors of DUX4-induced Myo1g upregulation, and a number of others had a modest effect ( Fig. 4A ). In addition to upregulated target genes, there are some genes that are downregulated by DUX4, and we have shown previously that one of the strongest of these is Myod1. 11 This effect is likely not secondary to the upregulation of a repressor of Myod1, as Myod1 is also a downregulated target of DUX4c, a variant of DUX4 lacking the C-terminal transcriptional activation domain, but rather represents an independent activity of DUX4. 19 Thus, we further tested all 100 purchased compounds for effects on Myod1 downregulation by DUX4 ( Fig. 4B ). One compound (SHC98) prevented both Myo1g activation and Myod1 repression. Two others (SHC40 and 75) prevented Myo1g activation only, and some others had weak effects. Although all of these purchased hits had previously been through a secondary screen for effects on the Tet-on system, we also evaluated levels of DUX4 transcript in the presence of selected inhibitors to determine whether the lack of effects on DUX4 target genes might have the trivial explanation of blocking the Tet-on system. Remarkably, all three compounds that inhibited Myo1g expression showed reduced dox-induced DUX4 expression, with the most potent being SHC98 ( Fig. 4C ). Thus, it is most likely that these three compounds do not directly inhibit the activity of the DUX4 protein, but rather prevent its expression by inhibiting Tet-on-driven transcription.
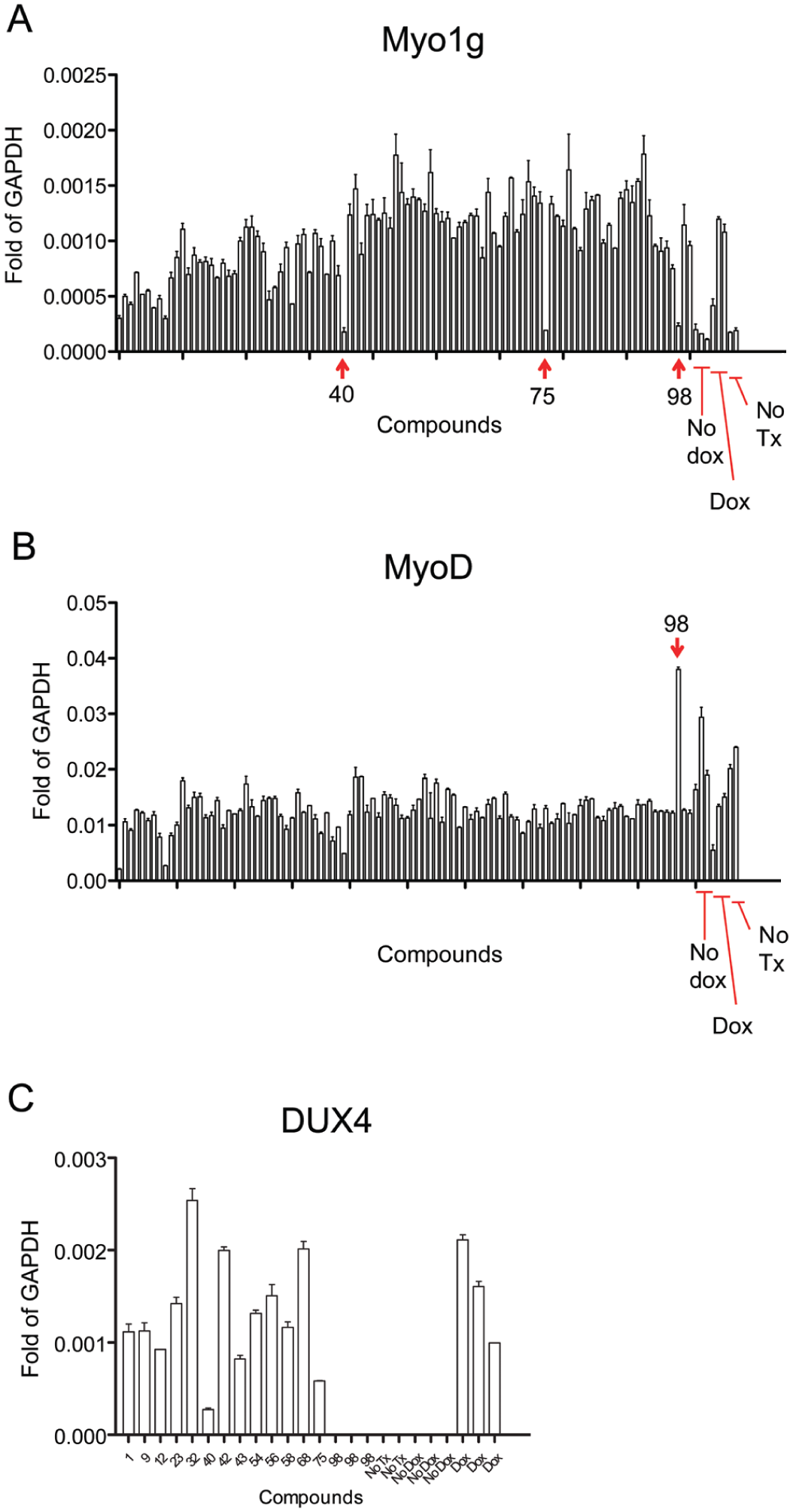
Transcriptional responses to DUX4 in the presence of selected repurchased compounds. (
Secondary Screen Based on Target Gene Expression Changes
Because none of the purchased compounds were able to prevent gene expression changes (except for those that prevented Tet-on-driven DUX4 expression itself), we went back the 640-compound hit set and performed one final secondary screen. In 384-well format, we tested every compound for its ability to inhibit DUX4-induced Myo1g upregulation. Compounds that prevented Myo1g upregulation ( Fig. 5A ) were then retested in conventional qRTPCR format ( Fig. 5B ). Because DUX4 downregulates MyoD in a manner that does not require its transcriptional activation domain,11,20,21 we also tested these compounds for potential relief of Myod1 repression ( Fig. 5C ). We purchased 10 of these compounds, including SHC161, a negative control determined to be a strong inhibitor of the Tet-on system, and tested this set of compounds first for effects on the induced DUX4 transcript ( Fig. 5D ). The Tet-on inhibitor control (SHC161) completely prevented DUX4 expression, as expected. Of the others, four showed moderate inhibition of the DUX4 transcript and five had no apparent effect on induced DUX4 expression. As an independent test of interference with the Tet-on system, we tested each compound over a dose series for inhibition of luciferase in the dox-inducible iC2C12-luc cell line ( Fig. 5E ). From this set, three compounds (SHC351, 540, and 572) were found not to inhibit the Tet-on system at the highest dose tested. Finally, we evaluated the potency of each of these repurchased compounds in the cell death protection assay. All three showed a dose-dependent inhibition of DUX4-mediated cytotoxicity ( Fig. 5F ).
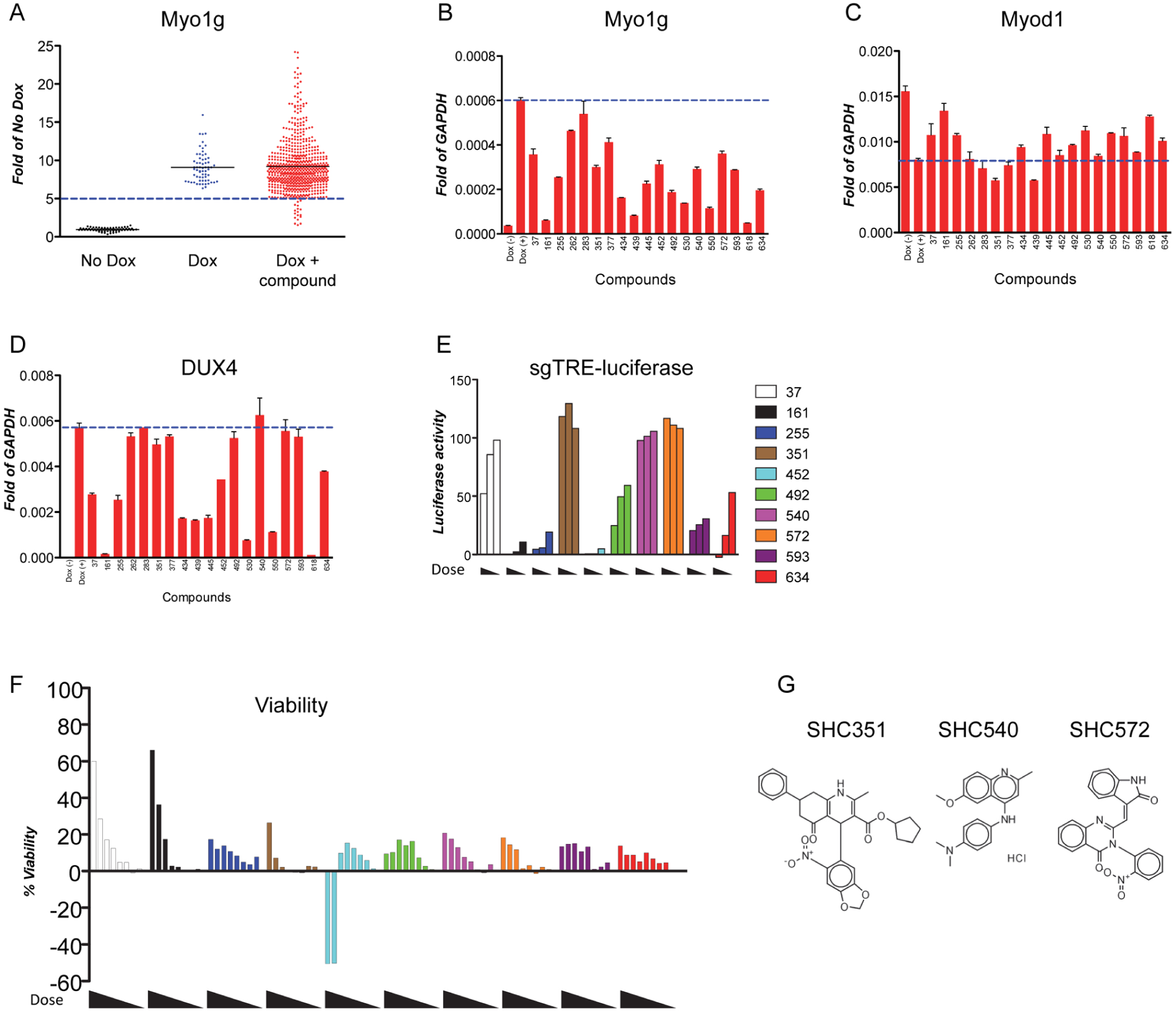
Identification of compounds with effects on transcription. (
We evaluated these three compounds for their performance in our secondary screens, and found that all failed the relatively stringent viability cutoff (30% viability) that we set in the first screen, the repeat of the primary assay. Compound SHC540 came the closest to passing (0.28). Interestingly, it passed the i3T3-DUX4 screen. Of the three compounds, only SHC351 belonged to a defined chemical series (series 4); however, no other members of this chemical series had been identified as actives after secondary screening, from either the 44,000 or 160,000 screen.
Discussion
DUX4 is the prime therapeutic target in FSHD. The rapid cell death caused by DUX4 in C2C12 myoblasts facilitates this system for screening of small-molecule inhibitors of DUX4 activity. A previous screen revealed that most compounds identified in this way were protective against oxidative stress,
17
which together with previous data on the pathological activity of DUX4,
11
as well as oxidative stress readouts in patient myoblasts
22
and in patients themselves,
23
highlights the importance of this pathway in the cell death phenotype. Likewise, this screen of a much larger library necessitating a more stringent hit cutoff identified many inhibitors of oxidative stress–induced cell death. In addition, two of the new oxidative stress protectors also protected against etoposide, a DNA damage–inducing agent. This may be due to linkage of these two pathways or to a more general anti–cell death activity, although these compounds were inactive against a host of other death-inducing insults. Of those compounds that did not act in any of the tested cell death pathways, it remained possible that some acted on DUX4 directly. However, of the 100 repurchased compounds, only 3 had activity against DUX4-induced Myo1g expression, indicating that the remainder were not active against DUX4 per se, but rather acted on downstream pathways. Outside of oxidative stress and DNA damage, what these pathways may be is unknown. For the three that did reduce Myo1g expression, they turned out to inhibit DUX4 transcription and, indeed, transcription of a dox-induced green fluorescent protein transgene (
We prioritized compounds that were identified in chemical series for purchasing and validation. This assumes that structures of the desired activity are sufficiently frequent to be represented multiple times in a 200,000-compound library. However, it has become recognized that transcription factors, perhaps because they generally do not have enzymatic active sites, are particularly difficult to target with small molecules, with some even labeling this class of protein essentially “undruggable.” With the recognition that our secondary screens severely refined the hit set, and that structures with the desired activity may be extremely rare, we went back to our 640-compound hit set and performed an alternative secondary screen, looking for compounds with activity against DUX4-mediated gene expression. Recognizing that such a screen would identify inhibitors of the Tet-on system, we then filtered the active compounds to remove those with activity against dox-induced DUX4 mRNA. This eliminated the majority, leaving three interesting compounds, discussed in more detail below.
Altogether, this study identified seven inhibitors of the Tet-on system: from the prioritized and purchased 100 compounds, SHC40, 75, and 98, and from the rescreened 640 phase II hit set, SHC161, 255, 452, and 634. There is no structural similarity within this set; however, three compounds are classified as PAINS:24,25 SHC75 and 98 were recognized by the original filters 18 and SHC634 because it has the potential to function as an irreversible inhibitor, which may react with cysteines. 26
This left three compounds, SHC351, 540, and 572, which protected cells from toxicity of DUX4, did not interfere with the Tet-on system, and prevented DUX4-mediated target gene upregulation. Looking at the structures of these compounds, one of these, SHC540, is classified as PAINS, and both SHC540 and 572 are predicted to be promiscuous toward targets using BADAPPLE (http://pasilla.health.unm.edu/tomcat/badapple/badapple). SHC540 was originally developed as an antimalarial, but its mode of action is not documented in the literature. 27 SHC351 is notably absent from the literature (SciFinder, 2016) though similar compounds have been reported as active against several diverse targets.28,29 Compound SHC572 is also absent from the published literature but, as is the case for all of these compounds, is available from compound vendors. Related compounds are known in the literature and have evidenced biological activity. 30 Whether any of these compounds interact with DUX4 itself versus factors necessary for DUX4 activity is presently unknown. It will be important to test these compounds on FSHD myoblasts. Because none of these three compounds strongly prevented DUX4-mediated downregulation of Myod1, it seems unlikely that they are completely inactivating DUX4, but rather acting in a way that is specific to transcriptional activation, for example, by antagonizing a coactivator, or the interaction with a coactivator. As much remains to be learned about mechanisms of transcriptional activation by DUX4, these compounds, which are commercially available, may be useful reagents to investigate this aspect of DUX4 function.
Footnotes
Acknowledgements
We thank the Dr. Bob and Jean Smith Foundation for their generous support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the NIH, R21 NS076671 and R01 AR055685; the Friends of FSH Research; the FSHD Global Research Foundation; and the FSH Society. D.B. was supported by a Muscular Dystrophy Association Development Grant (MDA 4361) and by a Marjorie Bronfman Fellowship and a research grant from the FSH Society (FSHS-MGBF-016 and FSHS-82010-01, respectively).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.