
Abstract
Abnormal accumulation of β-catenin protein, a key transcriptional activator required for Wnt signaling, is the hallmark of many tumor types, including colon cancer. In normal cells, β-catenin protein level is tightly controlled by a multiprotein complex through the proteosome pathway. Mutations in the components of the β-catenin degradation complex, such as adenomatous polyposis coli (APC) and Axin, lead to β-catenin stabilization and the constitutive activation of target genes. Since the signal transduction of Wnt/β-catenin is mainly mediated by protein–protein interactions, this pathway has been particularly refractory to conventional target-based small-molecule screening. Here we designed a cellular high-content imaging assay to detect β-catenin protein through immunofluorescent staining in the SW480 colon cancer cell line, which has elevated β-catenin endogenously. We demonstrate that the assay is robust and specific to screen a focused biologically diverse chemical library set against known targets that play diverse cellular functions. We identified a number of hits that reduce β-catenin levels without causing cell death. These hits may serve as tools to understand the dynamics of β-catenin degradation. This study demonstrates that detecting cell-based β-catenin protein stability is a viable approach to identifying novel mechanisms of β-catenin regulation as well as small molecules of therapeutic potential.
Keywords
Introduction
Wnt/β-catenin signaling regulates a diverse array of biological functions, including tissue homeostasis and tumorigenesis.1,2 While the diverse biological function is likely a manifestation of the large array of extracellular ligands and receptors that are expressed in various combinations in different tissues, the pathways converge, at least for the canonical pathway, at the level of β-catenin protein regulation.1,2 The cellular β-catenin protein level is normally tightly controlled by a multiprotein scaffold, including adenomatous polyposis coli (APC), Axin, and GSKα/β, which targets β-catenin for efficient degradation through the proteasome-mediated pathway. Mutations in the components of the β-catenin degradation complex, such as APC and Axin, or mutations in β-catenin itself, can lead to β-catenin stabilization and constitutive activation of target genes through its interaction with transcription factors TCF/LEF. The hyperactivation of the β-catenin pathway, in conjunction with mutations in the other cell growth regulatory genes, underlies tumor formation in a number of tissues, particularly in colon cancer.3–5 For example, 90% of colorectal cancer is associated with inactivating mutations in APC. And a hallmark feature of this type of tumor is the elevated level of the intracelullar β-catenin protein.
There is significant interest in searching for the chemical modulators of the Wnt pathways given the prominent role of Wnt/β-catenin signaling in normal development and human disease. Identification of tools to probe this pathway will help us understand the mechanism of signal transduction, and ultimately lead to the discovery of therapeutic agents for disease intervention.6–12 This has been particularly challenging because the key components of the Wnt pathway function through protein–protein interaction, a process that is proven difficult to perturb by small-molecule inhibitors. Therefore, using cell-based assays based on the distinctive cellular response of the Wnt pathway has gained popularity. Using cell lines incorporating a TCF promoter driving the luciferase reporter gene, two groups identified small-molecule inhibitors XAV969 and IWR that specifically promote β-catenin degradation.6,8 The cellular target for XAV969 and IWRs is tankyrase (TNKS), an enzyme that regulates Axin protein stability by promoting its ubiquitylation and degradation. Inhibition of TNKS by XAV969 therefore leads to Axin stabilization and β-catenin degradation. TNKS belongs to a large family of poly(ADP-ribose)polymerase (PARP) that transfers poly(ADP-ribose) to its acceptor protein substrates participating in diverse biological functions, including DNA repair. While the specificity of XAV969 and IWRs on TNKS remains to be defined, the TNKS itself is nonetheless not a dedicated component in β-catenin signaling. 13
To directly target β-catenin accumulation, here we took advantage of the human colon cancer cell lines, including SW480, which display an elevated β-catenin protein level due to mutations in the β-catenin pathway components. We have developed a 384-well high-content imaging-based screen using immunofluorescence to measure endogenous β-catenin protein abundance. We showed that the assay is specific and robust, and sufficient to screen a focused chemical library (~5000 compounds). 14 We identified a number of hits that reduce β-catenin levels without causing obvious cell death. This study demonstrates that the cell-based protein stability assay is a viable approach to identify novel mechanisms of β-catenin regulation as well as small molecules of therapeutic potential.
Materials and Methods
Cell Lines and Tissue Culture
Human colon cancer cell lines RKO (CRL-2755), HT1080, HCT116 (CCL-274), and SW480 (CCL-228) were derived at GlaxoSmithKline (GSK) from ATCC (Manassas, VA). The culture was expanded in RPMI-1640 media (supplemented with 10% fetal bovine serum [FBS] and 1% Glutamax) and cryopreserved in 1 mL aliquot of 5 × 107 cell density at −190 °C. The frozen cells were thawed and diluted into desired density in RPMI-1640 media with 1% FBS for plating on compound plates on the day of the assay.
Reagents
The ligands Wnt3a (cat. 1324-WN) and DKK1 (cat. 5439) were purchased from R&D (Minneapolis, MN). The rabbit anti-β-catenin antibody (C2206; Sigma, St. Louis, MO), goat-anti-rabbit secondary antibody labeled by Alexa Fluor 532 (A11009; Invitrogen, Grand Island, NY), and counterstain SYTO 63 red fluorescent nucleic acid stain (S11345; Invitrogen) were purchased. Ten percent bovin serum albumin (BSA) blocker solution was from Thermo Scientific (Waltham, MA; 37525).
Compound
GSK chemical compounds, TNKS inhibitors, and the fluorescent polarity (FP) ligand were synthesized by the Discovery Medicinal Chemistry Department (Research Triangle Park, NC) at GlaxoSmithKline. All compound stocks were prepared in DMSO at 10 mM.
Antibodies and Immunofluorescent Staining
Immunofluorescent staining of β-catenin is based on the standard protocol. Briefly, the SW480 cells from frozen stock are diluted in RPMI-1640 media supplemented with 1% FBS and plated at 5000 cells/well in 384 black-walled, clear-bottom poly-
Image Capture and Analysis
The β-catenin immunofluorescent images were captured by an Opera HCS system (PerkinElmer, Waltham, MA) with one exposure, and two lasers (532 nM for Alexa 532 and 635 nM for SYTO 63) were done as described 15 using 10× objectives. The instrument’s Acapella software image analysis algorithm parameters were optimized to select an object and quantify the signal outputs. The SYTO 63 counterstain signal was used to mask all cells and identify the cytoplasmic, membrane, and nuclei regions. The laser power and exposure times for image quality were adjusted to ensure maximum signal without saturation and cross talk between color channels. Three fields (about 700 cells/field) for each well were imaged. The median intensity was calculated based on the average intensity within the cytoplasm region or the membrane or the nuclei region per cell. The cell count for each field and the cell size (not shown) parameters were also captured, together with the fluorescent intensity. It takes approximately 20 min to complete the image acquisition for one 384-well plate.
Screen and Data Analysis
The library used in this screen is an internal collection of GSK. For the screen, the biologically diverse compound set (BDCS) of 5857 compounds were transferred to a 384-well Greiner plate at 300 nL/well using an Echo acoustic dispenser (Labcyte, Sunnyvale, CA). Column 6 (all 16 wells) contained DMSO only (low control, 0% inhibition). Column 18 (16 wells) contained TNKS inhibitor XAV at 1 mM concentration in DMSO (high control, 100% inhibition). Cells (30 µL) in the culture media were plated on compound plate (1:100 dilution of the compounds, 1% DMSO final concentration). Compounds were tested as dose response starting at a stock concentration of 5 or 10 mM and diluted serially threefold across 11 points, also in DMSO. The relative fluorescent intensity was normalized to the median fluorescent intensity of the control DMSO wells (0% inhibition) and median fluorescent intensity of the TNKS inhibitor–treated control wells (100% inhibition). The single-shot screen and dose–response curves of compounds were analyzed with AbaseXE (IDBS, Surrey, UK) and visualized by Spotfire (Tibco Software, Inc., Palo Alto, CA). Some dose–response curves were also analyzed by GraphPad software (La Jolla, CA). Z′ calculation was done with the following equation:
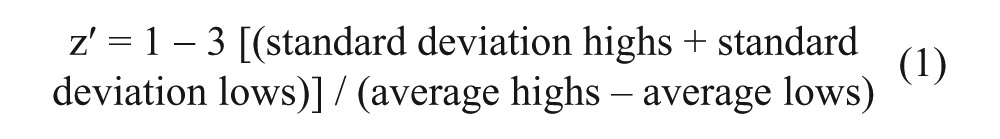
Robust Z′ values were calculated using the 16 wells of high and low control columns, where up to two outlier values were eliminated for the robust values.
FP Binding Assay for TNKS
FP assay was performed as described previously 8 using the Rhodamine-green-labeled XAV (made in-house) as the ligand for TNKS. The FP buffer contained 50 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM CHAPS, 1 mM DTT, 50 mM NaCl, 1 nM Rhodamine-green XAV, and 5 nM of TNKS1 enzyme. The compounds were plated at 0.1 µL with 100% DMSO in black polypropylene plates (NUNC 2677461). The enzyme/ligand mixture solution was plated at 10 µL/well in 384 wells using Multidrop Combi (Thermo Electron Corporation). After 2 h incubation in the dark, the plates were read using the Analyst GT reader from Molecular Devices (Sunnyvale, CA). The mP data were used in the 11-point dose–response curve fit using AbaseXE (IDBS) to calculate the compound IC50 data.
Results
Verify the Specificity of β-Catenin Antibody against Endogenous β-Catenin in Cancer Cell Lines
When detecting the endogenous signal, it is critical to verify the specificity of the antibody. In most cells, the endogenous β-catenin abundance can be increased by stimulation with its cognate soluble ligand Wnt, which is detectable by Western blot using an antibody specific to β-catenin (e.g., Zeng et al. 16 ). To demonstrate the immunohistochemical staining of β-catenin abundance is specific, we first tested RKO cells that were treated with known native extracellular modulators or small molecular tool compounds of the Wnt pathway. RKO is a colon cancer cell line with no mutations in the β-catenin pathway, and thus it has normal low levels of β-catenin protein. As expected, the immunofluorescent signal for β-catenin is undetectable in untreated cells. However, upon treatment with soluble ligand Wnt3a for 2 h, the β-catenin antibody staining signal is strongly enhanced two- to threefold ( Fig. 1A ). Upon quantification of the fluorescent intensity, the EC50 of Wnt3a on β-catenin accumulation is at 10 ng/mL ( Fig. 1B ). This effect is eliminated with the co-incubation of Wnt3a with its extracellular antagonist Dickkopf ( Fig. 1A ). 17 The IC50 of DKK1 inhibition in the presence of 100 ng/mL of Wnt3a is around 10 ng/mL ( Fig. 1C ). These data confirmed the specificity of the β-catenin antibody staining, which detects the endogenous β-catenin protein level in RKO cells that is modulated by Wnt3a. To further characterize the β-catenin antibody, we turned to three other cancer cell lines: SW480 (colon), HCT116 (colon), and HT1080 (fibrosarcoma, noncolon control). SW480 is known to carry APC mutations, which disrupt the scaffolding function of the APC protein and prevent β-catenin from phosphorylation and degradation. HCT116 has mutations in β-catenin on the GSK3β phosphorylation site, preventing it from recognition by the APC/Axin/GSK3β complex and degradation. As expected, SW480 ( Fig. 2D ) and HCT116 ( Fig. 2C ) showed an elevated basal level of β-catenin when comparing with RKO ( Fig. 2A ) in the absence of Wnt. After treatment with Wnt3a for 2 h, the β-catenin signal was increased by two- to threefold in RKO cells, as in Figure 1 , whereas HCT116 showed minimum change ( Fig. 2C , E ). Interestingly, SW480 still showed about 1.5-fold upregulation of β-catenin with Wnt3a treatment ( Fig. 2D , E ), suggesting that the β-catenin protein was not at saturated levels, even in the presence of APC mutations. Other components of the scaffolding complex, such as Axin, might help to keep the β-catenin level under check, which can be relieved by Wnt3a treatment. The control line, HT1080, shows a high basal level of β-catenin and is weakly responsive (less than onefold) ( Fig. 2B , E ) to Wnt ligand treatment, as reported. 18
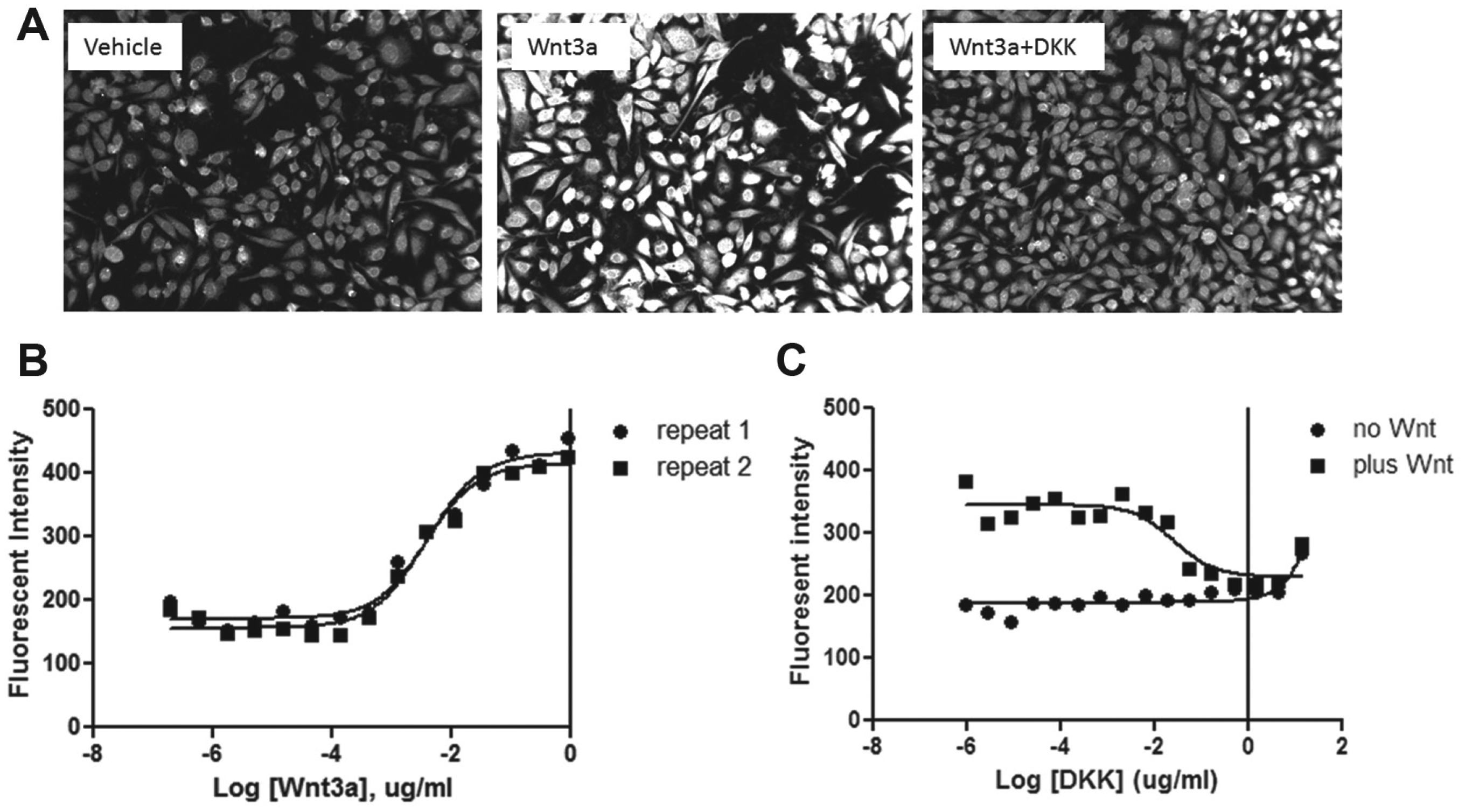
Verify the specificity of the β-catenin antibody in RKO cells in response to ligand treatment. (
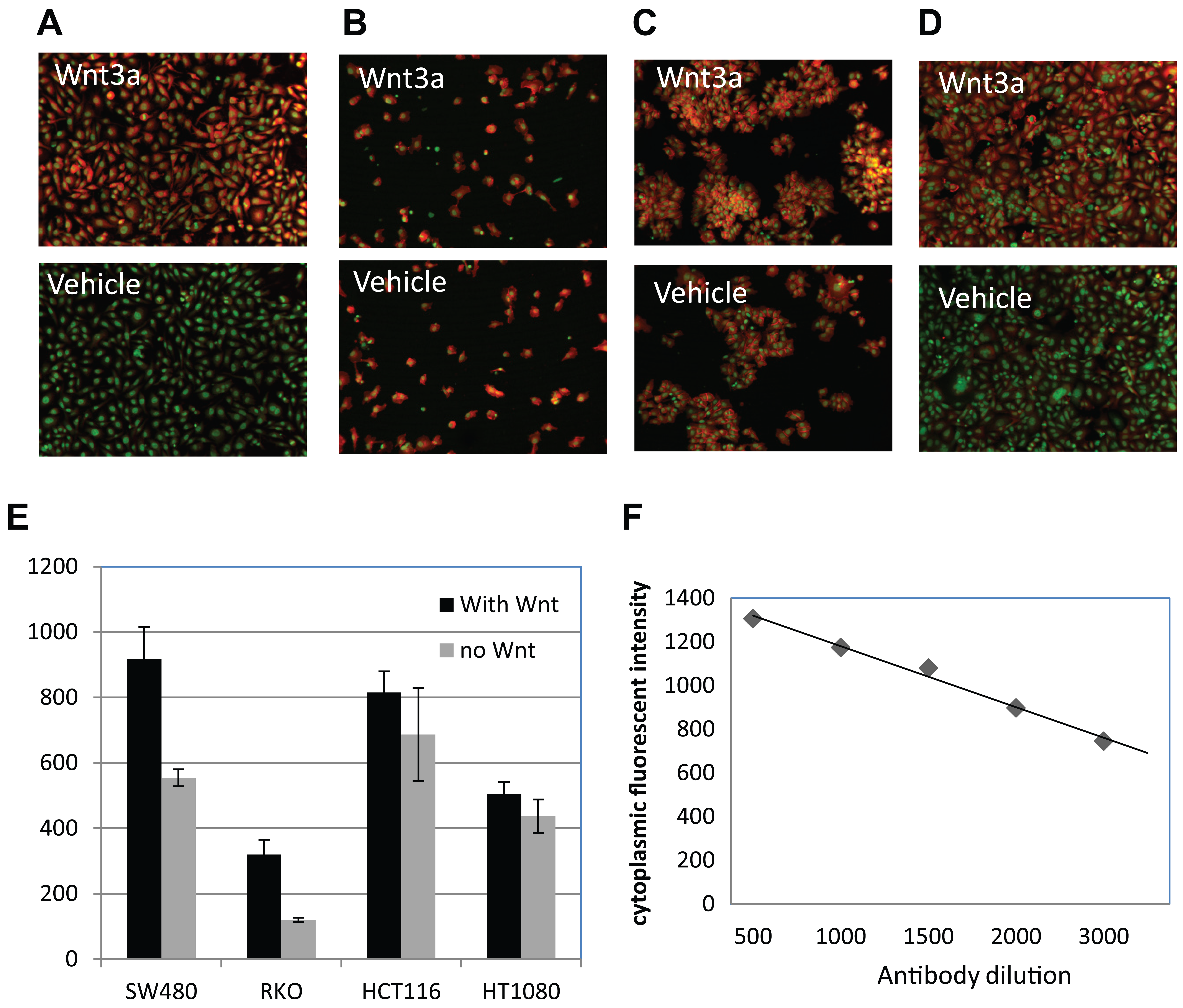
Validate the β-catenin antibody in cancer cell lines. The color overlay images show the β-catenin staining (red) and the SYTO 63 counterstain (green) in (
In addition to its role in the Wnt pathway, β-catenin is also a key component in the cell–cell adhesion through its association with the E-cadherin complex on the cell membrane. 19 It has been speculated that only the cytoplasmic pool of β-catenin is mobilized by Wnt signaling. It also has been reported that the β-catenin protein available for interaction with TCF in the nucleus could be regulated by cytoplasmic and nuclear shuttling.20,21 We tried to modify the fluorescence imaging analysis algorithm to quantify the signals from the cytoplasm, nucleus, and membrane pools separately based on SYTO 63 staining but failed to detect a significant difference in the ratio change between different subcellular compartments upon Wnt3a treatment in RKO or SW480 cells (data not shown). Therefore, all quantitative analysis output here represents the total signal from the cell body (cytoplasm plus nuclei). Given that SW480 shows high endogenous β-catenin signaling and APC mutations are directly relevant to human cancer, we decided to choose SW480 cells to proceed with the assay development to screen for small-molecule inhibitors of β-catenin stability. To optimize the signal-to-background ratio, we titrated the β-catenin antibody from 1:100 to 1:5000 and showed that the signal detection is linear without saturation ( Fig. 2F ). We chose 1:1000 dilution of the β-catenin antibody for optimum signal detection.
Assess the Activities of TNKS Inhibitors in β-Catenin Imaging Assay in SW480 Cells
To understand the robustness of the β-catenin protein imaging assay for compound screening, we tested TNKS inhibitors (XAV and IWR and their derivatives). These inhibitors were previously shown to reduce cellular β-catenin abundance by stabilizing Axin in Wnt-stimulated cells or cancer cells. However, since SW480 cells harbor APC mutations, it is unclear if stabilization of Axin by TNKS inhibition can override the effect of APC mutation. As shown in Figure 3 , when SW480 cells were treated with TNKS inhibitors for 24 h, β-catenin was reduced in a dose-dependent manner. This effect is not due to cytotoxicity, because as shown in Figure 3B , the β-catenin staining is significantly reduced by treatment of IWR I, yet the total cell number is not affected. We then further tested, in full dose response, a set of TNKS inhibitors (~70) that are derived from the three chemotypes represented by IWR I and II or XAV. As shown in Figure 3C , there is a good correlation between the pIC50 values of TNKS biochemical FP activity with the β-catenin protein reduction across all three chemotypes, confirming the effect of TNKS inhibition on β-catenin protein degradation, even in the presence of APC mutations. These data prove that the SW480-based β-catenin imaging assay is of sufficient sensitivity and displays the desired pharmacology and therefore is suitable for small-molecule screening.
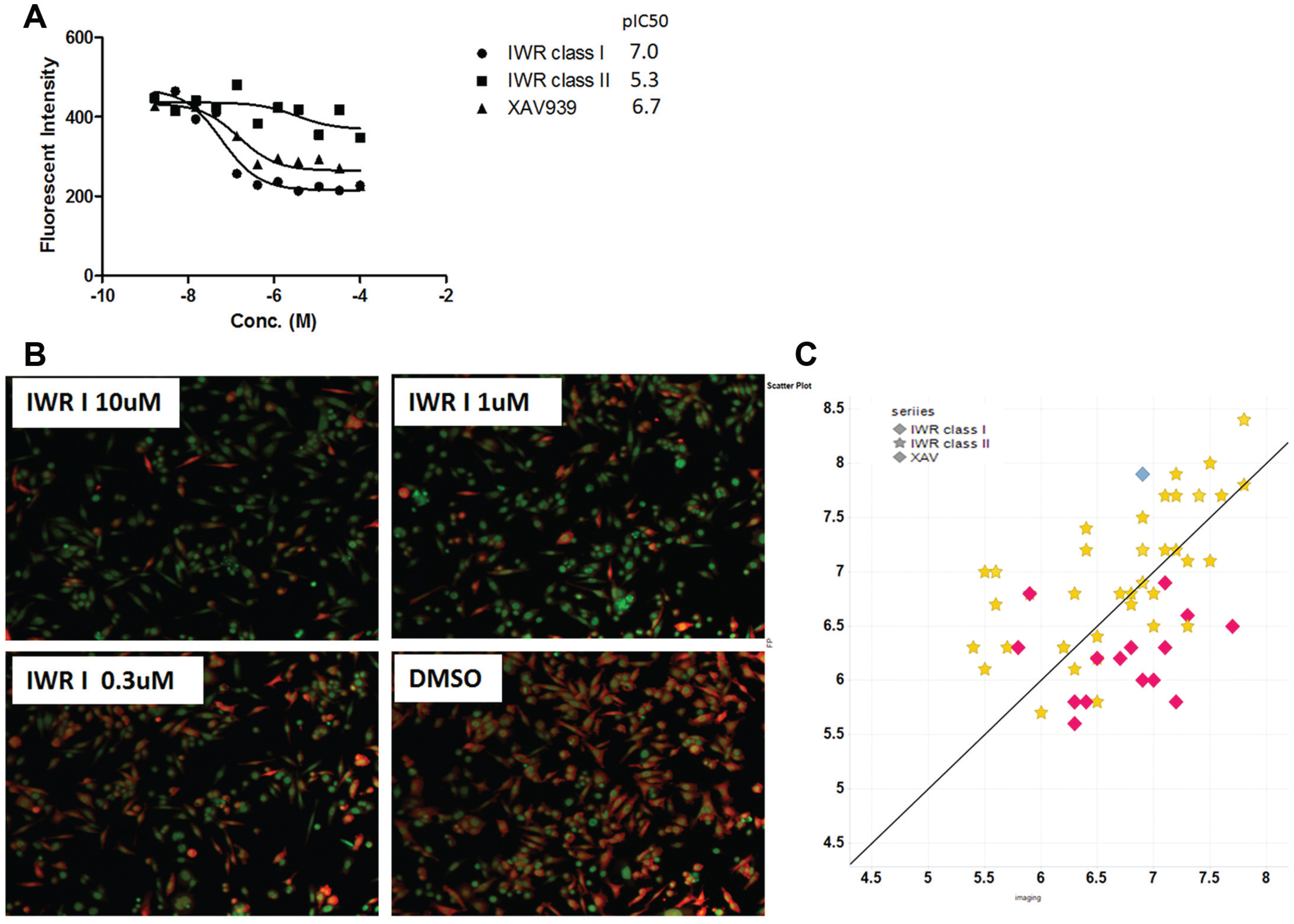
Test TNKS tool compounds in SW480 cells using the β-catenin imaging assay. SW480 cells were incubated with TNKS tool compounds for 24 h in a 384-well plate prestamped with compounds. (
Chemical Genetics Screen Using BDCS
To identify potential intracellular regulators and the small-molecule inhibitors of β-catenin protein stability, we screened a BDCS, an internal collection of a focused library of ~5000 compounds targeting ~700 unique proteins that play diverse cellular functions. Each target is represented by up to 10 selective compounds. The compounds were screened at single dose of 10 µM (final concentration) in SW480 cells (1% DMSO) using the β-catenin immunofluorescence as the readout in a 384-well format. After 24 h of treatment, the plates were processed for imaging assay. The response data from each compound are normalized to the negative control (DMSO, 0% inhibition) and the positive control (IWR3 at 10 µM, 100% inhibition) ( Fig. 4A ). The average Z′ across 15 assay plates was 0.4, which is typically seen in other cell-based imaging assays. The average robust cutoff is 46.2%, resulting in a hit rate of 9.8% (500 compounds), which is on the higher side but reasonable, given that this is an enriched set of compounds with biological activities. To remove the false positives caused by cytotoxic compounds, we filtered out compounds with a cell count reduction of 25% (arbitrary) or more ( Fig. 4B ). This left us with 155 hits, which were followed up by confirmation in full dose response. As expected, 8 out of the 10 TNKS inhibitors were identified, validating the robustness of the screen. Also included in the hit list are 7 (out of 10) PAK1 inhibitors.
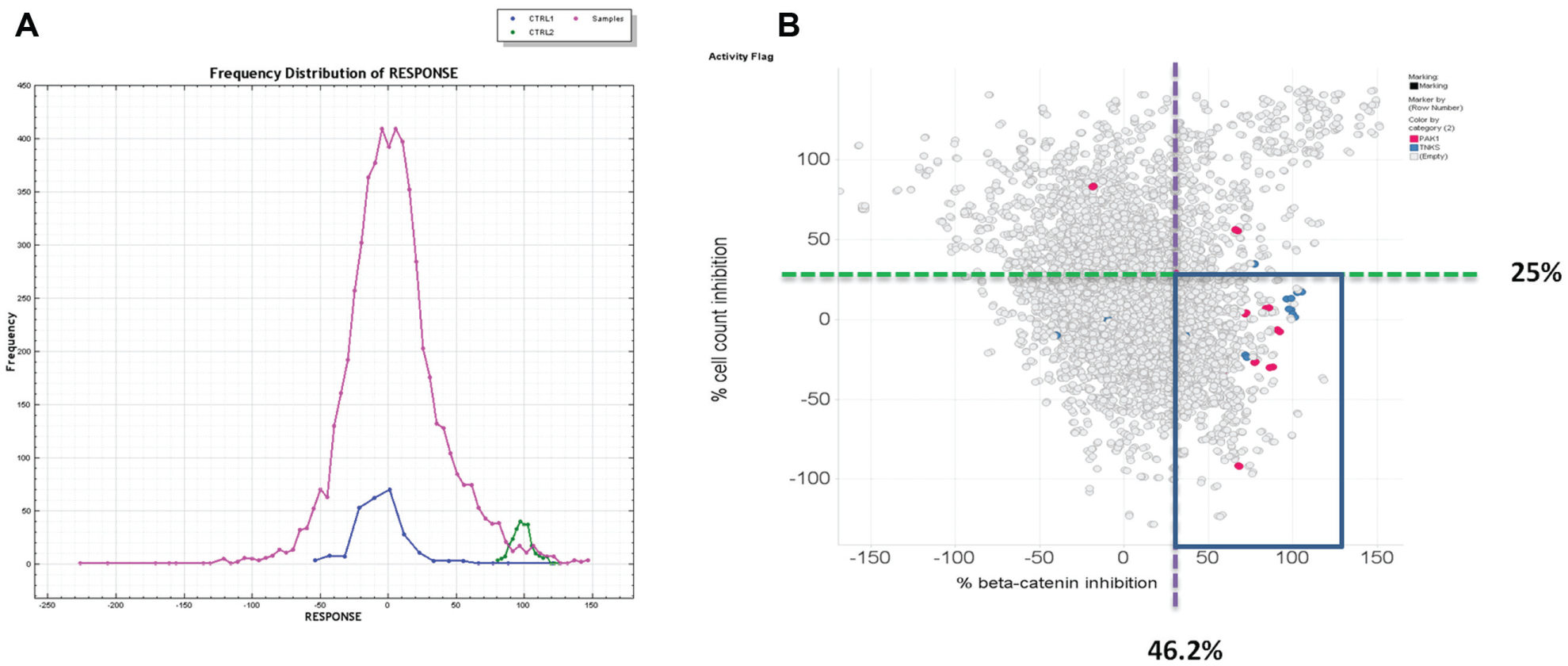
Hit distribution of BDCS screen in β-catenin imaging assay. (
The 155 hits were further evaluated by dose–response testing at 1:3 serial dilution for 11 points starting at a 100 µM top concentration. Seventeen compounds generated a dose-dependent inhibition curve fit and exhibited a pIC50 of >5.5 ( Fig. 5 ). Among these hits, other than TNKS inhibitors, five PAK1 inhibitors and one PAK2 inhibitor showed a pIC50 close to 6 or above, confirming the single-dose results. In addition to PAK1, two inhibitors of PKA catalytic subunit A (PKACA) were also identified with pIC50s above 6. We decided to focus on PAK and PKA inhibitors since each target was represented by more than two compounds in the hit list.
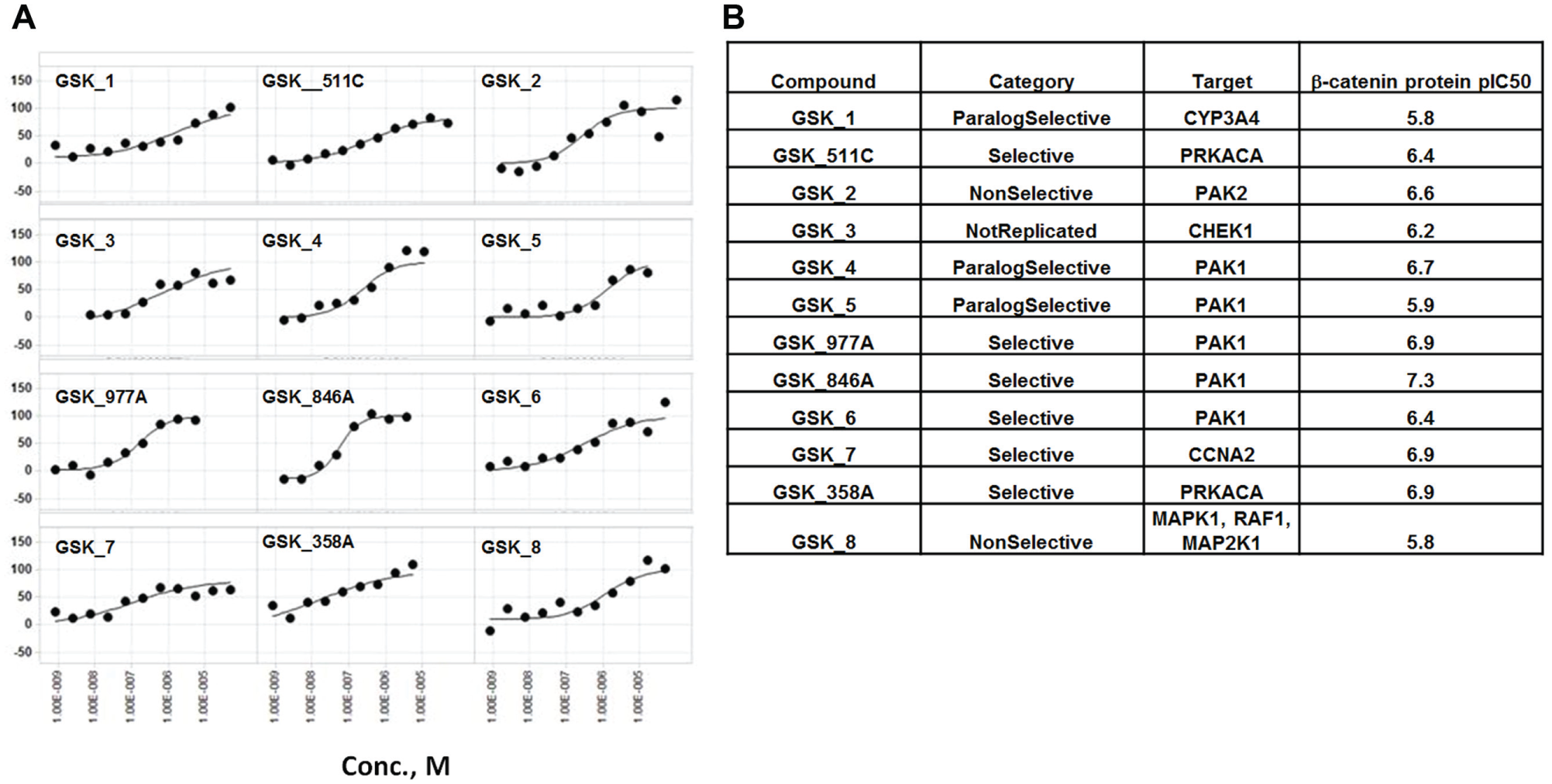
(
Further Characterization of the PAK and PKA Inhibitors
The annotated hits identified from BDCS provide some hints as to the potential targets involved in β-catenin regulation. Both PAK and PKA are ubiquitously expressed kinases and are involved in multiple biological processes. PAK and PKA inhibitor compounds included in BDCS, even though selected for its highest potency toward their annotated targets, also show cross-reactivity with other kinases (such as AKT, albeit at a lower potency; GSK internal data not shown), making it difficult to specifically pinpoint the kinase linking to β-catenin degradation in a cellular context. However, AKT-specific inhibitors included in this compound set did not show up as hits, suggesting that the AKT inhibition alone is not sufficient for β-catenin degradation. Nonetheless, with the molecular tools available, we can begin to address questions regarding the dynamics of β-catenin stability. The β-catenin imaging assay used 24 h of compound incubation time with SW480 cells. Potentially within a 24 h time frame, kinase inhibitors could act to downregulate β-catenin through indirect mechanisms, such as inhibition of protein translation or transcription, or affect other signal transduction pathways. We therefore reduced the incubation time to 4 h to minimize undesired mechanisms. As controls, we also included actinomycin D and cycloheximide as transcription and protein translation inhibitors, respectively. Interestingly, we found that with 4 h of compound treatment, while PAK inhibitors GSK_977A and GSK_846A and the TNKS inhibitor XAV failed to reduce the β-catenin level, PKA inhibitors GSK_511C and GSK_358A were still active ( Table 1 ), suggesting that these inhibitors may function through different mechanisms. In addition, since actinomycin D and cycloheximide were both inactive at 4 h, GSK_511C- and GSK_358A-induced β-catenin reduction is likely not due to general inhibition of transcription or translation.
Different Dynamics of BDCS Hits on β-Catenin Reduction and Cell Count after Compound Treatment.
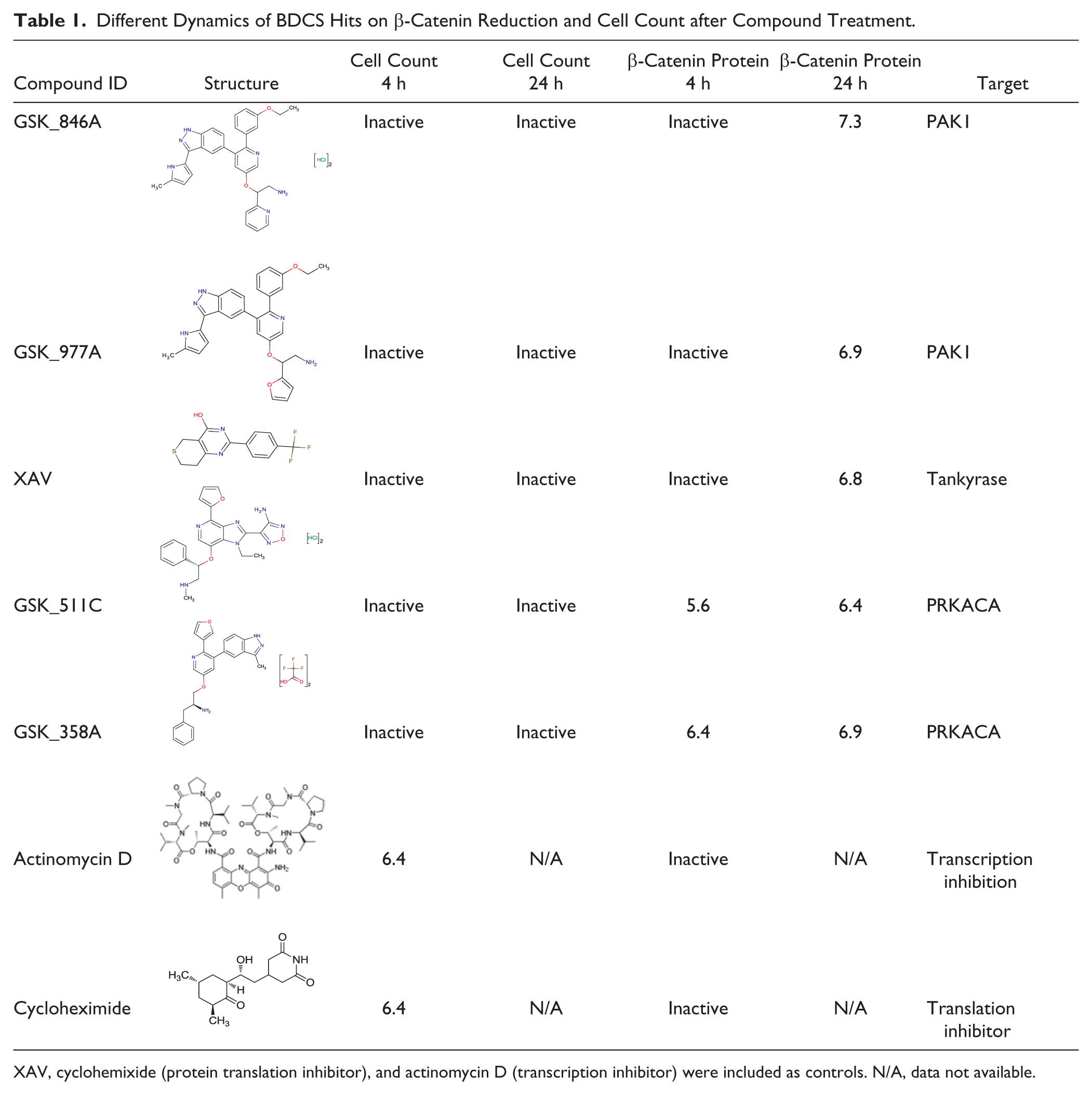
XAV, cyclohemixide (protein translation inhibitor), and actinomycin D (transcription inhibitor) were included as controls. N/A, data not available.
Discussion
Different from previous screens that use TCF reporter cell lines, the β-catenin protein imaging assay directly measures the endogenous β-catenin abundance in cancer cell lines that harbor human mutations of the β-catenin pathway, and therefore is disease relevant. Without using the artificially engineered cell lines, this approach is flexible to use on any cells, including the disease-derived primary cells (e.g., human patient–derived induced pluripotent stem cells (iPSCs) derived from neural progenitor cells 12 ). This is important because the reporter lines can be time-consuming to develop and most primary cells are not suitable for recombinant gene manipulation. Using an assay detecting the endogenous β-catenin protein level also has the advantage of having the readout proximal to the target of inhibition, reducing the chance of false positives that act through undesired mechanisms, such as the general transcription inhibition in TCF reporter–based screens.
The cellular imaging assay also has the advantage of distinguishing subcellular pools of β-catenin. β-Catenin is critical for cell–cell adhesion, a function that is not regulated by the Wnt pathway. In some cells, the membrane pool of β-catenin is so abundant that it masks the β-catenin change in cytoplasm in response to Wnt addition when β-catenin detection is in the whole cell lysate. With an imaging-based approach, this problem can potentially be resolved by separating the signal from different subcellular compartments based on colocalization with specific markers. In addition to degradation, it has been suggested that β-catenin can be regulated through other mechanisms, such as the cytoplasmic-to-nucleus shuttling. Using a β-catenin imaging assay could potentially identify modulators of these other mechanisms if the assay signal is sufficiently high enough for a screen.
When looking at the endogenous signal by immunostaining, it is critical to demonstrate the signal is not due to staining artifact. We showed specificity of the signal detection through three assays: (1) the increased β-catenin abundance on Wnt-treated cells, (2) the abundance of β-catenin signal in colon cancer cells with β-catenin pathway mutations in comparison with cells with intact pathway components, and (3) the inhibition by known pathways inhibitors, such as TNKS inhibitors. All three tests generated a robust signal of two- to threefold above the background, which is sufficient for a successful screen. One advantage of the imaging assay is the multiparameter output, which allows us to eliminate compounds that are cytotoxic under the same assay conditions. This is particularly important since many compounds included in the library were identified through biochemical screening. Their behavior in cell-based assays was not established. When screening for inhibitors of oncogenic pathways, while the target-dependent cytotoxicity is the ultimate goal, the off-target cytotoxicity often masks the desired activity, generating false positives and producing high hit rates. In the imaging assay, by modifying the script to output the fluorescent intensity signal normalized to the cell count, we ensured the fluorescent intensity signal is not impacted by cell count variation. Also, using a cytotoxicity filter, we reduced the hit rate to a manageable number without a separate cytotoxicity measurement. None of this would have been possible in a whole cell lysate detection format (e.g., enzyme-linked immunosorbent assay). The strategy is validated with the retrieval of 8 out of 10 known TNKS inhibitors in the screen.
Previous studies have indicated that PAK1 directly regulates β-catenin protein levels by phosphorylating β-catenin. 22 These studies mainly used RNAi knockdown and overexpression of the kinase dead form to reduce the kinase activity. These approaches normally take more than 24 h to take effect, making it hard to determine whether the effect on β-catenin protein reduction is direct or indirect, since PAK and PKA are involved in a broad spectrum of biological processes. Using small-molecule inhibitor tools, we were able to reduce the time course to 4 h, which could serve as a starting point to address the dynamics of the β-catenin protein stability and the on-target cytotoxicity. While it is still difficult to determine the specificity of these kinases on β-catenin given the broad activity of the PKA and PAK1 and the potential cross-reactivity of the inhibitors to other cellular targets, we nonetheless showed that these inhibitors act through different mechanisms to reduce β-catenin protein levels, and the rapid reduction of β-catenin within 4 h occurs in the absence of the inhibition of protein translation or transcription. It will then be interesting to understand whether this degradation pathway requires the APC/Axin/GSK3β complex.
In summary, we have established a robust assay to detect endogenous β-catenin protein changes in cancer cells upon treatment with small-molecule inhibitors. In addition to cancer, β-catenin signaling is also critical for stem cell self-renewal during development and in adults. The assay can be easily reformatted to detect the increase in β-catenin protein level, facilitating the discovery of reagents with potential applications to regenerative medicine.
Footnotes
Acknowledgements
The authors thank Ming Jiang and Julie Choi for assisting with the assay and screening. We thank Brandon Turunen and colleagues at Discovery Medicinal Chemistry Development who synthesized the TNKS tool compounds. The authors also thank their colleagues at SMTech for preparing compound plates for the screens.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.