
Abstract
Phenotypic screens are effective starting points to identify compounds with desirable activities. To find novel antifungals, we conducted a phenotypic screen in Saccharomyces cerevisiae and identified two discrete scaffolds with good growth inhibitory characteristics. Lack of broad-spectrum activity against pathogenic fungi called for directed chemical compound optimization requiring knowledge of the molecular target. Chemogenomic profiling identified effects on geranylgeranyltransferase I (GGTase I), an essential enzyme that prenylates proteins involved in cell signaling, such as Cdc42p and Rho1p. Selection of resistant mutants against both compounds confirmed the target hypothesis and enabled mapping of the compound binding site to the substrate binding pocket. Differential resistance-conferring mutations and selective substrate competition demonstrate distinct binding modes for the two chemotypes. Exchange of the S. cerevisiae GGTase I subunits with those of Candida albicans resulted in an absence of growth inhibition for both compounds, thus confirming the identified target as well as the narrow antifungal spectrum of activity. This prenylation pathway is reported to be nonessential in pathogenic species and challenges the therapeutic value of these leads while demonstrating the importance of an integrated target identification platform following a phenotypic screen.
Introduction
Phenotypic screens deliver more first-in-class therapeutics than any other high-throughput screening approach. 1 Various factors can contribute to this, including relevance of the model, the screening and counterscreening modalities, and an assay-intrinsic filter for cell-permeable compounds. Particularly in the area of anti-infectives and despite a wealth of genetic knowledge, targeted approaches have yielded high-affinity chemical probes that frequently fail to show efficacy in the whole cell.2,3 This can be attributed to several factors, such as lack of cell permeability, efflux of the compound, high nonspecific protein binding, or the existence of a compensatory pathway. Furthermore, a disease state might result from numerous target protein interactions that will not be addressed in a targeted approach. 4 Thus, a revival of phenotypic screens using whole cells as the starting point for novel drug discovery campaigns can be observed throughout the industry. 5 One disadvantage of the phenotypic screening approach is that leads often lack medicinal chemistry properties 6 that allow therapeutic development. The chemical properties of these suboptimal leads can be improved; however, this requires a detailed understanding of the molecular target. Despite the development of numerous genetic, chromatographic, and in silico target identification methodologies, 7 the identification of the primary binding protein and the mechanism of action of a compound is still a challenging scientific endeavor. Hence, establishing an effective target identification workflow is a prerequisite for any screening campaign using phenotypic readouts.
In this study, we aimed to identify novel antifungal agents. Increasing numbers of elderly, immune compromised, or hospitalized patients with fungal infections have led to high mortality rates, 8 demonstrating an inadequate arsenal of antifungal therapies and a significant unmet medical need. For example, in U.S. hematopoietic stem cell transplant recipients, 43% of severe invasive fungal infections (IFIs) are caused by Aspergillus species, 28% by Candida species, and 8% by Mucorales species. In North American solid organ transplants, the most frequent IFIs were candidiasis (59.2%), aspergillosis (24.8%), and cryptococcosis (7%). 9 However, as only three classes of antifungal drugs (polyenes, azoles, and echinocandins) are in current use, the treatment of IFIs is quite limited. 10
Thus, we set out to identify novel intervention points by screening for compounds that inhibit growth of the nonpathogenic fungus Saccharomyces cerevisiae. 11 S. cerevisiae was selected as a surrogate model for several reasons: safety, ease of handling, high conservation with the genomes of related pathogenic species, and the existence of high-quality, genomewide genetic collections and methodologies to enable target identification. 12 We identified two compounds with good potency, a favorable cytotoxicity window, and tractable chemical scaffolds. Lack of activity against pathogenic species called for chemical optimization requiring knowledge of the molecular target. Chemogenomic profiling highlighted geranylgeranyltransferase I (GGTase I) as a possible target of both these structurally diverse compounds. GGTase I is one of three enzymes responsible for transferring a prenyl group to the C-terminal cysteine of proteins bearing a CaaX-motive. GGTase I, GGTase II, and farnesyltransferase (FTase) are conserved between yeast and mammals. 13 GGTase I is a heterodimer consisting of an α-subunit, Ram2p, that is shared with FTase, and a β-subunit, Cdc43p.14,15 Both subunits of GGTase I are essential for yeast growth. 13 Posttranslational prenylation of proteins is often important for processes like signal transduction and intracellular trafficking pathways. 16 Two crucial substrates of S. cerevisiae GGTase I are the small rho-like GTPases Cdc42p and Rho1p that are involved in the establishment and maintenance of cell polarity. 17
Furthermore, we used a functional variomics tool 18 to confirm GGTase I as the molecular target of the compounds. This tool applies plasmid-based, high-complexity random mutagenesis libraries of the gene of interest and allows the systematic discovery of resistance-conferring single-nucleotide polymorphisms (SNPs), thereby defining amino acids that are critical for drug binding. Although the results we describe here indicate that the identified chemotypes have limited therapeutic value, the general strategy we present yields valuable chemical biology tools.
Materials and Methods
Fungal Strains, Media, and Chemicals
Commercially available fungal strains used for the antifungal susceptibility testing are listed in Table 1 . Complementation of S. cerevisiae GGTase I with Candida albicans GGTase I was performed in the BY4743 strain. For construction of the CDC43 and the RAM2 variome, as well as of the mutants, the BY4743Δ8 MATa/α strain, which is deleted for eight genes involved in drug resistance (efflux pumps SNQ2, PDR5, and YOR1 and transcription factors PDR1, PDR2, PDR3, YAP1, and YRM1), was used. The strains were grown either on 1% yeast extract, 2% peptone, and 2% dextrose (YPD) plates or in liquid medium or on synthetic complete (0.7 g/L Difco Yeast Nitrogen Base without amino acids, 0.79 g/L MPbio CSM amino acid mixture, 2% glucose) plates or in liquid medium. If plasmid selection was necessary, the strains were grown on synthetic complete medium lacking uracil or leucine. Compounds 1 and 2 were obtained from the Novartis compound store. All other reagents, chemicals, and buffer salts were purchased from Sigma-Aldrich Chemicals (St. Louis, MO) or Fluka (Buchs, Switzerland).
MIC Values for Tested Compounds.
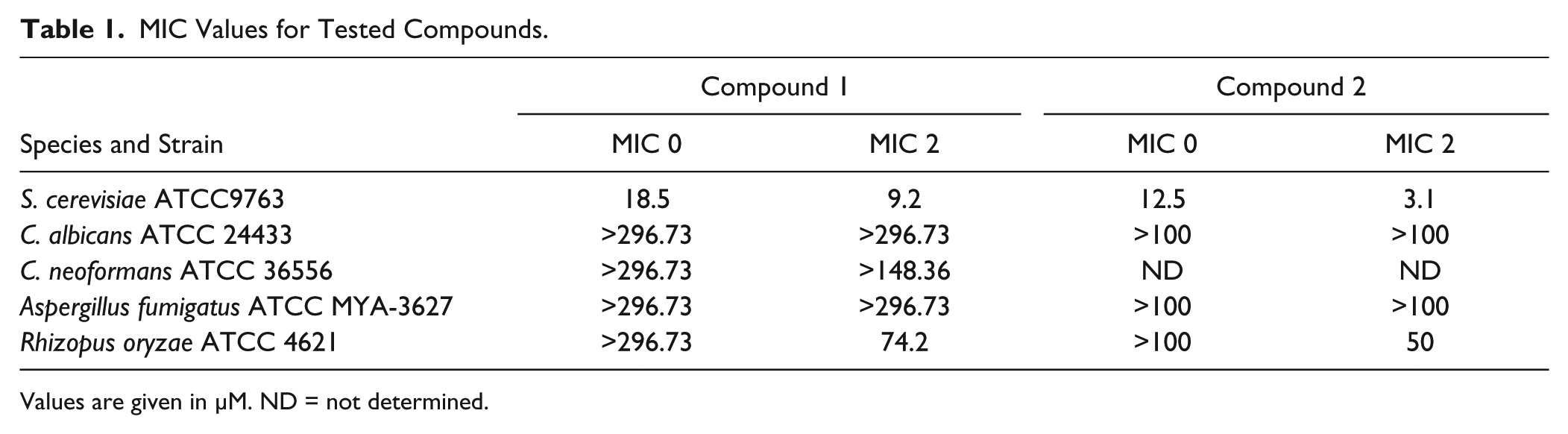
Values are given in µM. ND = not determined.
Chemogenomic Profiling (HIP and HOP)
The growth inhibitory potency of compounds was determined using wild-type S. cerevisiae BY4743 as described by Pierce et al. 19 OD600 values of exponentially growing cultures in rich medium were recorded with a robotic system. Twelve-point serial dilutions of the tested compounds were assayed in 96-well plates with a reaction volume of 150 μL, and the starting OD600 was 0.05. Solutions containing DMSO were normalized to 2%. IC50 values were calculated using logistic regression curve fits generated by TIBCO Spotfire v3.2.1 (TIBCO Software, Inc., Palo Alto, CA). Haploinsufficiency profiling (HIP), homozygous profiling (HOP), and microarray analysis was performed as described previously. 11 Sensitivity was computed as the median absolute deviation logarithmic (MADL) score for each compound–concentration combination. Z-scores are based on a robust parametric estimation of gene variability from >3000 different profiles and were computed as described in detail in Hoepfner et al. 11
Growth Curves
HIP and HOP profiles were validated by picking the individual strains from the HIP and HOP collections (OpenBiosystems, Huntsville, AL; cats. YSC1056 and YSC1055) and testing log-phase cultures in 96-well microtiter plates in YPD medium with serial dilutions of the compound. The assay volume was 150 μL/well, the starting OD600 was 0.05, and DMSO was normalized to 2%. Curves were calculated by taking the 14 h OD600 measurements and applying a logistic regression curve fit in TIBCO Spotfire v3.2.1. Strain HO/YDL228C was used as the wild-type reference. For evaluation of the compound competition, serial dilutions of geranylgeranyl pyrophosphate (GGPP) and farnesyl pyrophosphate (FPP) were added in addition to the tested compounds.
Functional Variomics
The variomic libraries were constructed like described before. 18 The promoter of CDC43 was amplified with primers VP08 and VP07 (see Table 2 for primer sequences). The terminator of CDC43 was amplified with VP06 and VP05. The promoter of RAM2 was amplified using primers VP04 and VP03, and the terminator using VP02 and VP01. The promoter and terminator fragments of CDC43 and RAM2 were fused by PCR and cloned into SacI and HindIII digested pXP597. 18 Error-prone PCR products of CDC43 and RAM2 were generated with the corresponding promoter and terminator primer. The plasmid containing the promoter–terminator fusion was then linearized with NotI and transformed together with the error-prone PCR products into BY4743Δ8 MATa/α. The cells were grown overnight. After back dilution, the variomes were plated on synthetic complete medium lacking uracil and containing 2.5 µM of each compound. Resistant mutants were picked after 3–4 days at 30 °C.
List of Used Primers.

Mutant Validation
To obtain single mutations, site-directed mutagenesis of the pXP597 plasmids containing the open reading frame, promoter and terminator of CDC43 and RAM2 was performed using Platinum Pfx DNA Polymerase (Life Technologies, Carlsbad, CA) and the following primers (see Table 2 for sequences): for Ram2p I25V, VP71 and VP70; for Ram2p L41P, VP69 and VP68; for Ram2p Y144H, VP61 and VP60; for Ram2p R234G, VP65 and VP64; for Ram2p S239G, VP63 and VP62; for Cdc43p F108S, VP79 and VP78; for Cdc43p D164N, VP83 and VP82; for Cdc43p Y169H, VP73 and VP72; for Cdc43p A173V, VP87 and VP86; and for Cdc43p E335D, VP81 and VP80. Plasmids were transformed in Escherichia coli DH5-α, purified, and transformed into S. cerevisiae BY4743Δ8a/α. Saturated cultures of the mutants were fivefold diluted and spotted on synthetic defined (SD) medium plates containing compound 1 or 2. The plates were grown 3–4 days at 30 °C.
Antifungal Susceptibility Testing
Assays for antifungal susceptibility testing were performed in RPMI 1640 (GE Healthcare HyClone, South Logan, UT; SH30011.03) with 2.05 mM glutamine and phenol red, without bicarbonate, and buffered with 0.165 mol/L MOPS [3-(N-morpholino)propanesulfonic acid] (AppliChem, Darmstadt, Germany; A1076,0250). The medium was adjusted to pH 7.0 and filter sterilized. Antifungal susceptibility testing was performed according to the Clinical and Laboratory Standards Institute (http://clsi.org/) guidelines for broth microdilutions M27-A3 (yeast) and M38-A2 (mold).
Complementation of S. cerevisiae GGTase I with C. albicans GGTase I
The coding sequence of C. albicans CDC43 was codon optimized for S. cerevisiae and generated by DNA synthesis containing a SacI restriction site at the 5′ end and an XhoI restriction site at the 3′ end. For cloning into the integrative plasmid pBYInt URA-pADH1-ADH-term, a PUC19 multiple cloning site was constructed into the backbone using primers VP10 and VP09. For transformation into the CDC43 HIP strain, the resulting pBYInt URA plasmid was cut in the TRP marker with Bsu36I. The RAM2 gene was amplified from C. albicans genomic DNA (ATCC 10231D-5) with primer VP88 bearing a KpnI restriction site and VP89 bearing a HindIII restriction site. CaRAM2 was cloned into the KpnI/HindIII linearized BYInt URA. For amplification of RAM2 with the ADH promoter, primers VP90 and VP89 were used. The resulting pADH-RAM2-ADH-term product was cloned into XmaI/HindIII digested BYInt LEU-LYS plasmid. The plasmid was cut in the LYS locus with BglII and transformed in the CDC43 HIP strain complemented before with C. albicans CDC43. A clone was picked and grown in YPD overnight. Cells were pelleted and resuspended in sporulation medium (1% KAc, 0.1% yeast extract, 0.05% glucose). The cultures were left rotating at RT for 14 days. For tetrad dissection 100 µL cell suspension was pelleted, resuspended in 50 µL H2O, and 2 µL of zymolyase (20 mg/mL) was added. Incubation time was 10 min at RT, and the digestion was stopped by placing the cells on ice. The cells were dropped on a YPD plate and tetrads picked and dissected. Spores were grown for 3 days at 30 °C. The right genotype was selected by plating the strains on medium plates containing G418 and on medium lacking uracil and leucine. Furthermore, PCR was performed with genomic DNA to test for haploidy and absence of S. cerevisiae wild-type CDC43.
Molecular Modeling and In Silico Docking
A homology model of S. cerevisiae GGTase I has been generated using the C. albicans 3D protein structure published in the Protein Data Bank (PDB ID: 3DRA) as template. This protein is a heterodimer, comprising Cdc43p and Ram2p. To build the homology model, the amino acid sequences of S. cerevisiae Cdc43p and Ram2p were merged into one amino acid sequence and then used to build the model, using the default setting of the MOE software (www.chemcomp.com). The homology model has been used to dock compounds 1 and 2. The protein binding site has been defined based on the GGPP binding mode in C. albicans. ICM docking software (in particular the 3D Editor 20 ) has been used for docking the two compounds in the putative binding site.
Results and Discussion
Yeast Chemogenomic Profiling
To identify the molecular targets of two compounds (Fig. 1a) with growth inhibitory activity against S. cerevisiae, HIP and HOP were performed. HIP can directly identify molecular targets by taking advantage of increased drug sensitivity when the gene dosage of a drug target is lowered from two copies to one copy in diploid yeast, whereas HOP reveals compensatory pathways by employing yeast with complete deletions of nonessential genes.
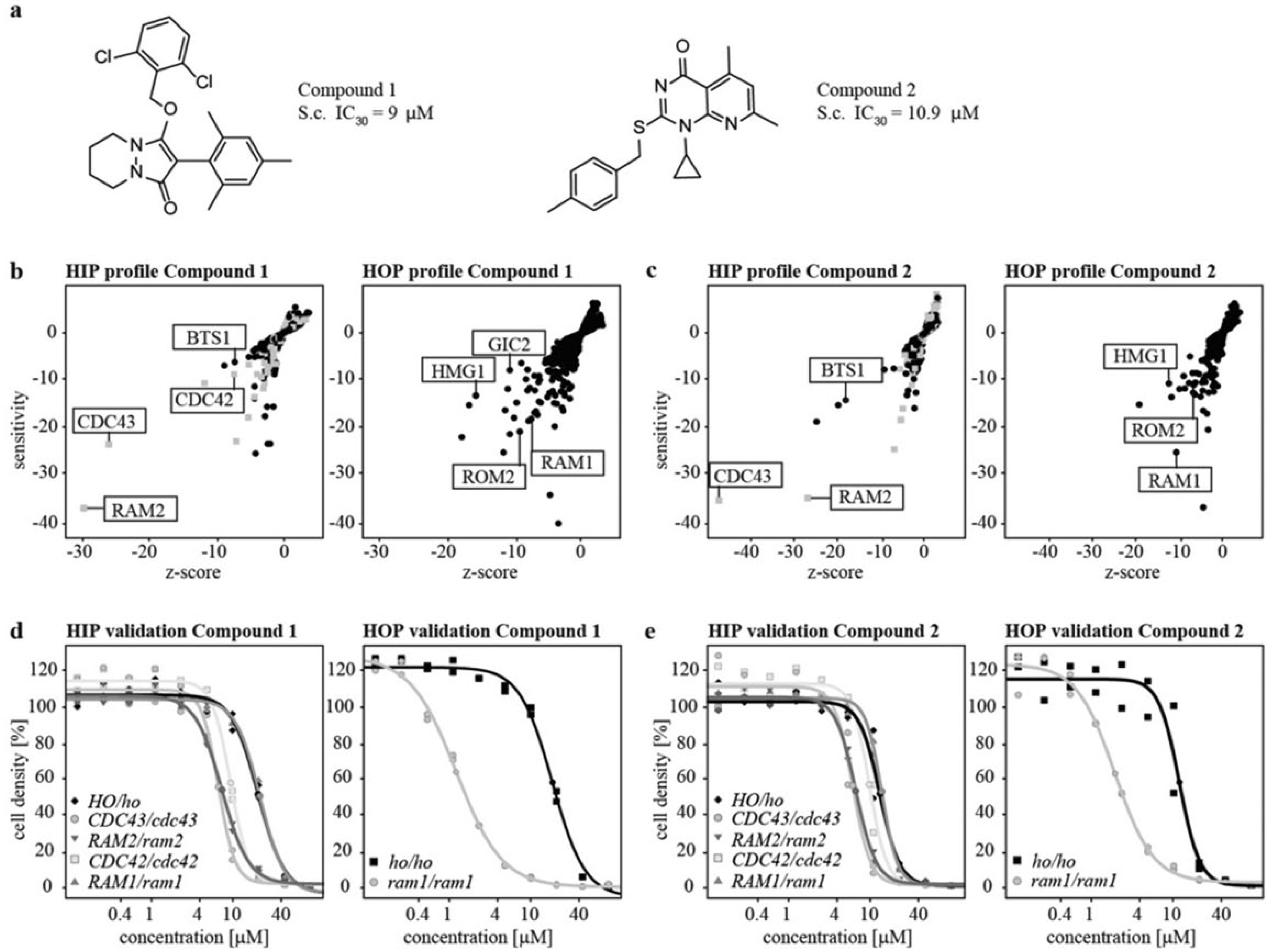
HIP and HOP results. (
At sublethal compound concentrations, strains carrying heterozygous deletions of RAM2 and CDC43, the α- and β-subunits of GGTase I, 15 showed decreased fitness ( Fig. 1b,c ). Furthermore, the heterozygous deletion of geranylgeranyl diphosphate synthase (BTS1), which acts upstream in the isoprenoid pathway, 21 was hypersensitive toward both compounds ( Fig. 1b,c ). For compound 1, the heterozygous deletion of the small rho-like GTPase CDC42, a GGTase I substrate, was also identified ( Fig. 1b ). The HOP highlighted a growth inhibition for the strain deleted for the α-subunit of farnesyltransferase RAM1. 22 Synthetic lethality was also observed for the strain with the homozygous deletion of HMG-CoA reductase (HMG1), a key enzyme in the mevalonate pathway. 23 Furthermore, deletion of the GDP/GTP exchange factor (GEF) for Rho1p and Rho2p, ROM2, 24 rendered the strain hypersensitive toward the compounds ( Fig. 1b,c ). In the case of compound 1, the homozygous deletion of the rho-like GTPase GIC2, a target protein of Cdc42p, 25 also led to reduced fitness ( Fig. 1b ). Growth curves of the individual HIP strains of CDC43, RAM1, RAM2, and CDC42, as well as the HOP strain of RAM1, confirmed the hypersensitivites toward compounds 1 and 2 ( Fig. 1d,e ). Furthermore, it was demonstrated that, in contrast to the homozygous deletion, the heterozygous deletion of RAM1 is unaffected by treatment with either compound. Taken together, the chemogenomic profiling suggests that compounds 1 and 2 impact the isoprenoid pathway potentially by directly modulating GGTase I.
A Functional Variomics Tool Identifies Compound-Resistant Mutants
To identify the amino acids involved in the binding of compounds 1 and 2, a functional variomics tool was applied. 18 As a minimal inhibitory concentration (MIC) on solid media for the compounds could not be identified up to 200 µM, the variomes of Cdc43p and Ram2p were constructed in an efflux-competent BY4743 background missing eight components involved in compound resistance (BY4743Δ8). Both collections were plated on compound. Ninety-six resistant colonies were picked for each condition and sequenced to identify the resistance-conferring SNPs. Sequence analysis of CDC43 revealed F108S and D164N as distinct and the most prominent SNPs for compound 1. F108S was found in 11 and D164N in 9 out of all analyzed clones ( Fig. 2a ). For compound 2, Y169H/N, A173V, and E335D were identified as specific SNPs. In the case of RAM2, five mutations were isolated as being resistant to both compounds: I25V, L41P, Y144H/C, R234S, and S239G ( Fig. 2b ). The mutation of tyrosine 144 was identified in all analyzed clones ( Fig. 2b ). As none of the identified SNPs for CDC43 and RAM2 were a single mutation, all of them were individually constructed via site-directed mutagenesis in the pXP597 background and resistance confirmed by spotting the strains on compound ( Fig. 2c,d ). For compound 1, the resistance of Cdc43p F108S and D164N ( Fig. 2c ), as well as Ram2p Y144H ( Fig. 2d ), was confirmed, and for compound 2, the resistance of Cdc43p Y169H and A173V, as well as Ram2p Y144H, was confirmed ( Fig. 2c,d ).
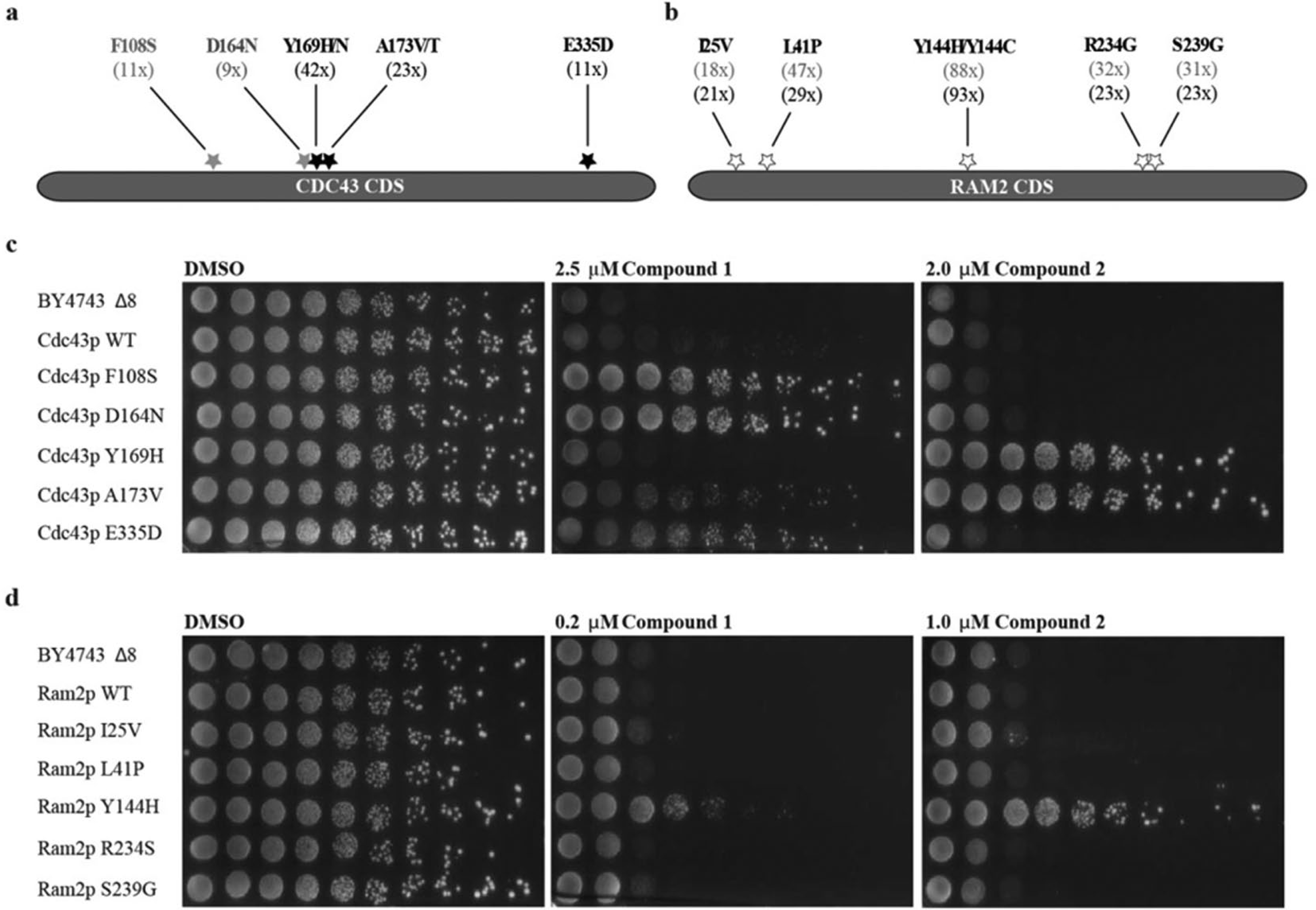
Identification and validation of resistant mutants. (
To locate the SNPs within the protein, a homology model of S. cerevisiae GGTase I was generated based on the published C. albicans crystal structure (PDB ID: 3DRA 26 ) ( Fig. 3a ), and the investigated compounds were docked into this model ( Fig. 3b ). According to the model, all identified Cdc43p mutations are situated within the substrate binding pocket. However, a satisfying docking model could only be retrieved for compound 2. Both mutated amino acids in Cdc43p could play a role for compound binding. Tyrosine 169 could be responsible for a stacking interaction with the compound that would be partially lost if the amino acid is changed to histidine. Alanine 173 is situated in close proximity to tyrosine 169 ( Fig. 3c ). If the alanine is exchanged for a larger amino acid, such as valine, it could result in a shift of tyrosine 169 such that the stacking interaction is affected. To evaluate the accuracy of the predicted docking model, we exchanged tyrosine 340 for a methionine, which is the corresponding amino acid in the C. albicans structure. Based on the model, tyrosine 340 forms a π-interaction with the pyridopyrimidone moiety of compound 2; however, exchange of this amino acid did not result in compound resistance (data not shown), suggesting that the proposed binding mode is only partially correct. The methyl-phenyl moiety of the compound could still interact with tyrosine 169, whereas the rest of the molecule might occupy another region of the binding pocket, for example, extending in the same direction as the GGPP observed in the C. albicans structure. Such a solution was not proposed by the docking since the binding site is quite narrow and the docking was performed using a rigid protein structure. In contrast to the identified Cdc43p mutations, tyrosine 144 of Ram2p does not locate to the GGPP binding site. This amino acid is situated at the entrance of the substrate binding pocket, and in the crystal structure of C. albicans it forms a water-mediated hydrogen bond with serine 212 of Cdc43p. This indicates that tyrosine 144 is responsible for the interaction between both GGTase subunits and thus for overall protein stability. Mutation of tyrosine 144 might lead to a structural change of the protein that explains the cross-resistance against both compounds.
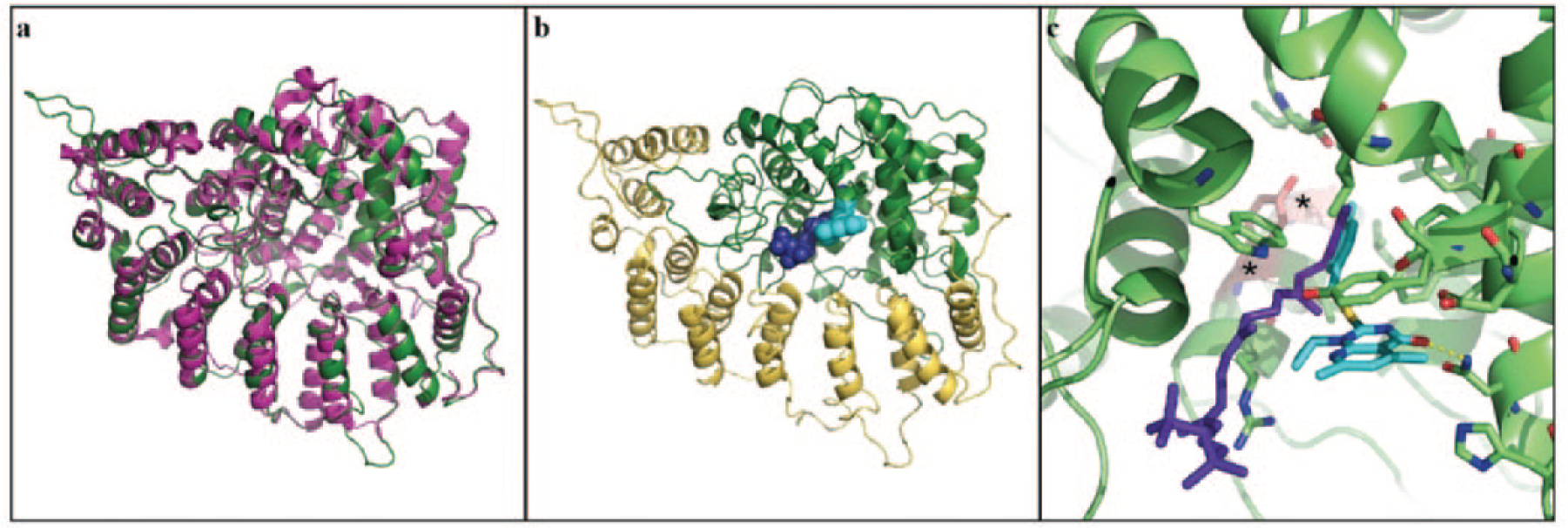
Docking studies of compound 2. (
Competition of GGPP and FPP
Next, we investigated if compound interactions interfere with substrate binding in the active site of the enzyme, as suggested by the in silico docking approach. As it has been described before that GGPP can cross yeast membrane and cell wall, 27 we performed a competition assay with various compound and substrate (GGPP and FPP) concentrations ( Fig. 4 ). In the case of compound 2, we could observe that at a compound concentration of 1.6 µM, increasing amounts of GGPP could rescue the growth inhibitory phenotype. At the highest tested GGPP concentration of 12 µM, the growth inhibition of compound 2 was completely abolished. Competition with FPP had a weak effect after 20 h of growth, which we attribute to the notion that FPP is able to occupy the same binding pocket as GGPP. For compound 1, the competition with both pyrophosphates did not result in a shifted IC50. Thus, the proposed docking in the active site could be corroborated for compound 2, but for compound 1, the target interaction mode remains to be determined.
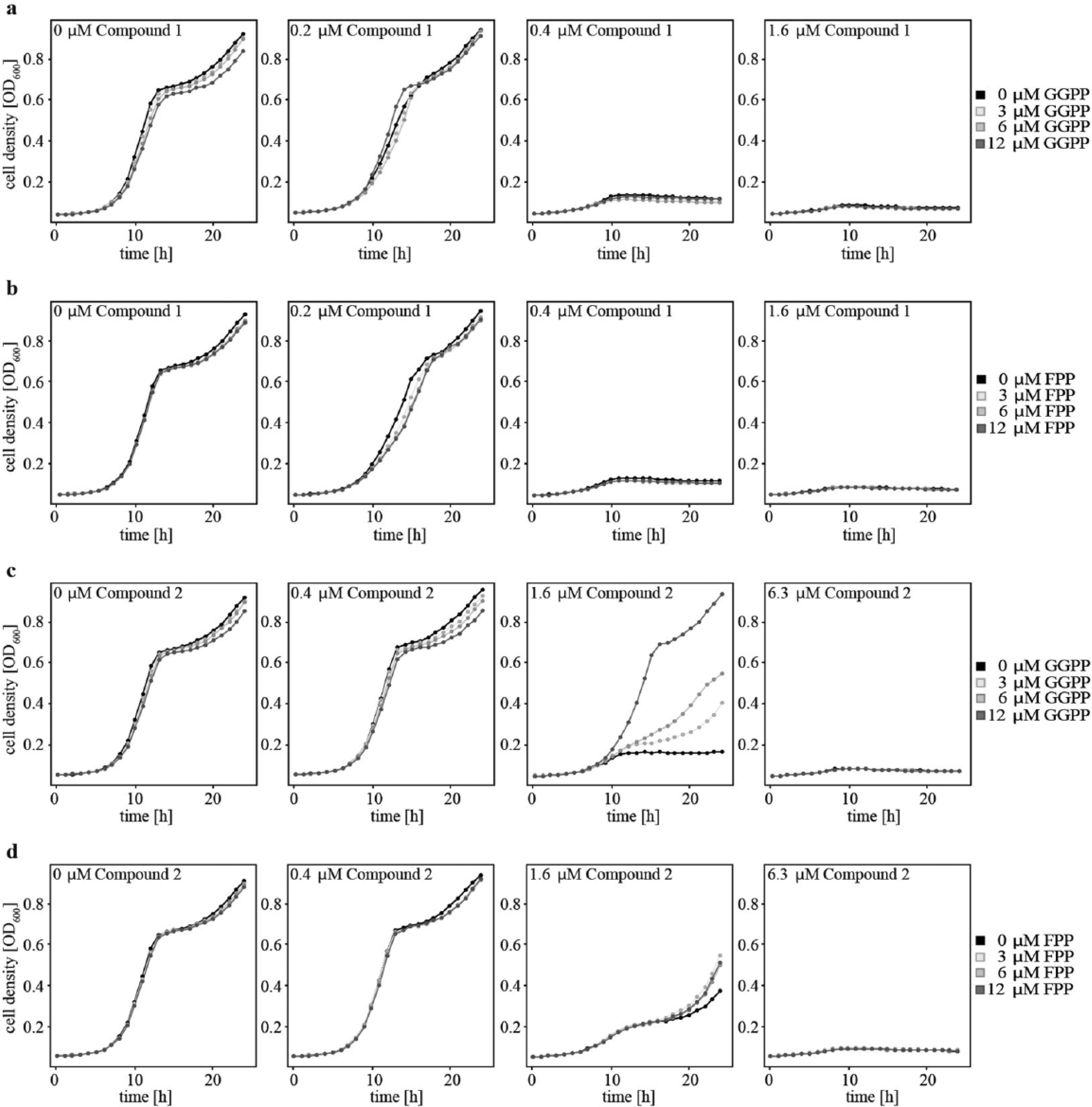
Compound competition with pyrophosphates. Four different concentrations of compounds 1 (
Complementation of S. cerevisiae GGTase I with C. albicans GGTase I
The subunits of GGTase I show several conserved regions among different pathogenic fungi such as C. albicans 28 and Cryptococcus neoformans. 29 However, no inhibition of growth by the tested compounds could be observed ( Table 1 ). There are reports that geranylgeranylation is not essential in those pathogens, as it can be compensated by FTase activity.29,30 Therefore, we were interested in whether the restricted antifungal spectrum was due to the nonessentiality of Cdc43p or a lack of inhibition of C. albicans GGTase. We complemented GGTase I of S. cerevisiae with Cdc43p and Ram2p of C. albicans. Tetrad analysis revealed that complementation yielded viable colonies, and two isolated clones bearing the desired genotype were grown on 150 µM of compounds 1 and 2 ( Fig. 5 ). Growth inhibition was completely abolished in the complemented yeast strains, indicating that the compounds did not inhibit C. albicans GGTase I. This observation validated S. cerevisiae GGTase I as the specific target protein and provided a molecular mechanism for the observed restricted antifungal spectrum of the compounds ( Table 1 ). The published lack of essentiality of GGTase I in relevant pathogenic species28,29 devalidates compounds 1 and 2 as promising starting points for further therapeutic development. However, the general understanding of the binding mode of compounds 1 and 2 in the S. cerevisiae enzyme could allow chemical optimization toward pathogen specific inhibitors. Therapeutic use of such compounds in combination with FTase inhibitors could increase the efficacy of FTase inhibitors. Combinatorial use of antifungal drugs might be favorable due to the emergence of rapid antifungal drug resistance when single agents are used. 31 Furthermore, it cannot be neglected that the substrate binding pockets of GGTase I and FTase show certain similarities. 32 Hence, the compounds identified within this work could be a fundament for the targeted design of selective FTase inhibitors. Such an approach was successfully described for the development of potent selective RabGGTase inhibitors on the basis of a FTase inhibitor scaffold. 33
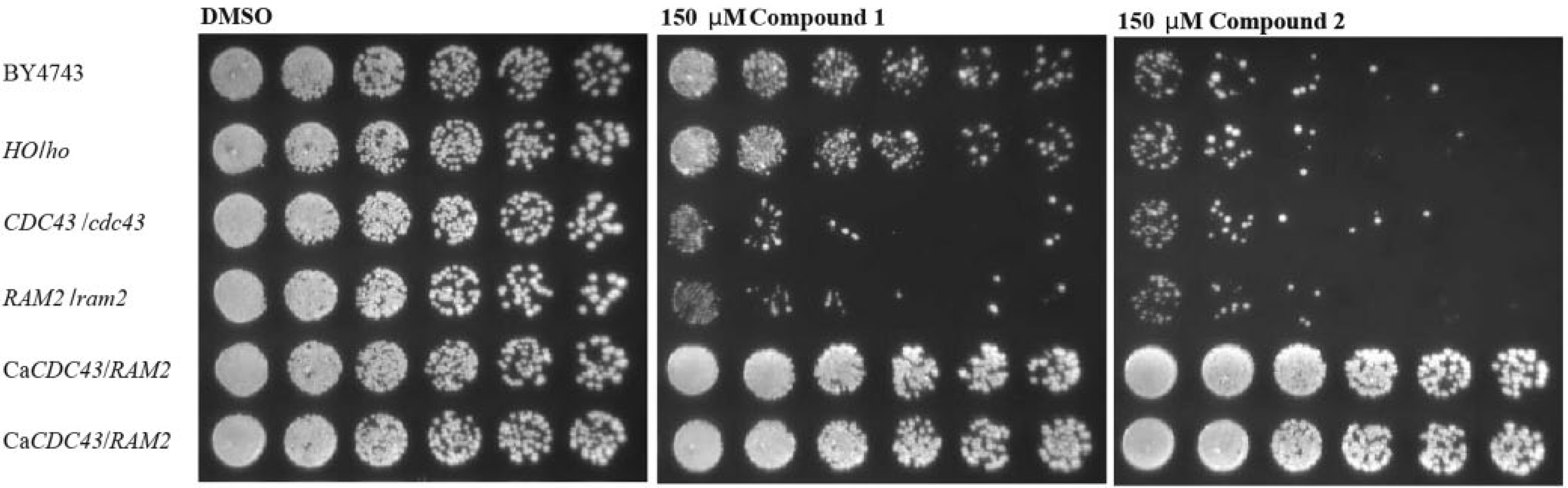
Complementation of S. cerevisiae GGTase I with the C. albicans enzyme. Fivefold dilutions of the complemented strains as well as two control strains and the heterozygous deletions of CDC43 and RAM2 were spotted on YPD plates containing 150 µM of each compound. The cells were grown for 3 days at 30 °C.
Here we report the identification of GGTase I as the primary binding protein of two chemically discrete compounds that are growth inhibitory to S. cerevisiae. Unbiased chemogenomic profiling in S. cerevisiae highlighted the α- and β-subunits of GGTase I as potential target proteins for the two compounds. This finding was corroborated by growth assays using the individual strains from the HIP collection. The implementation of a functional variomics approach, together with in silico docking, further validated the target hypothesis and enabled mapping of the compound binding site in the enzyme. A binding model for one compound is presented and was supported by substrate competition experiments. Despite the strong target validation, however, the nonessential role of the geranylgeranylation pathway in pathogenic fungi subverts the therapeutic development of compounds 1 and 2 as antifungal leads.
The approach presented here illustrates the conundrum that scientists frequently face when opting for a phenotypic screen: selection of the disease-relevant pathogen or cell line might result in pertinent leads, but handling and follow-up work might be seriously hampered due to lack of tractability and safety concerns. In contrast, selection of a tractable surrogate system can allow the rapid identification of promising leads and subsequent target identification, but as for the case presented here, the identified targets may lack conservation among different species. Despite the lack of translatability of the compounds presented in this study, the authors feel that the chosen path has its values, as rapid target knowledge allows for informed decisions concerning the next experiments and compound prioritization early in a project, even if this results in termination. This can be key, as illustrated by the example of novel antifungal inhibitors that were identified by the same strategy and showed conserved potency against pathogenic Aspergillus species. 34 Early identification of acetolactate synthase as the target and knowledge of its biology in an infection context devalidated this series and avoided cumbersome and costly in vivo experiments. It should also be noted that testing the approach in the other direction resulted in correct identification of the target of clinical antifungal compounds, including azoles, echinocandins, amphotericin, and AN2690, supporting the value of the strategy. 11 We feel the presented strategy allows effective identification and validation of molecular targets, although misses have to be factored in. But misses are not without value: even though they might not become medicines, they provide valuable tools for molecular biologists to dissect biological pathways and targets and may help them take better aim next time.
Footnotes
Acknowledgements
We would like to acknowledge Christian Studer, David Estoppey, and Britta Knapp for their advice on technical questions. Furthermore, we thank Heather Sadlish for discussion on the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.