
Abstract
There are nearly 2000 mutations in the CFTR gene associated with cystic fibrosis disease, and to date, the only approved drug, Kalydeco, has been effective in rescuing the functional expression of a small subset of these mutant proteins with defects in channel activation. However, there is currently an urgent need to assess other mutations for possible rescue by Kalydeco, and further, definition of the binding site of such modulators on CFTR would enhance our understanding of the mechanism of action of such therapeutics. Here, we describe a simple and rapid one-step PCR-based site-directed mutagenesis method to generate mutations in the CFTR gene. This method was used to generate CFTR mutants bearing deletions (p.Gln2_Trp846del, p.Ser700_Asp835del, p.Ile1234_Arg1239del) and truncation with polyhistidine tag insertion (p.Glu1172-3Gly-6-His*), which either recapitulate a disease phenotype or render tools for modulator binding site identification, with subsequent evaluation of drug responses using a high-throughput (384-well) membrane potential–sensitive fluorescence assay of CFTR channel activity within a 1 wk time frame. This proof-of-concept study shows that these methods enable rapid and quantitative comparison of multiple CFTR mutants to emerging drugs, facilitating future large-scale efforts to stratify mutants according to their “theratype” or most promising targeted therapy.
Keywords
Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR/ABCC7) is an ATP-dependent and phosphorylation-regulated plasma membrane chloride channel, arranged into two membrane-spanning domains (MSDs; six transmembrane helices in each), two intracellular nucleotide-binding domains (NBDs), and a regulatory (R) domain.1–3 Trans-membrane helices from MSD1 and MSD2 form the channel pore, whereas the NBDs form the catalytic dimer, which promotes channel opening and closing (i.e., gating); the R domain regulates CFTR gating.2,3 Mutations in CFTR cause the genetic disease cystic fibrosis (CF), and the major disease-causing mutations include the misprocessed mutation p.Phe508del (present on one or both alleles in 90% of cases) and the gating mutation p.Gly551Asp (approximately 3%–4% of cases).4,5 Excitingly, a small-molecule therapy (Ivacaftor, Kalydeco, or VX-770) has recently been developed by Vertex Pharmaceuticals and is Food and Drug Administration–approved for use in patients bearing the major CF disease-causing gating mutation p.Gly551Asp (as well as several other less common CFTR gating mutations: p.Gly178Arg, p.Ser549Asn, p.Ser549Arg, p.Gly551Ser, p.Gly970Arg, p.Gly1244Glu, p.Ser1251Asn, p.Ser1255Pro, and p.Gly1349Asp); VX-770 is a CFTR-specific potentiator capable of activating gating-incompetent channels in the clinical setting.6,7 Further, although a therapeutic compound is available for patients bearing several CFTR gating mutations, there still remains no targeted therapies for the remaining ~95% of CF patients.
Although approximately 2000 CFTR mutations have been identified to date (CFTR Mutation Database: www.genet.sickkids.on.ca), the molecular characterization of only a few (<10%) has been investigated. 5 Therefore, there is a need to generate these uncharacterized CFTR mutations for further in vitro study, in order to test the response of these mutants to emerging therapeutics as well as to discover targeted therapeutics to enhance the functional expression of CF disease-causing mutations. This could subsequently lead to the development of therapies earmarked for each genotype, rather than a one-size-fits-all approach, which is the current paradigm in the CF field (e.g., using pharmacologic potentiators to treat multiple mutations affecting channel gating or corrector compounds to target all mutations leading to CFTR misprocessing).
Site-directed mutagenesis has been a staple of molecular biology and genetic research, allowing for the generation of nucleotide substitutions to recapitulate disease-causing mutations in an artificial expression system to further understand the molecular consequences leading to pathophysiology.8,9 Polymerase chain reaction (PCR)–based site-directed mutagenesis has shed light on countless genetic diseases by allowing for the qualitative and quantitative analysis of the structure-function relationship of a vast number of disease-causing mutant proteins. 10 Most often, this is done in double-stranded plasmid DNA, which includes the complementary DNA (cDNA) of the exonic open reading frame sequence of the gene of interest. Subsequently, there are several approaches to obtain site-directed cDNA mutations using PCR, and traditional methods include overlap extension PCR, inverted PCR, megaprimer PCR, and recombination PCR. However, one of the first commercial mutagenesis kits (QuikChange by Stratagene) has become the method of choice over the past two decades for generating a countless number of engineered mutations, for further characterization using the tools of choice within any given laboratory. Other mutagenesis kits are commercially available, but most are variations of QuikChange. 11 Although QuikChange may be the most common site-directed mutagenesis strategy, it is not capable of performing deletion and insertion mutagenesis because it is designed and optimized for single- or multiple-site (primer-based) modifications. 11 In addition, insertion/deletion mutagenesis is incompatible with the one-step QuikChange approach as well as other one-step PCR-based site-directed mutagenesis kits, because of the relatively high error rate of their DNA polymerases (usually Taq DNA polymerase), which prevents high-fidelity replication of the template; DNA mismatch repair proteins are not included in PCR kits because they cannot survive thermocycling. 12 Therefore, to reduce the chance of second-site errors, molecular biologists typically perform site-directed mutagenesis in smaller constructs containing a fragment of their gene (cDNA) of interest, with the aim of subcloning the mutated fragment into an unadulterated full-length template plasmid.
Using site-directed mutagenesis methods, we have generated more than 100 CFTR missense mutant constructs to date, several of which have been recently reported,13,14 and the vast majority of these are critical tools within many ongoing projects. However, there is a large number of CF-associated mutations predicted to cause deletions, insertions, or truncations (i.e., stop mutations),
5
and generation of these mutations is relatively time-consuming (
To assess functional responses of CFTR variants in a high-throughput fashion, the recently described rapid and simple mix-and-read fluorometric imaging plate reader (FLIPR) membrane potential assay will be advantageous. 16 This method allows for kinetic resolution comparable to electrophysiological measurements, allowing for detection of rapid changes in membrane potential; however, FLIPR is a faster, less labor-intensive, and higher-throughput approach.17,18 In addition, the FLIPR system eliminates wash steps, which translates to healthier (and a higher number of) cells due to less manipulation, as well as shorter read times due to the simple mix-and-read protocol. Several other membrane potential–sensitive dyes exist (e.g., DiBAC), but they have slower response times (10× slower for DiBAC) and can be sensitive to temperature variations; therefore, FLIPR is more robust, allowing for high-quality screening results that yield high signal-to-noise ratios. 19 Importantly, this method has several advantages over traditional assays for CFTR function, such that FLIPR can measure both activation and inhibition, whereas CFTR assays using fluorescence-based halide dequenching (i.e., SPQ or MQAE dyes, halide-sensitive YFP) and iodide-selective probes are unidirectional and can detect only anion channel activation.20,21 A potential disadvantage of this method is that non-CFTR ion channels may also affect changes in membrane potential. However, specificity of the FLIPR response for CFTR can be assessed by confirming several signature features of the CFTR channel, including activation by agonists of cyclic AMP (cAMP), sensitivity to electrochemical anion gradients, and inhibition by CFTRinh-172.
In the present study, we developed an improved PCR-based site-directed insertion and deletion mutagenesis method to generate disease-causing mutations in CFTR cDNA. This technique is rapid, inexpensive, and simple. Importantly, it can be used for replicating large vectors (>10 kb) without the need for subcloning (a time-consuming yet previously necessary approach). Here, we incorporated our desired insertion into the primers (or omitted it for the deletion construct) and employed the KAPA HiFi HotStart PCR Kit, containing a high-fidelity (engineered B-family, proofreading) DNA polymerase, to generate four relatively complex CFTR mutants in a one-step PCR process. The deletion constructs removed 18 nucleotides (in-frame deletion of six amino acids in NBD2) rendering p.Ile1234_Arg1239del-CFTR, 405 nucleotides (in-frame deletion of residues 700-835 comprising the R domain) rendering p.Ser700_Asp835del-CFTR, and 2532 nucleotides (in-frame deletion of residues 2-846 comprising the MSD1-NBD1-R domain sequence) rendering p.Gln2_Trp846del-CFTR (i.e., MSD2-NBD2 construct). The insertion construct added 27 nucleotides (2 Gly, 6 His residues, and one stop codon), yielding p.Glu1172-3Gly-6His*-CFTR, a deletion mutant lacking 308 C-terminal residues (amino acids 1173–1480) and containing an engineered C-terminal poly-His-tag. When transiently expressed in human embryonic kidney (HEK)-293 GripTite cells (abbreviated to HEK in this study) and paired with a rapid, multiwell (384-well) plate reader assay of channel function (FLIPR, a membrane potential–sensitive dye-based system), these methods permitted comparison of the functional consequences of emerging CFTR therapies on these rare deletion mutants of CFTR.
Materials and Methods
Generation of Mutant CFTR Constructs
p.Ile1234_Arg1239del-CFTR (site-directed deletion mutagenesis), p.Ser700_Asp835del-CFTR (deletion), p.Gln2_Trp846del-CFTR (deletion), p.Glu1172-3Gly-6His*-CFTR (insertion), and p.Glu1172*-CFTR (point mutation) cDNA were generated using the KAPA HiFi HotStart PCR Kit (KAPA Biosystems, Woburn, MA) according to the manufacturer’s Standard PCR Protocol with high-quality (>300 ng/µL, 260/280 nm ratio of 1.8) plasmid DNA containing WT-CFTR cDNA (in pcDNA3.1) as the template. For p.Ile1234_Arg1239del-CFTR, the following PCR primers (synthesized by ACGT Corp, Toronto, Ontario, Canada) were used: 5′- CAT ATT AGA GAA CAT TTC CTT CTC A
Studies of CFTR Protein Expression
Human embryonic kidney (HEK)-293 GripTite cells (abbreviated to HEK in this study and kindly provided as a gift from Dr. Daniela Rotin, Hospital for Sick Children, Toronto, Ontario, Canada) 20 were grown at 37 °C in 24-well (clear, flat bottom; Sarstedt) plates to 50% confluence and transiently transfected with p.Ile1234_Arg1239del-CFTR, p.Ser700_Asp835del-CFTR, p.Gln2_Trp846del-CFTR, p.Glu1172-3Gly-6His*-CFTR, p.Glu1172*-CFTR or WT-CFTR (positive control) cDNA constructs (pcDNA3.1) using PolyFect Transfection Reagent (Qiagen), according to the manufacturer’s protocol. HEK cells transiently expressing CFTR proteins were maintained in DMEM (Wisent) supplemented with nonessential amino acids (Life Technologies) and 10% fetal bovine serum (Wisent) at 37 °C with 5% CO2 (HEPA incubator, Thermo Electron Corporation) and processed as previously described. 8 Briefly, following incubation at 37 °C for 24 h, HEK cells transiently expressing CFTR proteins were lysed in modified radioimmunoprecipitation assay buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 7.4, 0.2% [v/v] SDS, and 0.1% [v/v] Triton X-100) containing a protease inhibitor cocktail (Roche) for 10 min, and the soluble fractions were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on 6% or 4% to 12% Tris-Glycine gels (Life Technologies) as appropriate. After electrophoresis, proteins were transferred to nitrocellulose membranes (Bio-Rad) and incubated in 5% (w/v) milk, and CFTR bands were detected with human CFTR-NBD1–specific (amino acids 484–589) murine mAb 66015 (1:10,000, University of North Carolina, Chapel Hill, NC) for NBD1-containing constructs (i.e., p.Ile1234_Arg1239del-CFTR, p.Glu1172-3Gly-6His*-CFTR, p.Glu1172*-CFTR, or WT-CFTR) or human CFTR-NBD2–specific (amino acids 1204–1211) murine mAb 59615 (1:10,000, University of North Carolina Chapel Hill) for NBD2-containing constructs (i.e., p.Ser700_Asp835del-CFTR, p.Gln2_Trp846del-CFTR, or WT-CFTR) at 4 °C overnight, horseradish peroxidase–conjugated goat anti-mouse IgG secondary antibody (1:10,000, Pierce) at room temperature for 1 h, Amersham ECL (GE Healthcare), and exposure to film (Denville Scientific) for 0.5 to 5 min as required. Importantly, when blots containing p.Glu1172-3Gly-6His*-CFTR or p.Glu1172*-CFTR were probed with the CFTR-NBD2–specific (amino acids 1204–1211) murine mAb 59615 (1:10,000, University of North Carolina Chapel Hill), or p.Gln2_Trp846del-CFTR probed with the CFTR-N-terminus–specific (amino acids 25–36) murine mAb MM13-415 (1:1000; EMD Millipore) no signals were detected (the respective mAb epitopes are not expressed in these deletion mutants; data not shown). Calnexin was used as a protein loading control and detected with a Calnexin-specific rabbit pAb (1:10,000; Sigma-Aldrich) at 4 °C overnight, horseradish peroxidase–conjugated goat anti-rabbit IgG secondary antibody (1:10,000; Pierce) at room temperature for 1 h, Amersham ECL, and exposure to film for 0.5 to 5 min as required. Relative expression levels of CFTR proteins were quantitated by densitometry of immunoblots using ImageJ software version 1.46 (National Institutes of Health).
Studies of p.Gln2_Trp846del-CFTR Glycosylation Status
To evaluate protein glycosylation, HEK cells expressing p.Gln2_Trp846del-CFTR were grown at 37 °C for 24 h and then lysed in modified radioimmunoprecipitation assay buffer as described above and previously. 13 Lysates were then treated with either Endoglycosidase H (EndoH) or Peptide-N-Glycosidase F (PNGaseF; NEB, Ipswich, MA) endoglycosidases according to the manufacturer’s protocol. Samples were analyzed by SDS-PAGE using 4% to 12% Tris-glycine gradient gels (Life Technologies), and immunoblots were performed using the human CFTR-NBD2–specific murine mAb 596 as described above.
Studies of CFTR Function Using a Membrane Potential Assay
HEK cells transiently overexpressing p.Ile1234_Arg1239del-CFTR were grown at 37 °C to 90% to 100% confluence in 384-well (black, flat bottom; Greiner) plates. Following 24 h incubation in the presence of the pharmacologic corrector VX-809 (3 µM; Selleck Chemicals, Houston, TX), the cells were washed with phosphate-buffered saline, and blue membrane potential dye (dissolved in chloride-free buffer containing 136 mM sodium gluconate, 3 mM potassium gluconate, 10 mM glucose, 20 mM HEPES, pH 7.35, 300 mOsm, supplemented with 3 µM VX-809, at a concentration of 0.5 mg/mL; Molecular Devices), which can detect changes in transmembrane potential, was added to the cells for 1 h at 37 °C. HEK cells transiently overexpressing p.Glu1172-3Gly-6His*-CFTR, p.Ser700_Asp835del-CFTR, or WT-CFTR were grown and prepared in the same manner but did not require pharmacologic correction using VX-809. The plate was then read in a fluorescence plate reader (SpectraMax i3; Molecular Devices) at 37 °C, and after reading the baseline fluorescence (excitation: 530 nm, emission: 560 nm) for 10 min, CFTR was stimulated using the cAMP agonist forskolin (10 µM; Sigma) and the small-molecule potentiator VX-770 (1 µM; Selleck Chemicals); DMSO vehicle was used as a negative control. CFTR-mediated depolarization of the plasma membrane was detected as an increase in fluorescence and hyperpolarization (or repolarization) as a decrease. 16 To terminate the functional assay, the CFTR inhibitor CFTRinh-172 (10 µM; Cystic Fibrosis Foundation Therapeutics) was added to each well of the 384-well plate. Changes in transmembrane potential were normalized to the measurement taken at the time of agonist (i.e., DMSO, or forskolin and VX-770) addition and prior to activation.
Data Analysis
All data are represented as mean ± SEM. Prism 4.0 software (GraphPad Software, San Diego, CA) was used for statistical analysis (nonpaired Student t test), and p values less than 0.05 were considered significant. Each experiment was repeated at least three times.
Results and Discussion
The major advantage of mutagenesis methods described here is that the KAPA HiFi DNA polymerase (engineered B-family, proofreading) has a >100× lower second-site error rate compared with conventional DNA polymerases (e.g., Taq), allowing for direct PCR on the destination vector, rather than PCR with subcloning from an intermediate vector. 22 In addition, this approach does not require complex primer design (a complementary pair is used) or phosphorylation of primers/oligos (typical for mutagenic primers required for traditional primer-“tail”-based insertion/deletion), nor does it require subcloning or enzymatic ligation steps. Furthermore, this PCR-based mutagenesis method is minimalist on many levels, from its reaction volume (25 µL) and required DNA template (1 ng, up to 10 ng), to the time required for PCR cycling (approximately 3 h for a 10 kb vector), which can lead to positive clones identified (from primer design to plasmid sequencing) in as little as 4 to 5 d. Subsequently, these newly generated rare CFTR mutants can be functionally expressed and characterized in a high-throughput fashion within an additional 2 to 3 d, allowing for potential identification of disease-causing genotypes, which could benefit from current CFTR-specific small-molecule therapies (i.e., VX-809 and VX-770) within approximately a single week.
Generation of Site-Directed Insertion and Deletion Variants of CFTR
The strategies for site-directed insertion and deletion mutagenesis are illustrated in Figure 1 , and the characteristics of mutagenesis primers are noted in Figure 2 ; primers for p.Glu1172* (point mutation) are also included. For insertions (27 base pairs in this study, i.e., p.Glu1172-3Gly-6His*), the insert segment is included in the primer pair and directly incorporated into the template plasmid DNA by means of PCR cycling, whereas for deletions (18, 405, and 2532 base pairs in this study, i.e., p.Ile1234_Arg1239del, p.Ser700_Asp835del-CFTR, and p.Gln2_Trp846del-CFTR, respectively) the segment to be removed is excluded from the primer pair. This one-step site-directed insertion and deletion mutagenesis approach takes advantage of the secondary structures of the primers and template for insertion and deletions, respectively, such that a loop or hairpin structure of the noncomplementary sequence forms during PCR to generate the desired change ( Fig. 1 ). Following PCR cycling, the insert becomes integrated or the sequence to be deleted is removed. Although each amplified mutant construct becomes the major fraction present, the remaining starting template (i.e., methylated parental template DNA) is enough to contaminate downstream steps and therefore must be removed to enrich the synthesized mutant construct. To do this, PCR products are incubated with DpnI endonuclease; the mutant plasmid is then used to transform competent Escherichia coli cells, which allows for amplification and selection of positive clones. Furthermore, it must be noted that for the deletion mutagenesis, the actual Tm of the full-length primer can be used to determine the annealing temperature during PCR amplification; however, the annealing temperature for insertion mutagenesis must not take into account the sequence to be added (otherwise, the apparent annealing Tm will be too high). In this case, only the actual (or calculated) Tm of the complementary sequence of the primer pair should be used to determine annealing temperature.
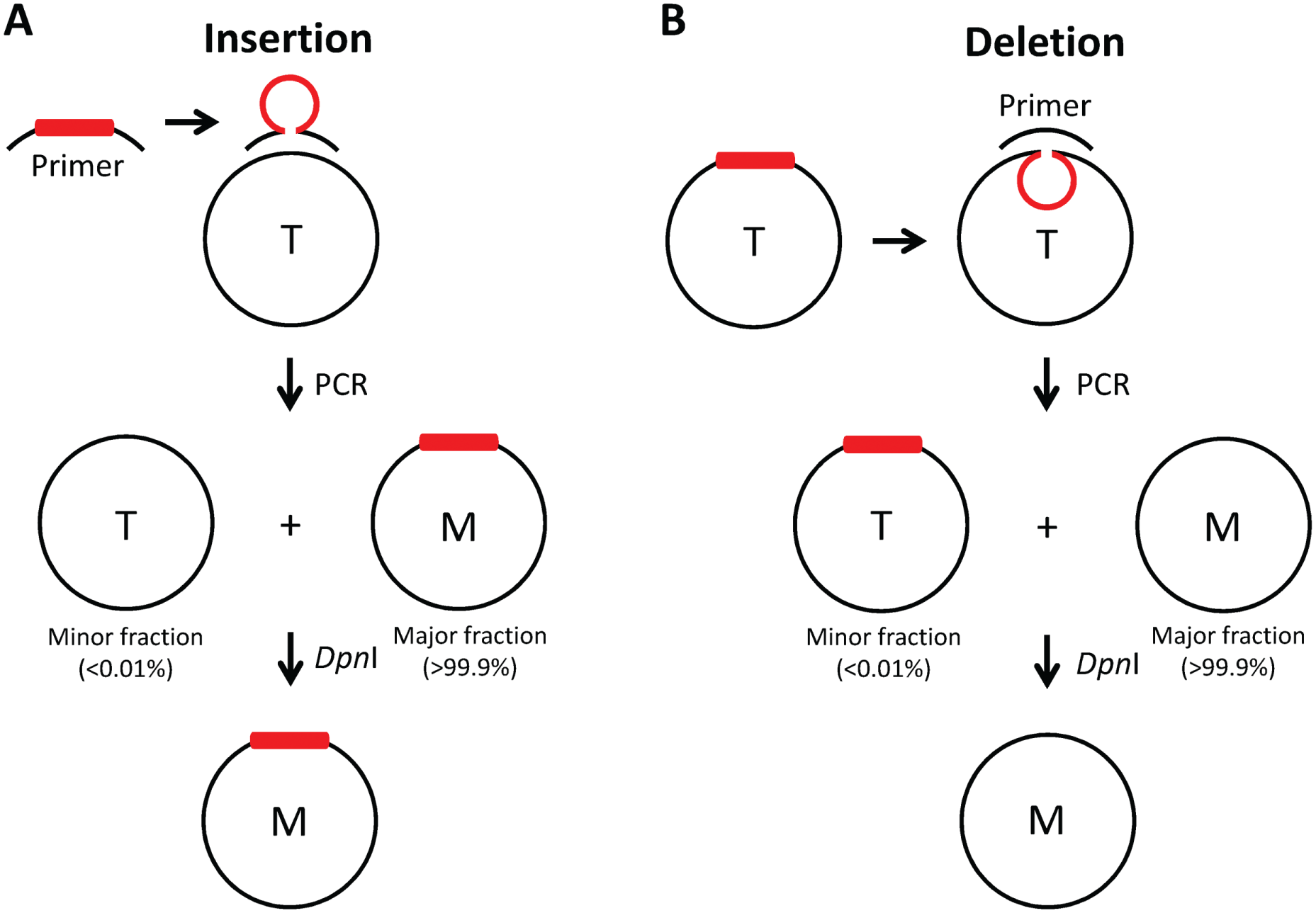
Schematic illustration of the PCR-based method for site-directed insertion and deletion mutagenesis. (
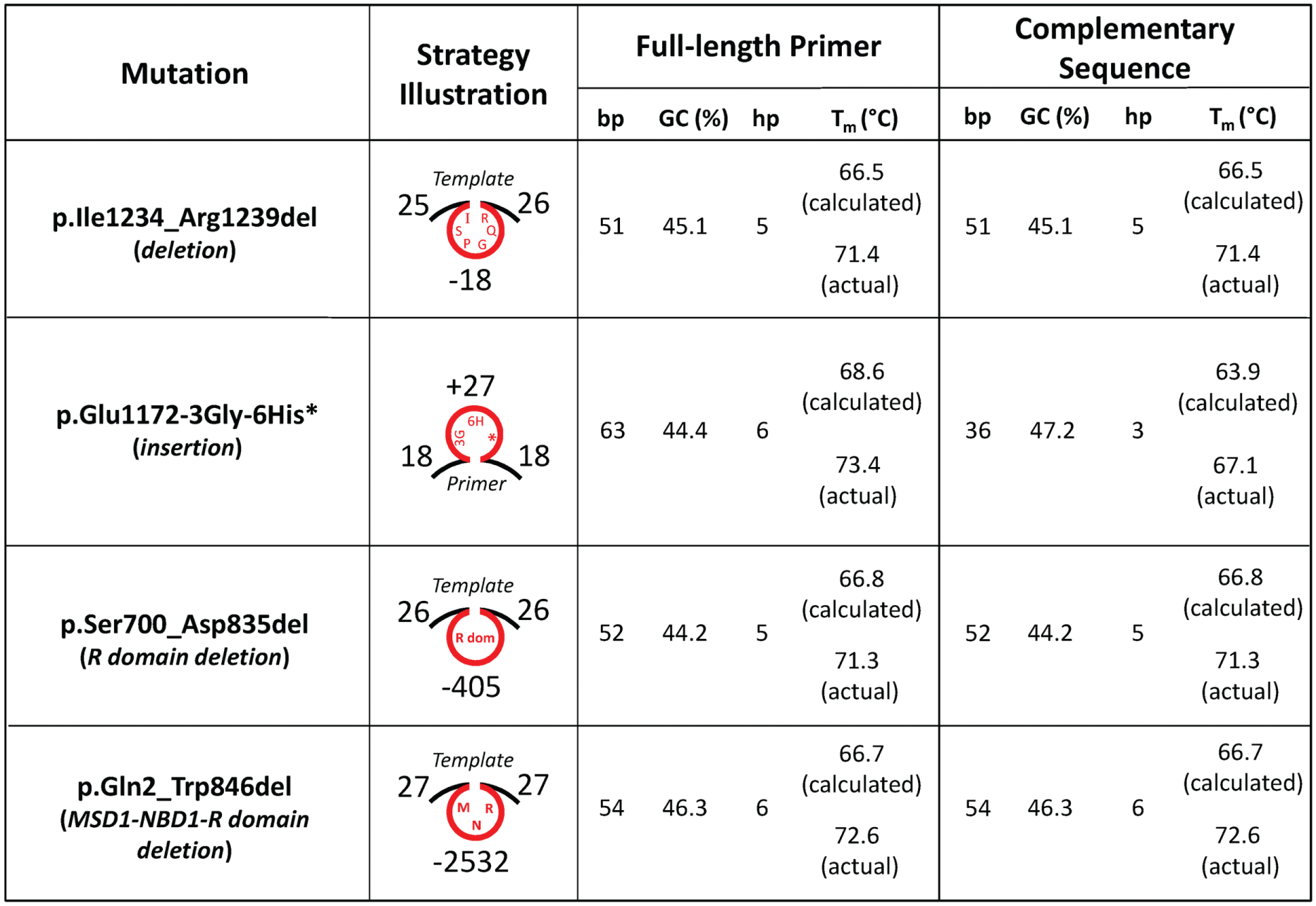
Characteristics of primers used in this study for PCR-based site-directed insertion and deletion mutagenesis. Sense and antisense primer pairs for p.Ile1234_Arg1239del (deletion), p.Glu1172-3Gly-6His* (insertion), p.Ser700_Asp835del (deletion), and p.Gln2_Trp846del (deletion) are described in the Materials and Methods section. Primer and template structures illustrating mutagenesis strategies are also shown (the number of complementary, deleted [–], and inserted [+] nucleotides are noted). For the full-length and complementary sequences of each primer pair, the number of base pairs (bp), percentage guanine-cytosine content (GC), number of calculated primer hairpins (hp; calculated using OligoAnalyzer, Integrated DNA Technologies, Inc., Coralville, IA), and the calculated and actual melting temperatures (Tm in °C; ACGT Corp., Toronto, Ontario, Canada) are tabulated.
Following PCR and DpnI digestion, 10 µL of each reaction was run on 0.8% agarose gels, and PCR products from site-directed deletion (p.Ile1234_Arg1239del) and insertion (p.Glu1172-3Gly-6His*) mutagenesis are shown in Figure 3 and Figure 4 , respectively. In Figure 3 , PCR for the deletion mutant was performed using 1 and 10 ng of template DNA at two annealing temperatures (Tm-10 °C and Tm-5 °C; Fig. 3A ). Tm-5 °C reactions did not yield a PCR product likely because the temperature (66.4 °C) was too close to the extension temperature of the DNA polymerase, and therefore successful primer annealing could not occur. However, the PCR products from the Tm-10 °C (i.e., 61.4 °C) reactions yielded a major band at ~10 kb (the expected size of the full-length vector containing CFTR cDNA; 500 ng template DNA was included as a control), which was the desired product. This PCR product was then transformed into competent cells, and the tabulation underneath the agarose gel shows that although many colonies grew on antibiotic selective agar plates, from the colonies that were picked (10 and 15 from PCR reactions starting with 1 ng and 10 ng template DNA, respectively), only a few produced high-quality DNA from minipreps. From the six successful DNA preps, only two had the desired mutation (33.3% efficiency in terms of the presence of the mutation in isolated DNA, whereas 8% efficiency in terms of colonies picked). Although these efficiency rates are low, it must be noted that one successful mutant clone is sufficient for all downstream structure-function applications; therefore, to isolate one successful deletion clone at this efficiency, we suggest that at least 12 colonies are grown from each site-directed deletion mutagenesis attempt. Furthermore, it must be noted that these relatively low efficiencies are at least partially caused by the direct transformation of PCR reaction mixtures, rather than transformation of gel purified PCR products; this would likely increase the efficiencies several-fold (as has been shown for other CFTR mutant constructs by our lab group, data not shown).
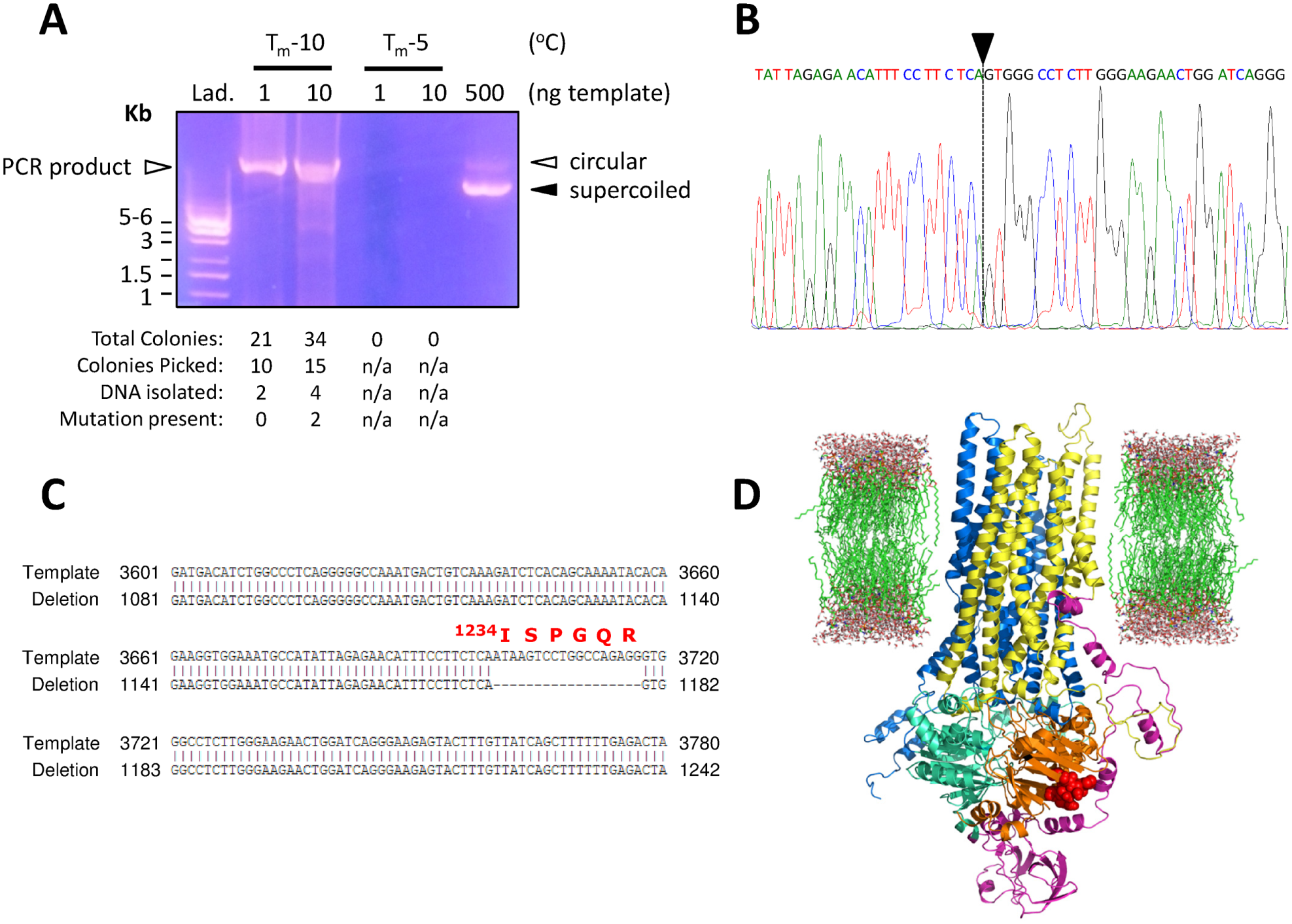
PCR amplification and analysis of deletion mutagenesis generating p.Ile1234_Arg1239del-CFTR cDNA. (
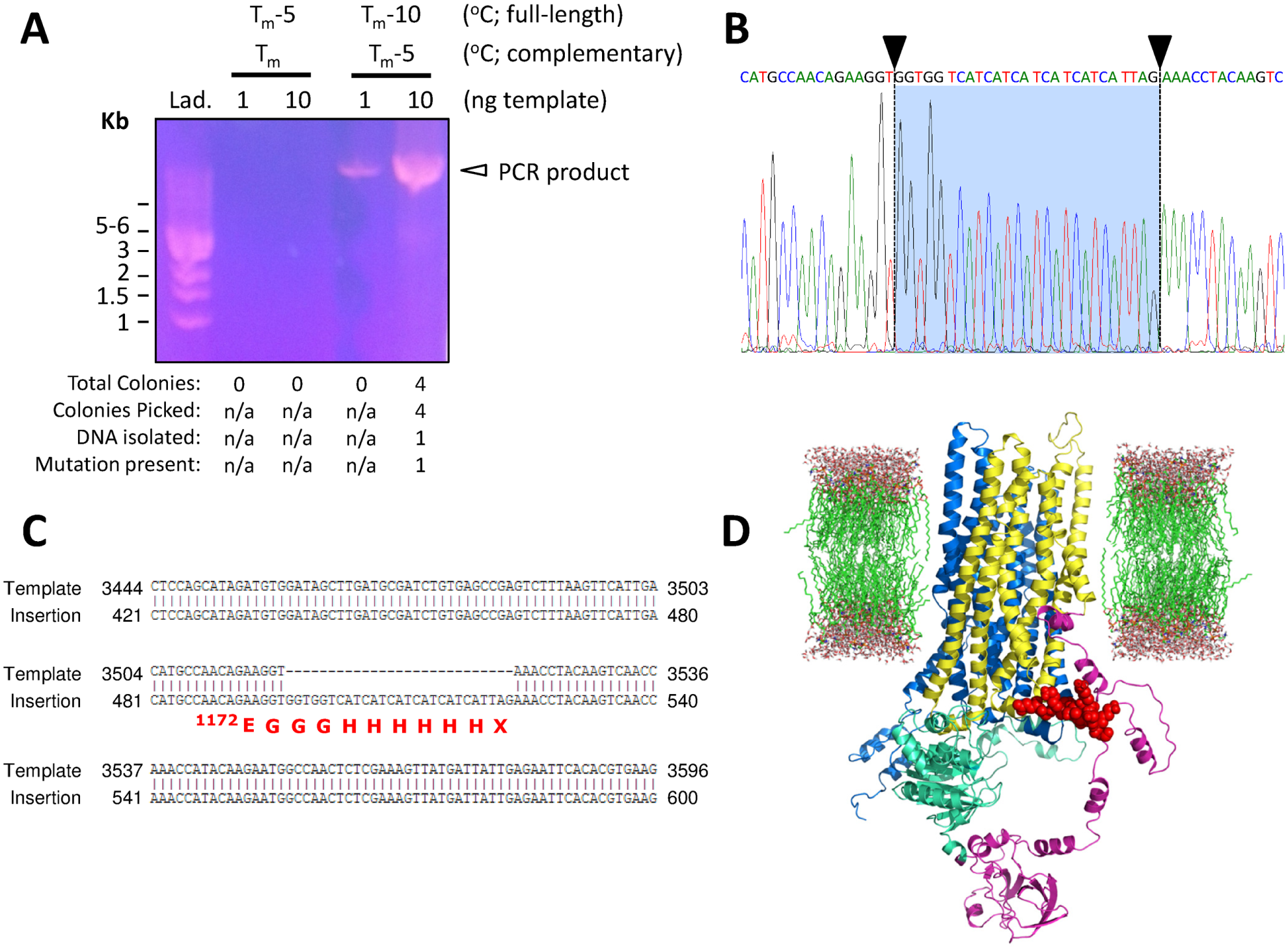
PCR amplification and analysis of insertion mutagenesis generating p.Glu1172-3Gly-6His*-CFTR cDNA. (
To further characterize the nucleotide sequence of the site-directed deletion mutant, we sequenced the entire cDNA of CFTR (nucleotides 1–4443, with stop codon) and show a short segment of the electrophoretogram containing the desired mutation ( Fig. 3B ). Here, the sequence omits the wild-type nucleotides, denoted by a black arrowhead and a vertical dashed line, suggesting that the deletion is present; this is more apparent when aligned with the cDNA sequence of wild-type CFTR ( Fig. 3C ). Further, we identify the location of the consequence of this 18 base pair deletion (i.e., p.Ile1234_Arg1239del) and highlight these six amino acids (within NBD2) in the tertiary structure of a protein homology model of wild-type CFTR, which is based on the structure of a related bacterial ABC transporter protein, Sav1866 ( Fig. 3D ). 23
Similarly, PCR for the insertion construct was performed using 1 and 10 ng of template DNA at two annealing temperatures (Tm-10 °C and Tm-5 °C in terms of the full-length primers; Tm-5 °C and Tm-0 °C in terms of the complementary primer sequences; Fig. 4A ). Tm-0 °C (complementary sequence of primers) reactions did not yield a PCR product because the temperature used (68.4 °C) was not below the Tm and therefore could not anneal to the template; this reaction acted as a control. However, the PCR products from the Tm-5 °C (complementary sequence of primers, i.e., 62.1 °C) reactions yielded a major band at ~10 kb (larger yield with 10 ng template). PCR products were then transformed, and the tabulation underneath the DNA gel shows that few colonies (4) grew on antibiotic-selective agar plates. When these colonies were picked and grown in liquid culture supplemented with antibiotics, only one grew, leading to the isolation of just one DNA miniprep clone. Fortunately, this clone was sequenced and found to contain the desired insertion mutation (25% efficiency in terms of colonies picked). Although this efficiency rate is low, again it must be emphasized that one mutant clone is sufficient for further biochemical analyses. Therefore, we suggest that in order to isolate one successful insertion clone at this rate of efficiency, screening of at least four colonies is necessary.
To further characterize the nucleotide sequence of the site-directed insertion construct, we sequenced the entire cDNA of CFTR (nucleotides 1–4443, with stop codon) and show a short segment of the electrophoretogram containing the desired mutation ( Fig. 4B ). Here, the sequence contains additional nucleotides that are not present in wild type, denoted by black arrowheads and vertical dashed lines (on a blue background), suggesting that the insertion is present; this is more apparent when aligned with the cDNA sequence of wild-type CFTR ( Fig. 4C ). Further, we highlight the location of the consequence of this 27 base pair insertion (i.e., p.Glu1172-3Gly-6His*) by coloring the poly-His tag in red (immediately following MSD2), as well as omitting NBD2 (because this mutation causes deletion of this domain; for reference, NBD2 is shown in orange in Fig. 3D ) in the CFTR protein structural model ( Fig. 4D ). 23
Finally, although not extensively reported in this work, p.Glu1172*-CFTR (point mutation) was also generated in this study (
Fig. 2
). Using an annealing temperature of Tm-5 °C (55.0 °C), 1 ng and 10 ng of template DNA yielded two and five colonies after transformation, respectively, which led to the successful growth of two and three antibiotic-selective liquid cultures, respectively, and sufficient miniprep DNA for sequencing (two from each). DNA sequencing determined that all four clones were positive for the desired point (stop) mutation, yielding a truncated CFTR variant lacking NBD2 and without a poly-His tag (57% efficiency in terms of colonies picked, 100% efficiency in terms of mutation within isolated DNA preps). These higher efficiencies are likely correlated to yield and probably due to the relatively simpler mutagenesis strategy involving a single nucleotide change (i.e.
Biochemical and Functional Analysis of Mutant CFTR Constructs
Following generation of the deletion (p.Ile1234_Arg1239del-CFTR) and insertion (p.Glu1172-3Gly-6His*-CFTR) constructs, we transiently transfected HEK cells and used immunoblots to evaluate steady-state levels of CFTR protein following incubation at physiological temperature (37 °C) for 24 h ( Fig. 5A ). Compared with wild-type CFTR, we found that p.Ile1234_Arg1239del-CFTR is misprocessed, as previously reported, 13 yielding immature, core-glycosylated protein as the major form of this CFTR variant, whereas p.Glu1172-3Gly-6His*-CFTR appears to have similar levels of mature and immature protein as that of wild-type CFTR, which is also comparable to previous reports 15 ; however, p.Glu1172-3Gly-6His*-CFTR lacks NBD2 (approximately ~30 kDa) and therefore appears to migrate faster on SDS-PAGE, having a lower apparent molecular weight than wild-type CFTR ( Figs. 5A and 5B ). When compared with the NBD2 deletion mutant lacking the poly-His tag (i.e., p.Glu1172*-CFTR), p.Glu1172-3Gly-6His*-CFTR appears to migrate slightly slower on SDS-PAGE ( Fig. 5C , denoted with asterisks). Although the tagged variant contains nine additional amino acids (apparent ~1 kDa increase in molecular weight), we speculate that this alone does not account for the difference in migration; instead, the nature of the amino acids (i.e., 6 His residues, which are positively charged during SDS-PAGE), together with the additional number of residues, collectively contributes to this difference.
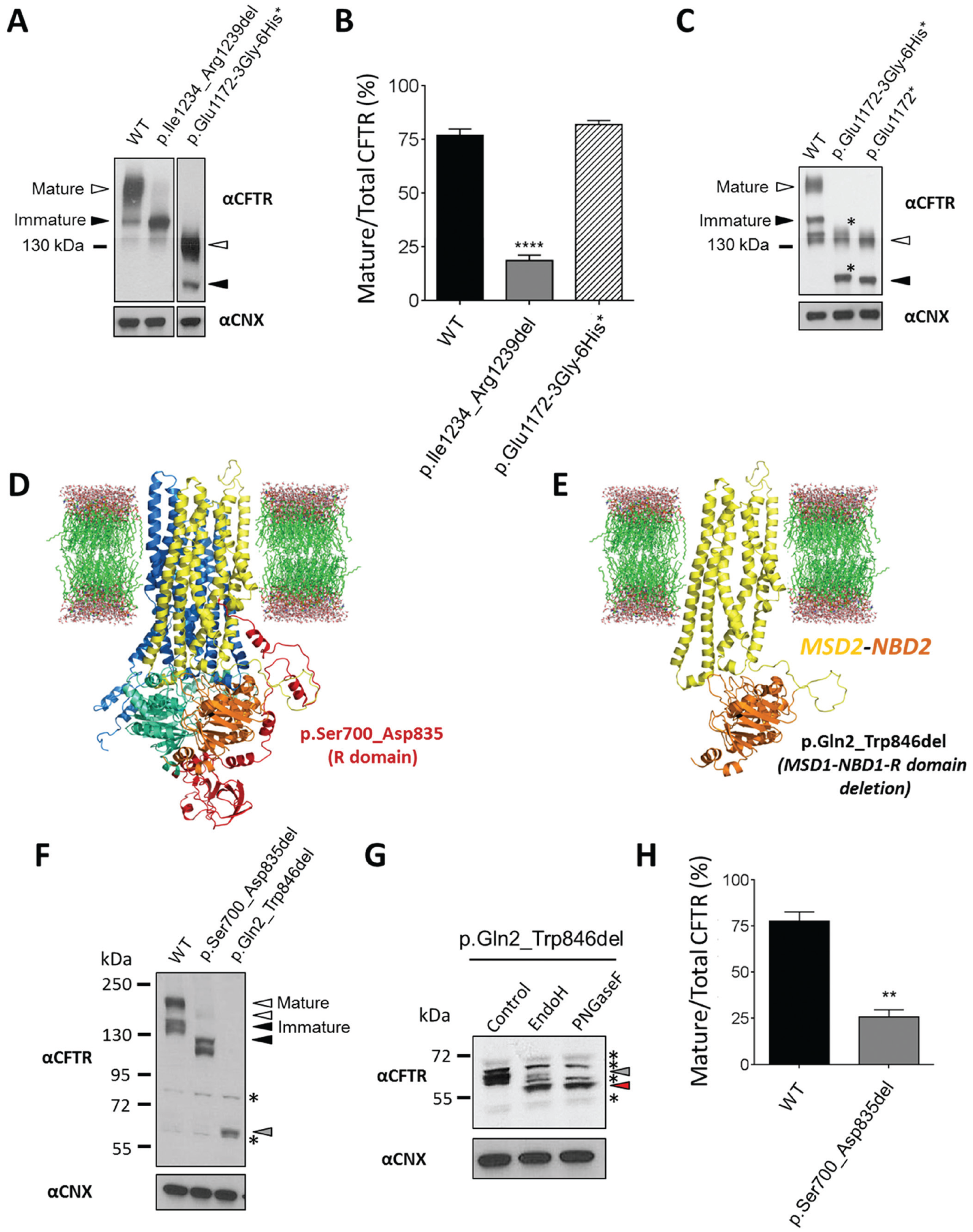
Relative expression levels of CFTR mutant constructs: p.Ile1234_Arg1239del-CFTR, p.Glu1172-3Gly-6His*-CFTR, p.Ser700_Asp835del-CFTR, and p.Gln2_Trp846del-CFTR. (
In addition, to assess whether our deletion mutagenesis strategy was robust, we generated p.Ser700_Asp835del-CFTR and p.Gln2_Trp846del-CFTR mutant constructs, which lack the R domain or MSD1-NBD1-R domain, respectively; these deletions are highlighted in the CFTR protein structural model ( Fig. 5D and 5E ). 23 We then transiently transfected HEK cells and used immunoblots to evaluate steady-state levels of CFTR protein following incubation at physiological temperature (37 °C) for 24 h ( Fig. 5F and 5H ). Compared with wild-type CFTR, we found that p.Ser700_Asp835del-CFTR migrates slightly faster (due to the loss of 135 residues) and is misprocessed, yielding immature, core-glycosylated protein as the major form of this CFTR variant, as previously described. 24 We also found that p.Gln2_Trp846del-CFTR appeared to have one major band that migrated substantially faster on SDS-PAGE (becuase of the loss of 844 residues), which we determined to contain both core- and complex-glycosylated forms due to relative endoglycosidase sensitivity to either EndoH (recognizes immature, core-glycosylation) or PNGaseF (mature, complex-glycosylation) glycosidases ( Figs. 5F and 5G ). However, we were not able to precisely discern and thus quantify the relative abundance of each form, but we speculate that the core-glycosylated form is more abundant based on these studies.
Using the FLIPR system, agonist-mediated (e.g., forskolin) CFTR activation is detected as membrane depolarization using a plate reader, whereas CFTR inhibition (mediated by CFTRinh-172) is detected as membrane repolarization (or hyperpolarization), both of which are probed using the same fluorescent membrane potential sensor dye within the same experiment. In Figure 6A , we evaluated the channel activity of p.Ile1234_Arg1239del-CFTR (pharmacologically rescued with the p.Phe508del-CFTR corrector VX-809), p.Glu1172-3Gly-6His*-CFTR, and p.Ser700_Asp835del, with HEK cells (without transfection of CFTR, as a negative control) and WT-CFTR (positive control) together on 384-well plates. 21 We tested the function of p.Gln2_Trp846del-CFTR, but it did not support chloride channel activity, presumably because this mutant protein lacks a substantial portion of CFTR, which is required for function (data not shown). These findings were similar to HEK cells not expressing CFTR ( Fig. 6B ) and therefore significantly different from responses of WT-CFTR ( Fig. 6C ). Because p.Ile1234_Arg1239del-CFTR is misprocessed, like the major CF disease-causing mutation p.Phe508del, we pharmacologically (partially) repaired the trafficking defect as previously reported, 13 to assess functional activity of mature, plasma membrane–associated p.Ile1234_Arg1239del-CFTR. Subsequently, we found that the adenylate cyclase activator forskolin, together with the CFTR-specific potentiator VX-770, activated both p.Ile1234_Arg1239del-CFTR ( Fig. 6D ) and p.Glu1172-3Gly-6His*-CFTR ( Fig. 6E ) to comparable levels; VX-770-dependent activation of the CFTR mutant lacking NBD2 is in agreement with recent reports. 25 In addition, we found that the CFTR inhibitor CFTRinh-172 inhibited both mutant proteins and reduced functional activation of both back to near baseline levels. Taken together, these findings suggest that each CFTR variant can be activated and inhibited with CFTR-specific modulators and that repurposing modulators of p.Phe508del-CFTR (i.e., VX-809) or p.Gly551Asp-CFTR and other CFTR gating mutations (VX-770) could be useful for other CFTR mutations. Importantly, for p.Ser700_Asp835del-CFTR, we found that the basal (forskolin- and VX-770-independent) channel activity was higher than that of wild-type CFTR (data not shown), which is in agreement with previous reports. 26 Interestingly, this mutant protein could be stimulated with forskolin and VX-770, suggesting that the R domain does not comprise the VX-770 binding site ( Fig. 6F ). These data suggest that p.Ser700_Asp835del-CFTR forms a partially unregulated chloride channel, which is sensitive to CFTR-specific modulators, and furthermore that this mutant protein may be a useful tool for identification of binding sites for certain small-molecule modulators. Furthermore, we found that the kinetics of inhibition by CFTRinh-172 were variable between CFTR mutants, and the mechanism of these responses is currently being investigated.
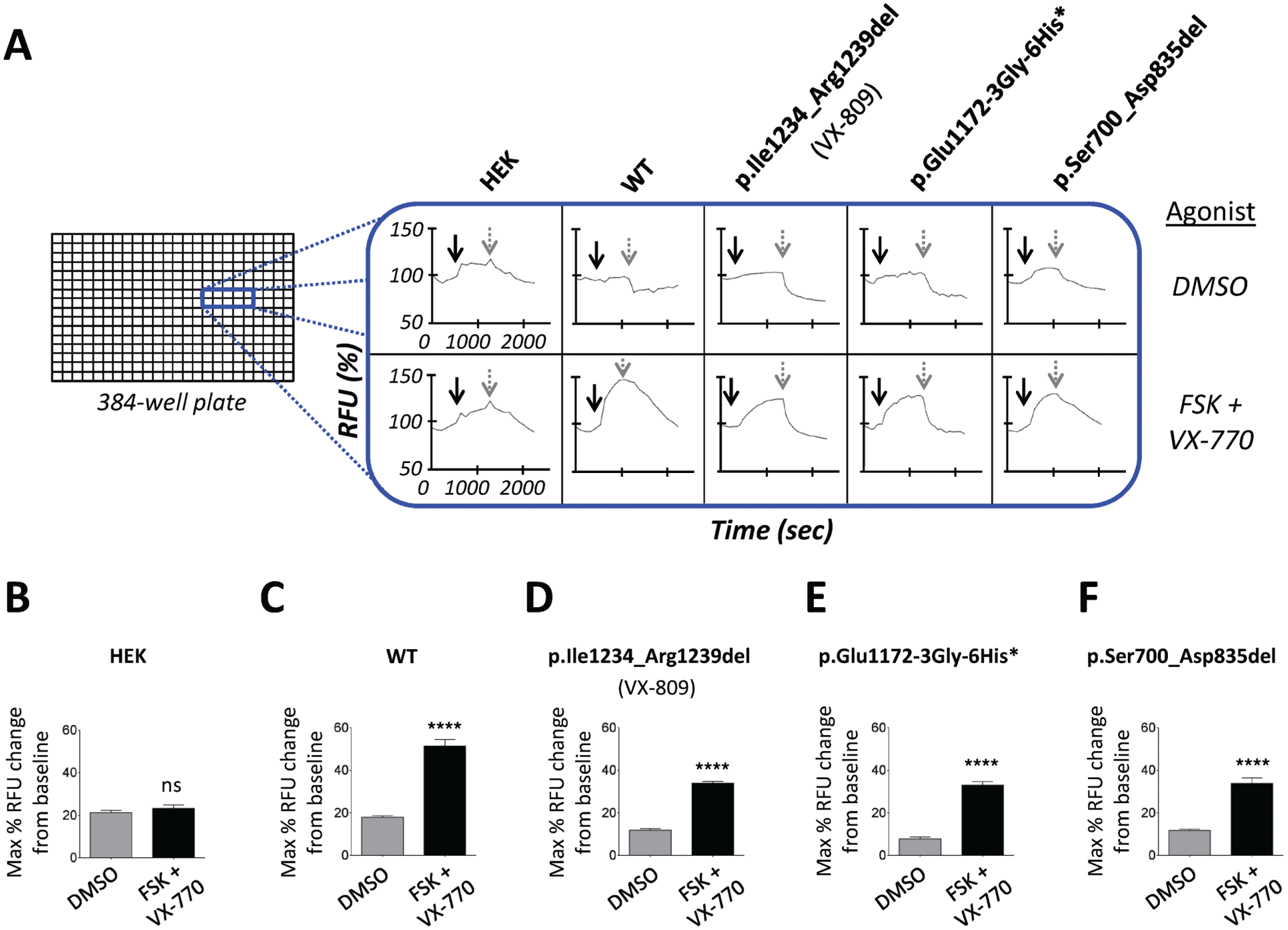
High-throughput functional analysis of CFTR mutants using the fluorometric imaging plate reader membrane depolarization assay. (
Using this high-throughput approach, future screening efforts may identify novel, potent, efficacious, and mutation-specific compounds for these and other rare and previously uncharacterized CFTR variants. In addition, this method has the potential to be sufficiently different yet equally advantageous when compared with the current “blind” drug-screening paradigm employed by academics and pharmaceutical companies (previously reviewed by our group27,28), such that instead of screening large libraries of compounds on a single CFTR mutant, we could now screen a cohort of rare CF disease–causing mutations against the most efficacious and CFTR-specific small molecules (i.e., VX-809 and VX-770) to identify genotypes that could potentially benefit from the current therapeutic regimen.
Application Toward Drug Discovery in Rare CF Disease–Causing Mutations
One major advantage of this work is the ability to rapidly screen multiple rare CFTR variants for compounds that functionally rescue mature CFTR protein in a high-throughput assay. Here, we validated our site-directed insertion/deletion mutagenesis and functional assay using four CFTR variants, by employing previously identified modulators of CFTR activity (i.e., those for p.Phe508del-CFTR, as well as p.Gly551Asp and other CFTR gating mutations, VX-809 and VX-770, respectively). It will be possible in the future to translate this strategy to evaluate the most efficacious CFTR-specific small molecules (i.e., VX-770 and VX-809) against all of the rare CF disease–causing insertion, deletion, and truncation mutations simultaneously (i.e., on 384-well plates), although it may soon be necessary to increase the throughput of our mutagenesis strategy, using tools that have been recently described. 29 This novel approach would allow for rapid determination of potential mutation-specific responders to the current therapeutic regimen for individuals with CF. Likewise, it may also be feasible to compare responses of the most common 100 to 200 missense and deletion mutations (e.g., those described in CFTR2: www.cftr2.org) to existing and/or emerging corrector and potentiator compounds. For example, by simultaneously evaluating novel translational “read-through” agents for CF disease–causing mutations conferred by premature termination codons (e.g., p.Gly542*, p.Arg553*, p.Arg1162*, and p.Trp1282*), these CFTR mutants can be readily generated and functionally characterized for responders within a short time frame. 30 Further, the necessary next steps would include functional analysis of primary tissues from patients with these rare CF disease–causing mutations, pending compound validation in our high-throughput overexpression system. However, studies involving a cohort of patient mutations in our HEK overexpression system will facilitate stratification into groups that may have potential therapeutic response, thereby justifying the acquisition of biopsies for in vitro analysis. Finally, this integrated mutagenesis method with functional screening strategy may also hold potential application toward other protein-folding diseases as well, by screening for novel therapeutic modulators and personalized medicines for patients with rare and uncharacterized mutations in various channelopathies (including those of K+, Na+, Ca2+, and other Cl− channels), which cause a wide variety of neurologic, cardiac, skeletal muscle, renal, and endocrine diseases.
Footnotes
Acknowledgements
We are grateful to Ling Jun Huan (Hospital for Sick Children, Toronto, Ontario, Canada) for providing helpful commentary on the article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These studies were supported by Operating Grants to C.E.B. from the Canadian Institutes of Health Research (CIHR MOP# 97954, CIHR GPG-102171) and Cystic Fibrosis Canada. S.V.M. was supported by J.A. Connolly and Ontario Graduate Studentships. S.A. was supported by the H.W.C. Clayton Paediatric Research Studentship.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.