
Abstract
Potassium (K+) channels, with their diversity, often tissue-defined distribution, and critical role in controlling cellular excitability, have long held promise of being important drug targets for the treatment of dysrhythmias in the heart and abnormal neuronal activity within the brain. With the exception of drugs that target one particular class, ATP-sensitive K+ (KATP) channels, very few selective K+ channel activators or inhibitors are currently licensed for clinical use in cardiovascular and neurological disease. Here we review what a range of human genetic disorders have told us about the role of specific K+ channel subunits, explore the potential of activators and inhibitors of specific channel populations as a therapeutic strategy, and discuss possible reasons for the difficulty in designing clinically relevant K+ channel modulators.
Potassium Ion Channels
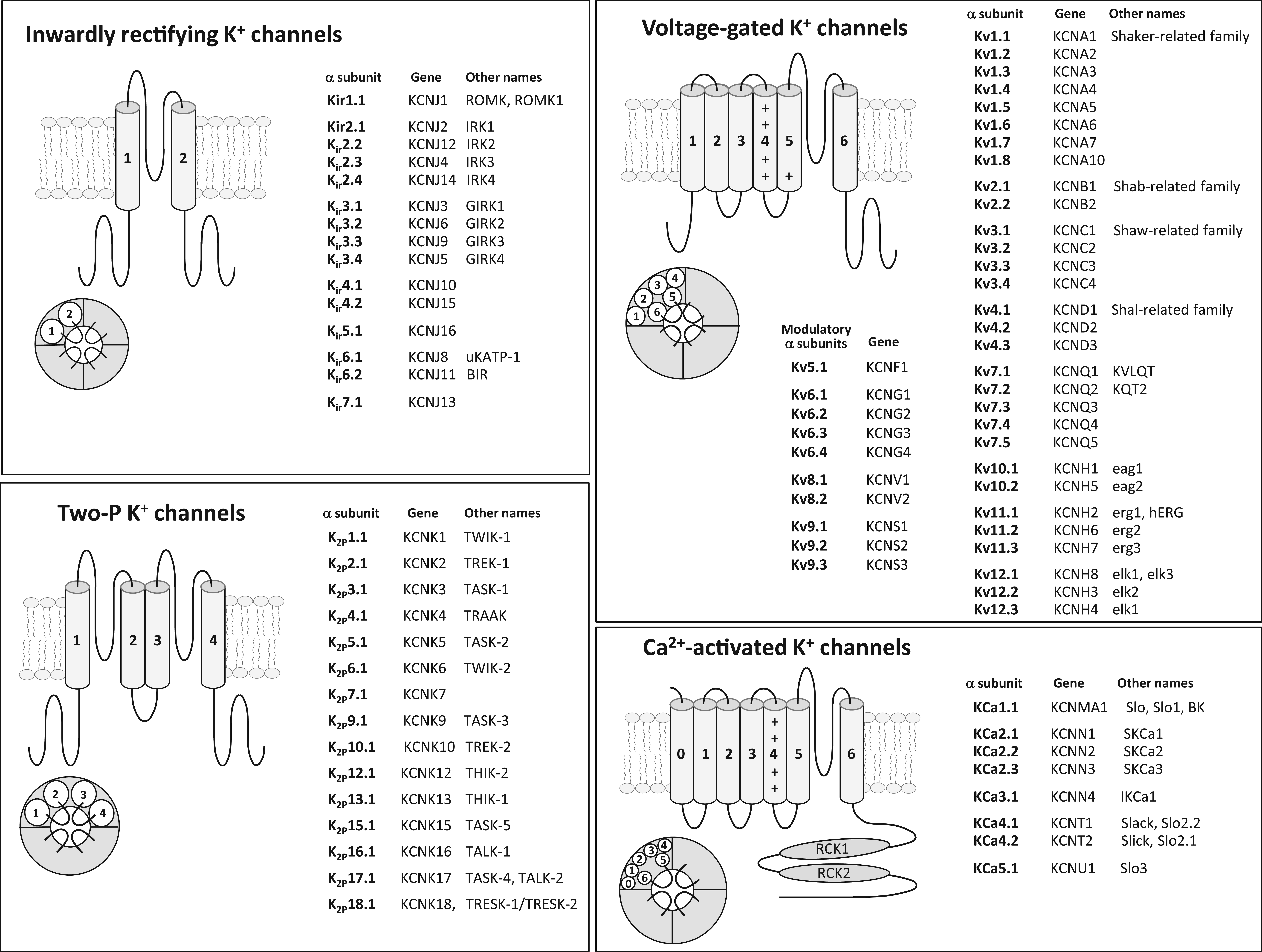
Potassium (K+) channels are a large family of integral membrane proteins that form aqueous pores in cell membranes through which K+ can flow. They are unique among ion channels in that they are found in virtually all types of cells in all organisms where they perform a range of biological functions. In the human genome, K+ channels are by far the largest and most diverse of the ion channel families, with almost 80 different genes encoding the principal pore-forming subunits ( Fig. 1 ).1–4 In most cells, they play an essential role in maintaining and stabilizing the resting membrane potential. The opening of K+ channels, which occurs in response to a range of different signals, leads almost universally to the efflux of K+ from the cell, causing the membrane potential to become more negative. In nerve and muscle cells, their ability to repolarize or hyperpolarize the membrane helps them control action potential frequency and duration, while other functions include regulation of neurotransmitter release and hormone secretion, potassium homeostasis, epithelial electrolyte transport, cell proliferation, apoptosis, and tumor progression.5–8
Disruption of genes encoding K+ channel subunits and subsequent loss or gain of channel function is linked to a range of human diseases, including hyperinsulinemia, disturbances of the heart rhythm, and some types of epilepsy.5,9,10 K+ channels can also be subject to pathological inhibition by autoantibodies, leading to diseases such as acquired neuromyotonia 11 and certain forms of epilepsy and encephalitis.12–15 These disorders have often helped to clarify the roles of particular channel populations within complex physiological systems and raise the possibility that activation or inhibition of selective K+ currents within cells could be a viable therapy. Indeed, K+ channel modulators are common medicines in certain diseases 16 ; for example, in the treatment of diabetes, the oral antihyperglycemics such as glibenclamide, nateglinide, and glipizide all inhibit adenosine triphosphate (ATP)–sensitive (KATP) channels. Most of the type III antiarrhythmics, including amiodarone, increase the cardiac refractory period by blocking several different types of K+ channel. 17 In both epilepsy and hypertension, there are examples of drugs that target K+ channels. 16 However, considering the scope for clinical impact, their membrane localization, diversity, and often defined tissue distributions, K+ channels remain underexploited as a target in drug discovery. This may be due to a number of factors. The sheer diversity of K+ channel subunits and their ability to form heteromultimeric complexes with different pore-forming subunits and accessory proteins means that the precise composition of functional channels within a particular tissue in vivo is often ill-defined. This makes predicting the functional outcome of channel loss or activation difficult and leads to unpredictable side effects even for specific activators/inhibitors. Drug screening programs also seem to find it difficult to identify selective channel activators. The direct measurement of ion channel activity by manual patch clamping is the best approach for assaying ion channel function but is time-consuming and unsuited to high-throughput screening (HTS). HTS techniques such as ligand binding or ion flux assays, on the other hand, lack the complexity or resolution to detect subtle shifts in channel-gating kinetics that functionally may have profound effects on channel activity. The introduction of screening methods based around fluorometric dyes, which measure changes in ion concentration or cell membrane potential, coupled to fluorescent plate readers with in-built electrical field stimulators, has improved temporal resolution but still represents an indirect measurement of channel activity. 18 More recent advances in automated electrophysiology using planar-array patch-clamp technology circumvent many of the problems of functional resolution and have the potential to screen large compound libraries, 19 and yet selective compounds remain elusive. One reason for this may be that channel modifiers often need to bind to relatively inaccessible sites within the channel pore or in clefts on or near regulatory domains. These sites may be relatively unforgiving to small structural changes within compounds, making chemical optimization of lead structures difficult. In this context, it is interesting to note that the K+ channel family that has the highest proportion of clinically used activators/inhibitors is the ATP-sensitive K+ (KATP) channel family where modifiers appear to work by interacting at exposed peripheral sites on accessory subunits. 20
Given their newly discovered roles in regulating cell proliferation and promoting tumor progression, drugs acting on K+ channels are likely to be of increasing clinical relevance.7,21–23 Here we review what disease-causing genetic disruption of specific K+ channel subunits in humans has told us about the functional role of K+ channels, explore the potential of selective activators and inhibitors as a therapeutic strategy, and further discuss possible reasons for the difficulty in designing clinically relevant K+ channel modulators. Due to the breadth of the field, we have limited our scope to K+ channels within cells of the human nervous and cardiovascular systems. A number of excellent recent reviews exist on disease-causing mutations in K+ channels within tissues such as the pancreas and nonvascular muscle and the role of drugs that target these channels. We will direct the reader to these, and others, throughout.
K+ Channel Diversity
Structurally, K+ channels form from the association of (usually) four pore-forming α subunits often in association with modulatory accessory subunits. They can be grouped into the following four major classes1–4 ( Fig. 1 ):
The diversity of K+ channels and ability for different subunits within a family to associate to form heteromers often makes it difficult to determine the molecular makeup of channel populations in vivo and thus to assign functional roles. This has been aided to some extent by investigations into human channelopathies, a range of diseases where the genetic disruption of channel subunit activity leads to a distinct phenotype.
Neuronal K+ Channels
Due to the equilibrium potential for K+ (mammalian cells ~–85mV), the opening of K+ channels generally mediates outward K+ currents that act to dampen cellular excitability. Loss of function of several types of K+ channel is thus associated with conditions characterized by neuronal hyperexcitability. This includes several forms of epilepsy ( Table 1 ), a disorder characterized by abnormal firing of neuronal networks within the brain due to an imbalance between network excitation and network inhibition.
Neuronal K+ Channelopathies.

EAST, epilepsy, ataxia, sensorineural deafness, and tubulopathy; NA, not applicable; SeSAME, seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance.
KV1.1-Containing Channels
The KV1 family includes mammalian homologues of the Shaker K+ channels originally cloned in Drosophila where mutation of the Shaker (Sh) gene leads to a characteristic trembling of the legs following etherization. 35 KV1.1 is known to associate with other Shaker-related channels (KV1.2, KV1.4) to form heteromeric channel complexes in various regions of the brain where they control neuronal excitability, action potential propagation, and synaptic transmission ( Fig. 2 ). 6 In humans, a single loss-of-function mutation of KV1.1 is associated with episodic ataxia type 1 (EA1), an autosomal dominant neurological condition characterized by continuous involuntary muscle quivering (myokymia) and bouts of severe contractions of head and limb muscles, leading to loss of coordination and balance. 36 Seizures have been linked to dysfunction of neuronal networks within the hippocampus, a region of the brain located in the medial temporal lobe and often associated with epileptic seizure. Channels containing the KV1.1 subunit have been identified in both the axons and synaptic terminals of hippocampal neurons in the rat, and mutations associated with EA1 in humans cause reduced current amplitude and a positive shift in voltage activation of KV1.1-containing channels, consistent with reduced channel activity.37,38

Differential localization of K+ channel subtypes in neurons. KV1.1-containing channels are expressed in the axon and presynaptic terminal, where they regulate neuronal excitability, action potential propagation, and synaptic transmission. KV4.3-containing channels are expressed in dendrites and are involved in regulating the spread of back-propagating action potentials in the dendritic tree. KV7.2/7.3 channels, which form the M-current, are expressed in the axon initiation segment and are active at subthreshold membrane potentials where most voltage-gated K+ channels are closed. They act to dampen excitability and repetitive firing in neurons. KCa1.1-containing channels are expressed in the presynaptic terminal, where they localize with voltage-gated Ca2+ channels. They are activated by the Ca2+ influx that occurs in response to action potential–induced terminal depolarization and act to terminate the action potential and generate after-hyperpolarizations that close Ca2+ channels and reduce neuronal excitability. Kir4.1-containing channels are expressed in glial cells, where they are potentially involved in the redistribution of K+. See text for details.
KV1 Modulators as Drugs
Researchers at Wyeth, now part of Pfizer (New York, NY), have published on novel small-molecule inhibitors of protein-protein interactions that act to modulate KV1.1 activity by blocking channel inactivation. 39 In the hippocampus, KV1.1 is coexpressed with accessory KVβ1 subunits, which convert KV1.1 from a slowly inactivating delayed rectifier-type current into a fast inactivating current. This increases neuronal excitability by reducing the sustained hyperpolarizing current that flows through KV1.1 and increasing the ability of the neuron to fire repetitively. A novel approach to reducing neuronal excitability is therefore to prevent inactivation of KV1.1 channels. A yeast two-hybrid screen identified a number of small-molecule “disinactivators” that most likely interact with sites on the KVβ1 N-terminus or its receptor site on KV1.1 and prevent it binding and inhibiting KV1.1 channels. 39 It is suggested that drugs based on these disinactivators may ultimately be useful for preventing inactivation of KV1.1 channels in the brain and thus reducing neuronal hyperexcitability in diseases such as epilepsy. A key advantage of this approach is that, unlike existing anticonvulsants, it would not prevent a neuron from responding to excitatory stimuli but would instead act predominantly to dampen repetitive firing.
There is also therapeutic potential in blocking the activity of functional KV1 channels and increasing neuronal excitability. The organic compound 4-aminopyridine (4-AP, fampridine) has been used extensively as a pharmacological tool to study the functional properties of KV1 channels for which it is a reasonably selective blocker. 40 4-AP (marketed as Ampyra in the United States and Fampyra in Europe) was approved by the Food and Drug Administration (FDA) in 2010 and licensed in the United Kingdom in 2011 for use in the treatment of multiple sclerosis, having been shown to improve walking speed in patients with multiple sclerosis in two clinical trials. 4-AP’s therapeutic effect has not been fully elucidated, but it most likely functions by blocking the prolonged hyperpolarizing currents that flow through KV1 channels, shortening the relative refractory period and increasing axonal conduction.
KV3-Containing Channels
Channels in the KV3 Shaw subfamily activate rapidly at high-voltage thresholds (~−10 mV) and have very fast deactivation rates. This allows them to open during the peak of the action potential to speed up membrane repolarization and enable repetitive neuronal firing at high frequencies. 41 KV3.3 is expressed throughout the central nervous system, particularly in cerebellar Purkinje neurons. Mutations in KV3.3 cause the autosomal dominant neurological disorder SCA13 (spinocerebellar ataxia type 13), which leads to degeneration of the cerebellum and the spinal cord. 42 The four main disease-associated mutations in KV3.3 lead to either reduced channel expression or channels with altered gating properties when expressed in Xenopus oocytes. 43 The reduction in protein levels arises due to a reduction in protein half-life as SCA13 mutations generate unstable proteins that are rapidly degraded. Interestingly, mutant KV 3.3 protein levels could be partially restored by treatment with trimethylamine N-oxide, a chemical chaperone that stabilizes the mutant protein and helps folding. 43 This suggests that identification of small-molecule chaperones may be a novel approach to partially rescuing channel activity.
KV4-Containing Channels
Channels containing KV4 Shal subunits mediate the fast-inactivating “A-type” current in dendrites of hippocampal neurons ( Fig. 2 ). 44 These channels are active at subthreshold potentials and are believed to regulate firing frequency and the spread of excitability in the dendritic tree. Action potentials initiated in the axon hillock propagate down the axon but also invade the soma and dendrites (back-propagating action potentials) to inform dendritic synapses that the neuron has fired. 45 Summation of back-propagating action potentials and excitatory synaptic inputs received by the dendritic tree is believed to be the basis of dendritic signal integration. The activity of dendritic A-type channels limits the spread of back-propagating action potentials and the regulation of KV4 expression, localization, and kinetics, thus modulates certain aspects of dendritic signal processing. 44 Truncation of the KV4.2 subunit, resulting in a decrease in dendritic A-current density, has been associated with temporal lobe epilepsy. 46 The most common form, mesial temporal lobe epilepsy (MTLE), arises in the medial aspect of the temporal lobe where the hippocampus, parahippocampal gyrus, and the amygdala are located. A reduction in dendritic A-current would be expected to lower the firing threshold for action potentials as well as increasing the spread of back-propagating action potentials. Interestingly, seizures have been shown to induce surface recruitment of KV4.2 subunits in thalamocortical neurons, which relay sensory information to the cerebral cortex, presumably in a feedback response to reduce excitability. 47 Pharmacologically, KV4 currents are selectively inhibited by several spider toxins that modify gating kinetics 48 and can be activated by the NeuroSearch (Hellerup, Denmark) compound NS5806. 49 NS5806 increases current flowing through KV4 channels by enhancing peak amplitude and slowing current decay. This latter effect on current inactivation depends on the presence of KChiP2, a cytosolic accessory protein that interacts with the intracellular N-terminus of KV4 channels. 49 Also see the section on cardiac KV4 below.
KV7-Containing Channels
Four of the five members of the KV7 family are expressed in the nervous system, where they form homomeric and heteromeric K+ channels. In many regions of the brain, channels composed of KV7.2 and 7.3 subunits underlie the slow voltage-gated “M-current,” so called because the current is inhibited by neurotransmitters acting via Gq-coupled muscarinic receptors. 50 These channels localize predominantly to the axon initiation segment in neurons and are open at membrane potentials (from around −60 mV) that are the subthreshold for most voltage-gated K+ channels. They do not inactivate and thus generate a steady outward current that stabilizes the membrane in the face of depolarizing currents (reviewed by Brown and Passmore 51 ). Since their activation is slow, they tend not to contribute to the repolarization phase of the action potential but act to subdue excitability and repetitive firing in neurons. A number of different mutations of KV7.2 and KV7.3 have been shown to be associated with idiopathic generalized epilepsy, including benign familial neonatal seizures (BFNS), a disorder characterized by recurrent seizures in newborns.52–54 Not all the mutations have been functionally characterized, but most result in reduced amplitude of the M-current, which would be consistent with neuron depolarization and increased burst activity. Seizures in BFNS usually spontaneously stop within the first 15 weeks, although the susceptibility to seizures in later life is increased in BFNS-diagnosed infants (~16% compared with 1%–2% in the general population 52 ). It is unclear why seizures cease. One possibility is that the expression of KV7 subunits is developmentally regulated, and the neonatal brain is most dependent on the stabilizing effect of the M-current.
Loss of function mutation of KV7.3 is also associated with autism spectrum disorders (ASD). In this context, it is probably an M-current formed by the association of KV7.3 subunits and KV7.5 subunits that is important. 55 The control these channels exert over neuronal excitability may be important in the generation of synchronous oscillations of networks of neurons, which is believed to be involved in memory formation and storage and, potentially, emotional processing and behavioral monitoring, which are all affected in individuals with ASD.56–59 The causal gene for autosomal dominant nonsyndromic sensorineural hearing loss (DFNA2) has been identified as KCNQ4, which encodes the KV7.4 protein.60,61 This progressive form of hearing loss is thought to result from a decrease in K+ efflux from sensory hair cells, potentially leading to damage over time due to K+ overload.62–64
KV7.2-7.5 Modulators as Drugs
Recently, retigabine (Potiga in the United States and Trobalt in the European Union), a first-in-class KV7.2-7.5 opener, has been approved for the use of drug-resistant epilepsy with partial-onset seizures.65–67 Retigabine is derived from flupirtine, a drug with longstanding use as a nonopioid analgesic with known relaxant/anticonvulsive properties. Retigabine generates a hyperpolarizing shift in the voltage dependence of channel activation, thus enhancing the stabilizing M-current and limiting neuronal excitability. It shows little selectivity between neuronal KV7.2/7.3 channels and other KV7.2-7.5 channels and may also affect neurotransmission involving the major inhibitory transmitter gamma-aminobutyric acid (GABA). Retigabine has been shown to increase the concentration of GABA in the brain, by either enhancing GABA synthesis or blocking GABA metabolism, and increases GABA-induced current in rat cortical neurons. 68 A concerning side effect of retigabine is bladder voiding possibly due to relaxation of bladder smooth muscle (detrusor muscle) or loss of excitability in sympathetic neurons in the bladder. 69 Other reported side effects include dizziness, 65 cardiovascular disorders such as prolonging of the QT interval (see later), 70 and eye pigmentation color change.70,71 Interestingly, studies on the retention of patients taking retigabine in the open-label extension study show 60% discontinuation of retigabine treatment at 28 months. 72 In somewhat different study cohorts, a trial undertaken at University College London predicted a possible near 100% discontinuation of retigabine treatment at 2 years. 73 ICA-27243, a compound from Icagen (Durham, NC), is reported to be a selective KV7.2/7.3 opener that binds to a site in the voltage sensor domain. 74 In the folded protein, the binding pocket appears to be formed from residues in both the C-terminal end of the S2 domain and the N-terminus of the S3 domain, regions of the channel protein with a high degree of variability between KV7 subfamily members. 74 This highlights the need for improved information regarding the 3D structure of K+ channels to identify variable regions that can be targeted by selective modulators.
KCa1.1-Containing Channels
KCa1.1 (BK) subunits are widely distributed in the axons and at presynaptic terminals of excitatory neurons in the cortex and hippocampus.75,76 At synaptic terminals, they are localized in close proximity to voltage-gated Ca2+ channels and are activated in response to the Ca2+ influx that occurs in response to action potential–induced terminal depolarization.77,78 Their activation serves to terminate the action potential and generate after-hyperpolarizations that close Ca2+ channels and dampen neuronal excitability. Subunit mutations resulting in loss of channel function would therefore be expected to heighten neuronal excitability consistent with epilepsy. Interestingly, a KCa1.1 mutation discovered by Du et al. 79 is a gain-of-function mutation associated with generalized epilepsy with paroxysmal dyskinesia (GEPD). This mutation is thought to cause seizures in two possible ways: either KCa1.1-containing channels are expressed in inhibitory neurons, or this gain of K+ channel function allows quicker release of Na+ channels from inactivation, therefore increasing burst firing of neurons.80,81
KCa1.1 Modulators as Drugs
KCa1.1 channels have proved particularly challenging for drug design (reviewed by Nardi and Olesen 82 ). Clinically prescribed drugs such as hydroflumethiazide (Saluron) and chlorothiazide have antihypertensive effects probably because they activate KCa1.1 channels in vascular smooth muscle (see later), but these compounds are essentially diuretics that inhibit Na+/Cl– reabsorption from the distal convoluted tubules in the kidneys. 83 The NeuroSearch activator NS1619 has been used extensively as a pharmacological tool to study KCa1.1 channel function but has poor potency and many off-target effects, most significantly the inhibition of L-type Ca2+ channels. 84 NS11021 is a more selective activator that functions by shifting the voltage activation curve to more negative potentials. 82 The activation of neuronal K+ channels to decrease excitability and neurotransmitter release has been seen as a novel approach for targeting acute ischemic stroke. The activator BMS 204352 (Bristol-Myers Squibb, New York, NY) appeared initially promising and has neuroprotective effects in animal models, significantly reducing cortical infarct volume in a stroke model in spontaneous hypertensive rats. 85 It reached phase 3 clinical trials for treatment of acute ischemic stroke but failed to show a significant effect over placebo. 85
Kir4.1-Containing Channels
Kir4.1 channel subunits are found primarily on nonneuronal cells within the brain, mostly glial cells within the hippocampus, cortex, thalamus, and brainstem.86,87 Kir4.1 can form homomeric channels or complex with Kir5.1 or Kir2.1 to form heteromeric channels. These heteromeric channels show strong inward rectification, unlike the Kir4.1 homotetramer. In common with other inward rectifiers, Kir4.1 controls the resting membrane potential of astrocytes, and their ability to allow K+ to move reasonably freely both into and out of the cell has led to the idea that they help control the microenvironment around neurons by assisting in spatial K+ buffering.88,89 The restricted extracellular space around neurons means that the repolarization of a single action potential can cause a significant increase in extracellular [K+], with high-frequency firing potentially raising extracellular K+ by several millimolar. 90 Due to the high K+ permeability of membranes, a prolonged increase in extracellular K+ would depolarize neurons and alter excitability. Excess extracellular K+ therefore needs to be efficiently siphoned from the immediate vicinity of the neuron, and it is postulated that Kir4.1-containing channels allow K+ influx into glia at sites of high extracellular K+. This K+ is then potentially shuttled via a network of gap junction–connected glia and released by efflux through homomeric Kir4.1 channels at sites of low extracellular K+. 91 Disruption of this ability to clear K+ would have profound effects on neuronal excitability, and there is increasing interest in the role of Kir4.1 in epilepsy. Interestingly, glial cells taken during surgery from patients with intractable epilepsy have reduced Kir currents. 92 Loss-of-function mutation of Kir4.1 has also been shown to be associated with seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME) and epilepsy, ataxia, sensorineural deafness, and tubulopathy (EAST) syndromes.93–95 No current modulators are available for these channels.
Kir6.2-Containing Channels
Kir6.2 is the pore-forming subunit of the neuronal KATP channel. These channels respond to fluctuations in intracellular levels of adenine nucleotides and are inhibited by ATP but activated by Mg2+-bound nucleotides, particularly MgADP. 96 This ability to sense intracellular ATP/adenosine diphosphate (ADP) levels ensures that changes in cellular metabolism are translated to changes in membrane K+ permeability and thus membrane potential and excitability. A number of Kir6.2 gain-in-function mutations give rise to a group of syndromes known as DEND (development delay, epilepsy, and neonatal diabetes). 97 Twenty percent of DEND patients have neurological disorders such as generalized epilepsy, delay of motor development, and speech and learning disabilities. All characterized mutations share the common feature that they decrease the ability of ATP to close that channel or increase the ability of Mg2+ nucleotides to active it. The role of KATP channels is perhaps best understood in pancreatic β cells, where they are involved in glucose-dependent insulin secretion. 98 Due to the subunit composition of these pancreatic channels (believed to be Kir6.2 in combination with the regulatory subunit SUR1) and the relatively low [ATP]/[ADP] in pancreatic β cells during fasting, pancreatic KATP channels are constitutively active under basal conditions and help maintain the β-cell resting membrane potential. Elevation of blood glucose results in increased glucose uptake by β cells and its subsequent metabolism, leading to a rise in the intracellular levels of ATP and a fall of ADP. This closes active KATP channels, resulting in a reduction of K+ efflux, depolarization, and activation of L-type voltage-dependent Ca2+ channels, which increases Ca2+ influx and triggers the Ca2+-dependent secretion of insulin. Increased KATP activity in pancreatic β cells would reduce insulin secretion, explaining the diabetes in these patients, but how hyperactivity of neuronal KATP channels induces neurological effects is unclear. KATP channels are expressed predominately in inhibitory GABAergic neurons, with KATP channel openers causing decreased firing of pyramidal cells in substantia nigra.99,100 Drugs that modulate KATP channel activity are one of the few K+ channel clinical success stories and have been extensively reviewed elsewhere. 101 KATP channel blockers include the first-generation antidiabetic sulfonylurea tolbutamide, now largely fallen into disuse due to side effects, and the second-generation antidiabetic sulfonylureas glibenclamide, glipizide, gliclazide, glimepiride, and gliquidone. KATP channel openers include the antihypertensives diazoxide, minoxidil, pinacidil, and the vasodilator nicorandil, which is largely prescribed for the treatment of angina.
Cardiac K+ Channels
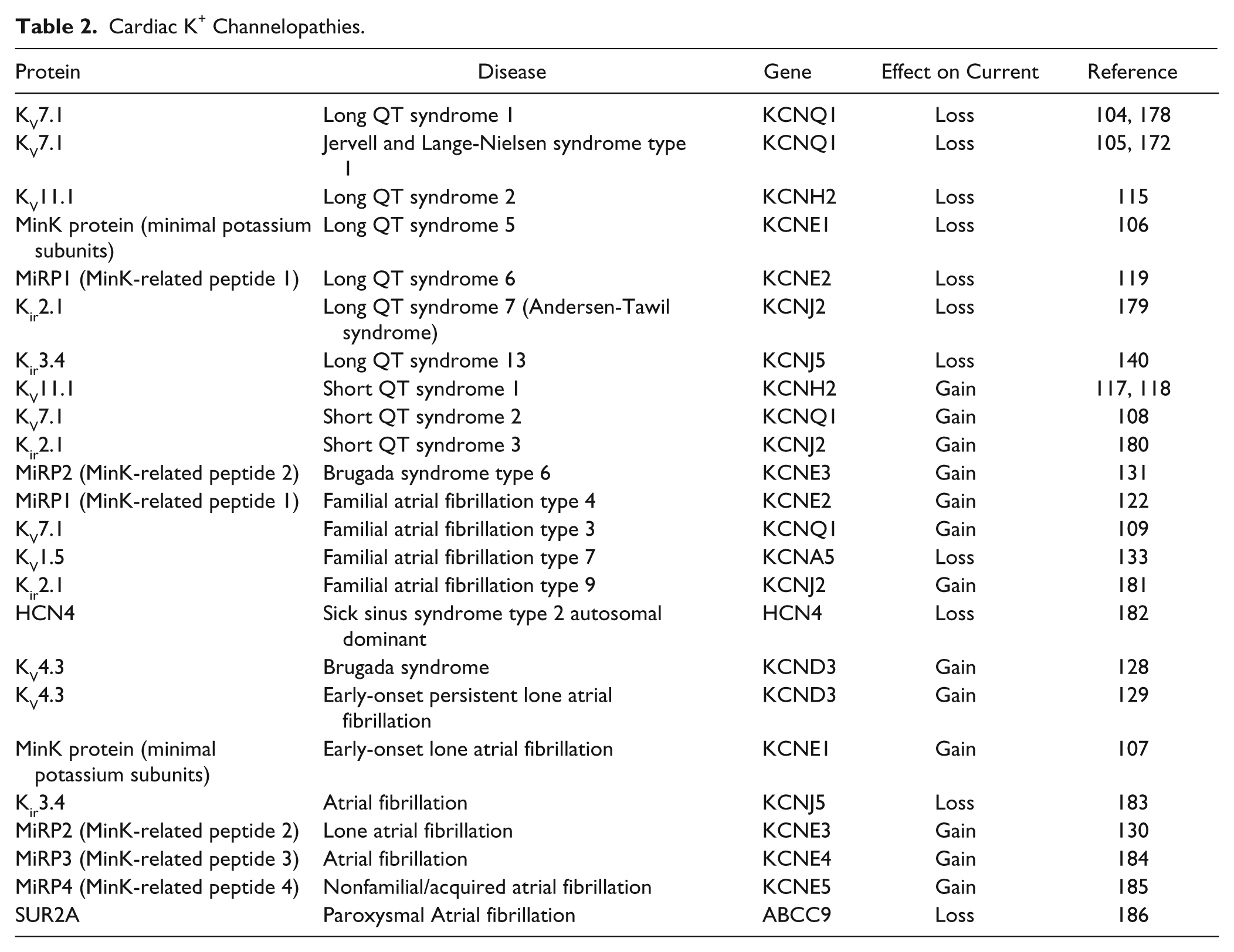
Disorders of the heart relating to K+ channel dysfunction mostly involve disruption of the cardiac action potential and thus the rhythmic contraction of the heart muscle. Alteration of K+ channel function results primarily in repolarization disorders such as long QT syndrome (LQTS) and short QT syndrome (SQTS). The synchronized electrical activity of cells within the heart can be recorded on the body surface as the electrocardiogram (ECG). The time elapsed from the beginning of the QRS complex to the end of the T wave on the surface ECG is defined as the QT interval and is largely determined by the length of the ventricular action potential ( Fig. 3 ). Prolonged QT interval can produce early after-depolarizations, leading to Torsades de Pointes (TdP), which is the typical arrhythmia associated with LQTS; TdP can in turn generate ventricular fibrillation, a lethal arrhythmia. LQTS results from delayed repolarization of ventricular cells due to a reduction in repolarizing (outward) currents or an increase in depolarizing (inward) currents. It is caused by either loss of function of K+ channels or gain of function of Na+ or Ca2+ channels. SQTS, on the other hand, is caused by gain of function of K+ channels. 102 Mechanisms underlying loss or gain of function vary. Mutations of K+ channels have been shown to reduce the number of functional channels at the cell surface by altering trafficking or by affecting the kinetic properties of channel behavior. 103 LQTS can arise from the disruption of several genes encoding K+ subunits (see Table 2 ). These mostly involve mutation of subunits encoding the channels involved in repolarization (IKs, IKr, IK1; Fig. 3 ). Repolarization of the atria also involves atrial-specific channels such as KV1.5, Kir3.1/3.4, and KCa2.2/2.3, which underlie IKuR, IKACh, and ISK respectively. The limited distribution of these channel subtypes makes them interesting and potentially important targets for the development of novel treatment for atrial fibrillation that do not affect ventricle function.
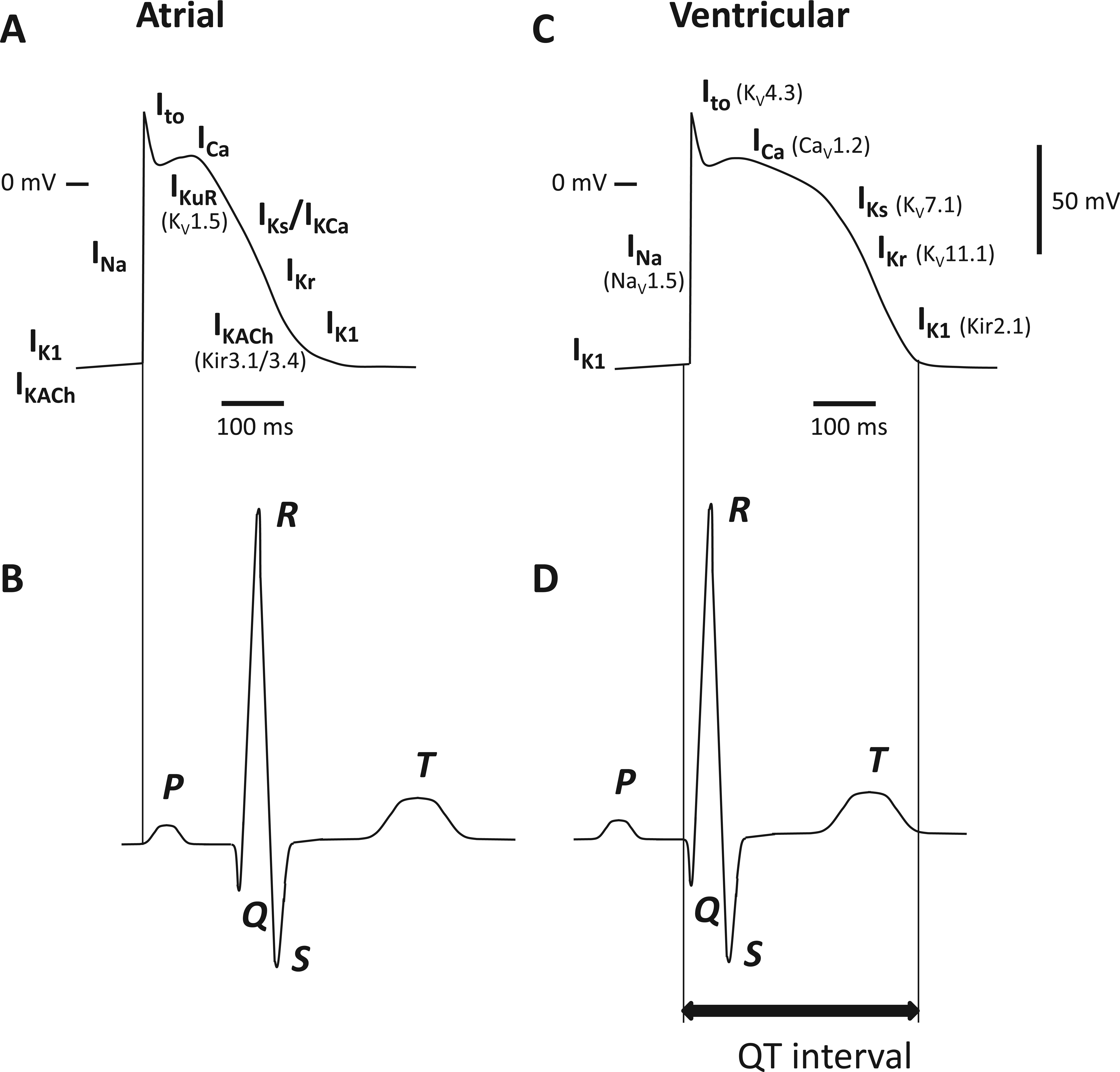
Electrical activity in the atrium and ventricles of the heart. (
Cardiac K+ Channelopathies.
KV7.1-Containing Channels (IKs)
Unlike the other four members of the KV7 family, KV7.1 is not widely expressed in the CNS but instead is found predominantly in cardiac myocytes and inner ear neurons. In cardiomyocytes, KV7.1 (KVLQT) combines with the accessory β subunit MinK (KCNE1) to form channels that mediate the slow delayed rectifying K+ (IKs) that contributes to the repolarization phase of the cardiac action potential. Loss-of-function mutations of KV7.1 lead to long QT syndrome 1 and Jervell and Lange-Nielsen syndrome type 1.104,105 These mutations are usually single amino acid missense mutations that cause protein misfolding and early degradation of the channel subunit. In addition to cardiac rhythm defects, patients with Jervell and Lange-Nielsen syndrome have deafness from birth. 105 Mutation of the accessory protein MinK has been shown to be associated with long QT syndrome 5 106 and atrial fibrillation, 107 while gain-of-function mutations of KV7.1 are associated with short QT syndrome 2 and familial atrial fibrillation type 3.108,109
KV7.1 Modulators
IKs is a target of interest for the development of antiarrhythmic drugs. Amiodarone, a licensed class III antiarrhythmic, is a nonspecific channel inhibitor that prolongs the cardiac action potential via block of both IKr and IKs. 110 KV7.1 is insensitive to the anticonvulsant retigabine, which activates other KV7 family members, because it lacks a tryptophan residue in the S5 transmembrane domain that is required for retigabine action 111 (see above). Pharmacologically, KV7.1 can be selectively activated by the benzodiazepine L-364,373 (R-L3), which is a partial agonist and increases the amplitude of KV7.1 currents as well as slowing the rate of channel activation and deactivation. 112 Interestingly, most mutant channels associated with long QT syndrome 1 respond similarly to wild-type channels, suggesting that the disease-associated channels would be susceptible to activation. 112 To our knowledge, the therapeutic benefit of L-364,373 has not been tested. A number of KV7.1 selective blockers are known, including L-768,673, HMR1556, and JNJ282 (Johnson & Johnson, New Brunswick, NJ). These have generally shown promising results in animal models, prolonging cardiac action potentials and reducing the incidence of arrhythmias, but have also triggered debate regarding the extension of their usage into humans. 113 Potential side effects include hearing loss, inappropriate vasoconstriction (see vascular KV7.1 below), and the potential to generate TdP and ventricular fibrillation. 113 We are unaware of any studies investigating selective KV7.1 blockade in humans.
KV11.1-Containing Channels (IKr)
KV11.1 (hERG1a) subunits associate with subunits produced by an alternative transcript of the KCNH2 gene, termed hERG1b, to form channels that mediate the rapid delayed rectifier current (IKr). 114 IKr represents the most important of the repolarizing currents for action potential termination in the ventricles, atria, and cells of the cardiac conduction system. Loss-of-function mutations of KV11.1 reduce the amplitude of IKr and lead to long QT syndrome 2 (LQTS2). 115 About 300 different KV11.1 mutations are linked to LQTS2. 116 These mutations cause loss of KV11.1 channel function by a range of mechanisms, including reducing channel synthesis, suppressing trafficking to the cell membrane, altering channel gating kinetics, or suppressing ion permeation through the channel pore. 116 Most mutations appear to affect trafficking. Gain-of-function mutations of KV11.1 and an increase in repolarizing IKr current are associated with short QT syndrome 1.117,118 When KV11.1 is coexpressed with the β subunit MiRP1 in Xenopus oocytes, MiRP1 suppresses KV11.1 trafficking to the cell surface and accelerates channel deactivation. 119 In the healthy human heart, MiRP1 is predominantly expressed in the conducting Purkinje fibers, although protein levels have been detected in human ventricles.120,121 A loss-of-function MiRP1 mutation is associated with long QT syndrome 6119, while gain-of-function mutations of MiRP1 are associated with familial atrial fibrillation type 4. 122
KV11.1 Modulators
Limitations in the ability of HTS methods to monitor the complex behavior of the channel has restricted the discovery of activators. Several small-molecule activators of KV11.1 have, however, been identified. Type 1 activators such as RPR260243 (originally synthesized by Aventis now part of the Sanofi group) increase KV11.1 currents by dramatically slowing channel deactivation (reviewed by Zhou et al. 123 ). Type 2 activators such as A935142 (Abbott, Abbott Park, IL), NS1643 (NeuroSearch), ICA-105574 (Icagen), and PD118057 and PD307243 (both Pfizer) primarily impair channel inactivation by binding near the selectivity filter and shifting the voltage dependence of inactivation (reviewed by Zhou et al. 123 ). Mallotoxin, a naturally occurring extract from the tree Mallotus philippinensis, and KB130015 in contrast accelerate the rate of channel activation. The therapeutic potential of these activators as antiarrhythmics has not been demonstrated clinically. They appear to have off-target effects and may be proarrhythmic and increase the risk of ventricular fibrillation. 124 Interestingly, some low-affinity KV11.1 blocking agents appear to paradoxically restore IKr by acting as chaperones to transport mutant KV11.1 subunits to the membrane. 125
KV4.3
Channels containing KV4 subunits underlie the fast-inactivating “A-type” current Ito ( Figure 3 ). Ito is formed of fast and slow recovering components, Ito1,f and Ito1,s, respectively. 126 The channel responsible for Ito1,f is formed by assembly of KV4.2 subunits, KV4.3 subunits, or a combination of the two, while the channel responsible for Ito1,s is composed of KV1.4 subunits. The extent to which alteration of Ito can generate arrhythmic activity in the heart has been difficult to ascertain due to a lack of selective blockers or activators. Dynamic clamp of human atrial myocytes, where a current mimicking Ito but of opposite polarity was injected into cells, selectively reduced Ito and significantly prolonged atrial action potential duration. 127 In the same study, reduction of Ito by dynamic clamp of rabbit atrial myocytes during β-adrenergic stimulation triggered abnormal membrane potential oscillations (after-depolarizations). These could be abolished by dynamic-clamp increases in Ito or by application of the β1-antagonist atenolol. 127 This suggests that changes in Ito can potentially provoke arrhythmias. Loss-of-function mutation of the channel subunits underlying Ito have not been reported but gain-of-function mutation of KV4.3 results in Brugada syndrome 128 and persistent lone atrial fibrillation. 129 Mutations of MiRP2, a normally inhibitory β subunit that associates with KV4.3, are also linked with lone atrial fibrillation 130 and Brugada syndrome. 131 Consistent with this, exposure of ventricular myocytes and ventricular wedge preparations from normal canine heart to NeuroSearch’s KV4-selective activator NS5806 mimics the symptoms of Brugada syndrome. 132
Atrial KV1.5 (IKuR)
KV1.5 underlies the ultrarapid delayed rectifier K+ (IKuR) current in the atrium involved in the early stages of atrial repolarization ( Fig. 3 ). It represents a potentially important target in treating atrial fibrillation (AF), primarily through the prolongation of the atrial effective refractory period (ERP). The ERP represents the period of time after an action potential has been initiated in which a new action potential cannot generate. During this period, depolarization of cells in the myocardium will not produce significant depolarization in surrounding cells, and the ERP thus acts as a protective mechanism to prevent arrhythmias. Antiarrhythmic agents often act to prolong the ERP, but agents designed to treat AF by prolonging the ERP usually also affect the ventricles, inducing other forms of arrhythmia. Functional currents involved in repolarization of the atrium, but not the ventricles, are thus promising new targets for the development of treatments for AF, and several pharmaceutical companies are currently actively exploring this route. It must be mentioned, while many companies are exploring IKuR inhibitors (see below) for treatment of AF, loss-of-function Kv1.5 mutations have been associated with atrial fibrillation.133,134 Loss of Kv1.5 protein has been detected in chronic AF patients; therefore, inhibiting this remaining current may not produce significant effects on ERP in this particular disease state. 135
IKuR Modulators
Brivaness (formerly known as Vernakalant or RSD 1235) is a new antiarrhythmic drug recently approved in Europe that inhibits the atrial-specific channels KV1.5 and Kir3.1/3.4. It has been shown to be effective in terminating acute-onset atrial fibrillation but is relatively nonspecific and can also have some inhibitory effects on Ito and IKr currents. 136 Bristol-Myers Squibb has a KV1.5 inhibitor BMS-919373 in phase 1 trials to study the effects on atrial ERP in patients with a pacemaker (NCT02153437) and in phase 2 trials to assess the effect of BMS-919373 on the time spent in AF (NCT02156076). Pierre Fabre Medicament (Paris, France) has F373280, a novel docosahexaenoic acid derivative and blocker of KV1.5 in phase 2 clinical trials for the treatment of persistent AF (NCT01831856). Xention (Cambridge, UK) has the KV1.5 blocker XEN-D0101 in a phase 1 proof-of-mechanism electrophysiological study and, in partnership with Servier (Neuilly-sur-Seine, France), the more potent and selective XEN-D0103 in two phase 2 clinical studies.
Atrial Kir3.1/3.4
The acetylcholine-activated K current (IKACh) carried by Kir3.1/3.4 channels is also a candidate for the development of atrial-specific antiarrhythmics. 137 The novel compound NTC-801 has been shown to inhibit IKACh with a selectively >1000-fold over other major cardiac currents. 138 NTC-801 reversed action potential shortening induced by carbachol in isolated guinea pig atrial myocytes but had no effect on ventricular action potential duration. It was also shown to prolong the atrial ERP in a rapid atrial pacing model. 138 The benzopyrane derivative, NIP-151, is also reported to selectively block IKACh and be capable of atrial-specific ERP prolongation and stopped AF in two animal models of AF. 139 In contrast, the same study found that dofetilide, a class III antiarrhythmic used in the treatment of AF, significantly prolonged both atrial and ventricular ERP but had little effect in terminating AF in either model. 139 While IKAch is predominately thought to be an atrial-specific current, recent research has shown involvement of IKAch in ventricle repolarization, along with a mutation of Kir3.4 being associated with LQTS.140,141 This may limit the potential of IKACh as a therapeutic target for AF.
Atrial KCa 2.x
The calcium-activated K+ current IKCa mediated by KCa2.x has recently been shown to be atrial specific in human hearts142. Blockade of IKCa produces an increase in ERP in sinus rhythm human atrial preparations, whereas in longstanding AF, IKCa blockade has no effect, probably due to the downregulation of KCa2.2/2.3 in longstanding AF. 142 Interestingly, other studies have found an upregulation in KCa2.x in AF, which leads to speculation that KCa2.x expression is initially increased in AF before downregulation takes place. 143 Therefore, inhibition of IKCa in recent-onset AF may prove beneficial, as has been shown in paced guinea pig hearts. 144 To this end, Acesion Pharma (Copenhagen, Denmark) is currently undertaking studies into KCa2.x modulation for the treatment of AF.
Vascular K+ Channels
The primary role of K+ channels in the vasculature is to control the resting membrane potential and thus the activity of voltage-gated Ca2+ channels, a major Ca2+ influx pathway. 145 In vascular smooth muscle cells, loss or reduction of K+ channel activity results in membrane depolarization, increased open probability of voltage-gated Ca2+ channels, increased Ca2+ influx, and thus contraction and increased vascular tone. An array of different K+ channels from all the major families contributes to this role of regulating tone through membrane potential, with channel type and distribution varying markedly with vascular bed and vessel diameter. Mutations in a number of K+ channel subunits have been linked with human disease ( Table 3 ).
Vascular K+ Channelopathies.

KV1.5-Containing Channels
The major voltage-gated K+ channels expressed in the vasculature are KV1.2, KV1.5, KV2.1, and KV7.4/7.5.146,147 Their distribution varies considerably with vascular bed, and there is some controversy over their relative contribution to the regulation of the resting membrane potential. Inhibited gene transcription and/or decreased stability of KV1.5 mRNA has been implicated in the reduction of functional KV current in pulmonary artery smooth muscle cells (PASMCs) from patients with primary pulmonary hypertension (PPH).148,149 PPH is a relatively rare disease characterized by increased pulmonary vascular resistance and arterial pressure that can ultimately lead to right heart failure. Dependent on the contribution of KV1.5 to the resting membrane potential in PASMCs, channel dysfunction might be expected to lead to a membrane depolarization and increased Ca2+ influx via activated voltage-gated Ca2+ channels. Given the role of KV1.5 in the atrial ERP (see above), it seems unlikely that systemically targeting these channels with activators, which would be expected to reduce the ERP, would have significant beneficial effects in these patients over more traditional Ca2+ channel-blocking strategies. In this context, it is interesting that targeted introduction of KV1.5 into the rat pulmonary circulation by nebulization of an adenovirus carrying the human KV1.5 gene reduced pulmonary hypertension. 150
KCa1.1 Channels
Increases in intravascular pressure induce a graded depolarization of the smooth muscle cell membrane, which increases the activity of voltage-gated Ca2+ channels, raising global Ca2+ and initiating contraction. 151 Although in smooth muscle, the precise mechanism is unclear, Ca2+ influx through voltage-gated Ca2+ channels also activates ryanodine-sensitive Ca2+ release channels (RyRs) located on regions of the sarcoplasmic reticulum in close proximity to the inner side of the plasma membrane. 152 Localized Ca2+ release from single or tightly clustered groups of these channels (subsurface Ca2+ sparks) can increase contractility by directly contributing to global Ca2+ or by increasing Ca2+ entry through membrane depolarization by activating Ca2+-activated chloride channels. Ca2+ sparks also have a significant negative-feedback effect that acts to limit pressure-induced vasoconstriction. 153 This is achieved through the activation of plasma membrane KCa1.1 channels. Increases in KCa1.1 channel activity and resultant outward current (spontaneous transient outward currents or STOCs) induce membrane hyperpolarization, which decreases Ca2+ entry via voltage-gated Ca2+ channels, lowering global Ca2+ and exerting a vasorelaxing effect. 152 A recent study reports that KCa1.1 current density and STOC activity are significantly decreased in vascular smooth muscle cells from patients with hypertension. 154 While KCa1.1 levels are similar in normotensive and hypertensive individuals, mRNA and protein levels of the β1 subunit KCNMB1 are reduced in arterial tissue from patients with hypertension. This is consistent with previous animal models of hypertension where similar findings have been reported. 155 Population-based genetic epidemiological studies have also revealed essential hypertension-related genetic variants in the human KCNMA1 gene. 156 Although no functional deficiency in the KCa1.1 protein was found to explain the association of KCNMA1 genetic variation with an increased risk of systolic severe hypertension, one polymorphism potentially disrupts a binding site for proteins regulating translation and may affect KCa1.1 mRNA levels. 156 Interestingly, a single-nucleotide substitution in the KCNMB1 gene, leading to a channel gain of function through increased Ca2+ sensitivity, is associated with a decreased prevalence of diastolic hypertension. 157 As mentioned above, the clinically prescribed diuretics hydroflumethiazide (Saluron) and chlorothiazide have off-target antihypertensive effects because they activate KCa1.1 channels in the vascular smooth muscle. 83 Modulators against these channels would clearly have clinical value, but their broad distribution and history of failed drug design may ultimately make them less attractive targets. 81
KCa2.3 and KCa3.1 Channels
A link between genetic variation in the Ca2+-activated SK (KCa2.x) and IK (KCa3.x) genes and cardiovascular disease is not well established. These channels are particularly important in the endothelium, where their opening mediates vasorelaxation via the endothelium-derived hyperpolarizing factor (EDHF) pathway (reviewed by Edwards et al. 158 ). Here, a rise in endothelial Ca2+ induced by the binding of vasodilating mediators to endothelial receptors opens KCa2.3 channels on the endothelial cell surface and KCa3.1 channels located on endothelial projections that protrude through small holes in the internal elastic lamina to make contact with the underlying smooth muscle. 159 The K+ currents that flow out through these open channels induce endothelial hyperpolarization that can spread to subjacent smooth muscle via myoendothelial gap junctions 160 ; alternatively, the effluxing K+ can activate inwardly rectifying K+ (Kir) channels or Na+/K+-ATPase on smooth muscle cells to induce smooth muscle hyperpolarization and ultimately vasorelaxation. 161 Selective activation of these channels thus has therapeutic potential for the treatment of conditions such as hypertension, although due to subunit expression in tissues such as the heart (see Atrial KCa2.x section), there are likely to be significant issues with systemic activation. Population analysis has identified several single-nucleotide polymorphisms (SNPs) in both coding and noncoding regions of the KCa2.3 and KCa3.1 genes. 162 Currently, the only suggestion of genetic linkage to cardiovascular dysfunction is the finding that an intronic SNP in the KCa3.1 gene was significantly less prevalent in a cohort of 313 Japanese patients who had myocardial infarctions than in a control group. 163
K2P3.1 (TASK1) Channels
A number of members of the K2P family, including TASK-1/2, TREK1/2, TWIK1/2, THIK-1, and TRAAK, have been shown to be present in the vasculature. 164 These channels are believed to underlie the poorly defined “leak” or background currents and are subject to extensive regulation. Whole-exome sequencing of members of a family with pulmonary arterial hypertension without identifiable mutations in any of the genes usually associated with the disease identified a novel missense variant in KCNK3, which encodes K2P3.1 (TASK1). 165 Five further missense variants in KCNK3 were subsequently identified in unrelated patients with familial pulmonary arterial hypertension and idiopathic pulmonary arterial hypertension. 165 Functional studies revealed that all these missense mutations resulted in loss of channel function, which could be reversed in most mutants by application of the channel activator, and phospholipase A2 inhibitor, ONO-RS-082. Drugs that pharmacologically inhibit TASK channels include bupivacaine, methanandamide, and Sanofi-Aventis (Paris, France) A293, but as yet there has been little development of selective activators, although a number of patents have been filed focusing on screening and assays using the channel proteins. 101 The K2P family is a relatively recent discovery and as such represents an area of considerable scope and opportunity for the development of therapeutics.
Summary
In conclusion, K+ channels occupy distinct physiological niches within the human body and have an accessible cell surface location, considerable subunit variability, and often tissue-defined distribution yet have largely evaded successful drug discovery. With the exception of the antidiabetic sulfonylureas and antihypertensives that target KATP channels, most K+ channel modulators in clinical use today are poorly selective and have significant off-target toxicities. One of the reasons for this comparative failure in drug discovery is that these protein complexes are not easy to study. They often gate very quickly, have complex inactivation kinetics, and can be subject to elaborate regulation by voltage and intracellular and extracellular ion concentrations. Many HTS methods rely on indirect measurement of channel activity (ion flux, fluorometric dyes, luminescence) and lack temporal resolution over a physiologically relevant range. The introduction of automated planar-array patch-clamp technology has significantly improved the capacity to track physiological channel activity in response to compound libraries, but potent selective modulators remain elusive. Optimization of lead structures appears difficult, perhaps due to structural restraints imposed by modifiers binding to relatively inaccessible or spatially restricted sites in the channel pore, in regulatory domains, or at the interface with modulatory subunits. Here, in silico modeling and advances in structural biology techniques to crystallize channel proteins within lipid matrices to mimic in vivo open and closed states should generate important data. In addition, crystals of ion channels in complex with modulatory ligands/accessory subunits may reveal key interaction sites and interfaces that can be targeted in drug design. Indeed, targeting interaction interfaces with compounds that either mimic or disrupt the regulatory influence of accessory subunits (see, e.g., the KV1.1 “disinactivators” 39 ) may ultimately be a more fruitful approach to modulating channel behavior than directly targeting the ion-conducting subunit. It is also worth noting that most disorders associated with loss-of-function mutations in K+ channel genes originate not from direct defects in channel activity but from problems with protein folding that lead to early degradation and a reduction in functional channels at the cell surface. An alternative approach may be to identify chaperone agents that stabilize these mutant subunits and allow enhanced trafficking to the membrane. In this context, recent applications have been filed to the FDA and European Medicines Agency (EMA) for a combination therapy for cystic fibrosis using lumacaftor and ivacaftor. Lumacaftor promotes folding of mutated cystic fibrosis transmembrane conductance regulator (CFTR) subunits and increases expression of this Cl– channel at the cell surface. Ivacaftor then acts to increase Cl– conductance in CFTR channels by increasing their open probability. 166 The applications follow on from two successful phase 3 studies (TRAFFIC and TRANSPORT) that demonstrated significant and sustained improvement in lung function in people with the most common (F508del) form of cystic fibrosis. 167 The opportunity and need for novel, effective ion channel modulators exists but now need to be matched with innovative design and discovery.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: E.S.A.H. is funded by the British Biotechnology and Biological Sciences Research Council (BBSRC). C.D. is funded by the British Heart Foundation (BHF).