
Abstract
For the past decade, cardiac safety screening to evaluate the propensity of drugs to produce QT interval prolongation and Torsades de Pointes (TdP) arrhythmia has been conducted according to ICH S7B and ICH E14 guidelines. Central to the existing approach are hERG channel assays and in vivo QT measurements. Although effective, the present paradigm carries a risk of unnecessary compound attrition and high cost, especially when considering costly thorough QT (TQT) studies conducted later in drug development. The
Introduction
On July 23, 2013, a think-tank meeting sponsored by the Cardiac Safety Research Consortium (CSRC), Health and Environmental Sciences Institute (HESI), and the US Food and Drug Administration (FDA) was held at the FDA headquarters in Silver Spring, Maryland, to discuss a novel approach to assess the proarrhythmic potential of drugs that prolong the QT interval. Prolongation of the QT interval is used as a surrogate marker for predicting the risk of a compound to induce a potentially fatal ventricular cardiac arrhythmia called Torsade de Pointes (TdP). However, the link between QT interval prolongation and TdP appears multifaceted and is influenced by a number of underlying factors including age, gender, underlying disease state, electrolyte imbalance, concomitant medication, and more.1,2 Although the majority of compounds that can induce TdP are known to inhibit cardiac potassium channels encoded by human-ether-à-go-go Related Gene (hERG),3–5 block of ionic current (IhERG) carried by recombinant hERG channels alone is not always predictive of delayed repolarization or proarrhythmic risks. For example, the L-type calcium channel blocker verapamil is a potent blocker of hERG current 6 but does not prolong the QT interval or pose a risk of TdP. 7 For small chemically manufactured molecules, the current preclinical (ICH S7B) and clinical (ICH E14) safety guidelines require first a preclinical electrophysiology test against hERG (or the native cardiac equivalent, the rapid delayed rectifier current IKr; known also as hERG) and an in vivo QT measurement followed, for drugs that pass preclinical testing, by a thorough QT (TQT) study.8,9 All of these measures are surrogates for, rather than automatic predicators of, TdP proarrhythmia, and following a decade of testing using this approach, it has become clear that the strong initial focus on a single ion channel (hERG) favors a conservative approach that may have led to the attrition of potentially useful drugs, mainly on the grounds of their activity on hERG rather than due to arrhythmia induction per se. There is good reason, therefore, to reconsider approaches to the evaluation of drug-induced arrhythmia.
This article will focus on the preclinical aspects of the proposed CiPA paradigm and will not address in detail any potential impact on either the E 14 guidance document or the dedicated TQT study. To touch on those briefly, collaboration between the Consortium for Innovation and Quality in Pharmaceutical Development and the CSRC was established to design a clinical study in healthy subjects demonstrating that the TQT study can be replaced by robust electrocardiogram (ECG) monitoring and exposure-response analysis of data generated from first-in-human single-ascending-dose studies. Results from that study were presented at a meeting held in Silver Spring, Maryland, in December 2014, and readers should see Darpo et al. 10 for more information.
What Is CiPA?
The new paradigm, termed the
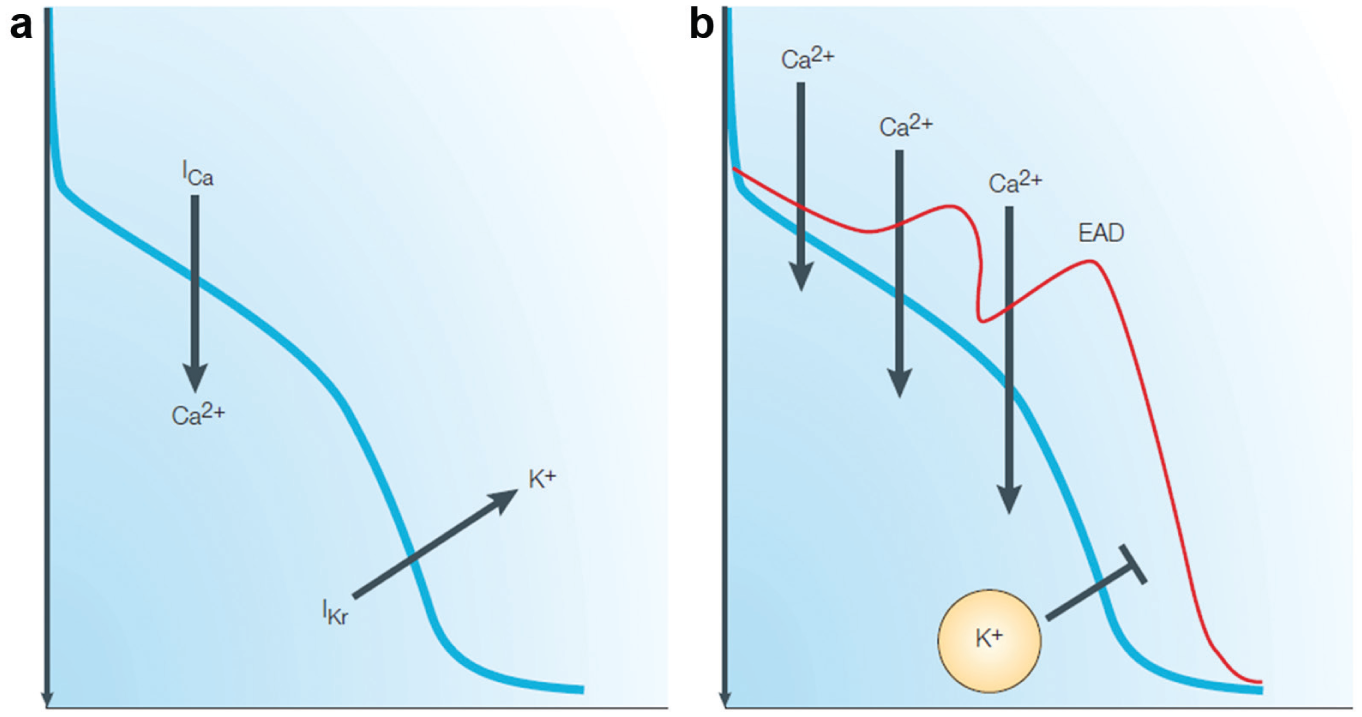
The balance between inward and outward currents determines the morphology and duration of the cardiac action potential (AP) and, consequently, the duration of the QT interval. Drug-induced inhibition of the IKr/IhERG current can delay repolarization and prolong the AP duration and the QT interval. Lengthening repolarization enables reactivation of calcium channels, resulting in late inflow of calcium ions and contributing to the propensity of developing early afterdepolarizations (EADs). When generated in the presence of transmural heterogeneity in ventricular repolarization, EADs are believed to contribute to the generation of extrasystoles that can trigger Torsades de Pointes. The red AP trace highlights delayed repolarization and generation of EADs. Adapted with permission from ref. 13: Nat. Rev. Drug Discov.
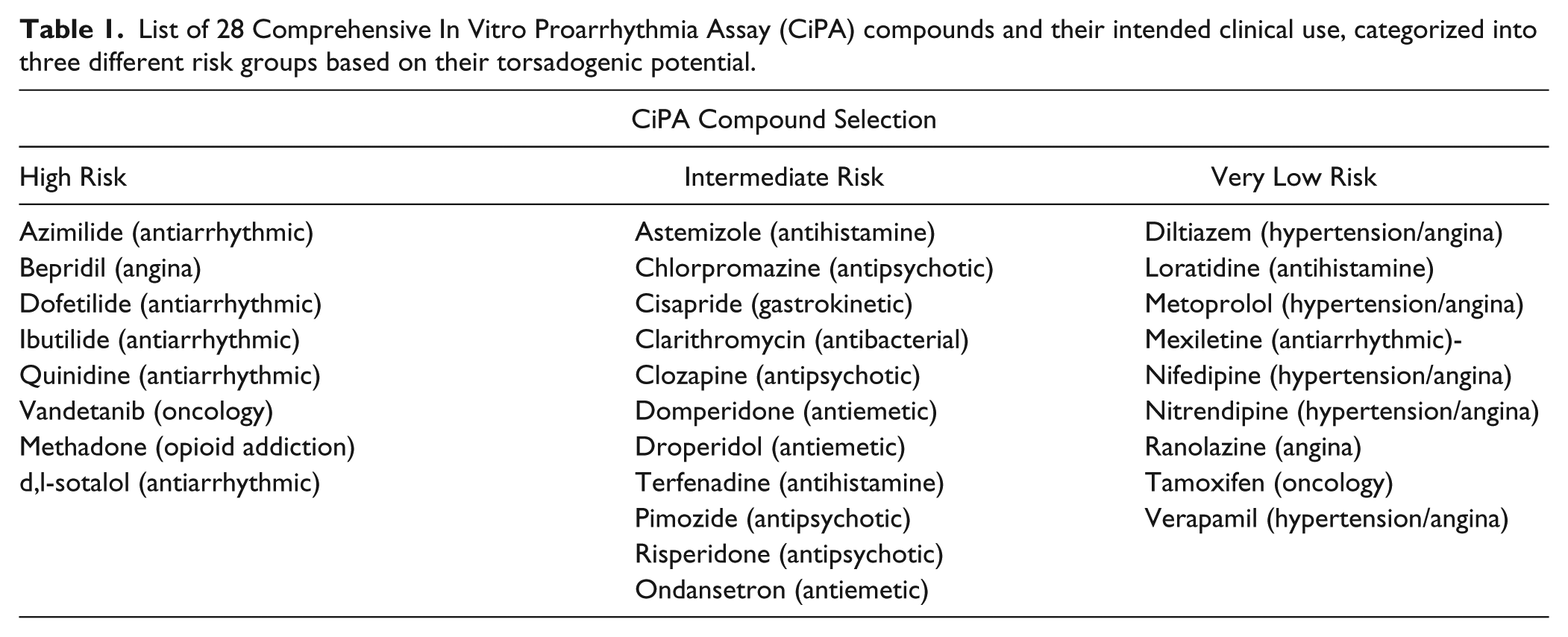
The CiPA initiative is being driven by a consortium comprising a number of collaborators including the FDA, HESI, CSRC, Japan National Institute of Health Sciences, Health Canada, European Medicines Agency, Pharmaceutical and Medical Devices Agency (Japan), Japan iPS Cardiac Safety Assessment, academics, in silico modelers, and partners from contract research organizations, the pharmaceutical industry, stem cell providers, and device companies. Because of the complexity of the task proposed, several work streams have been established in support of CiPA. As such, the Safety Pharmacology Society (SPS) has positioned itself as an important contributor to this effort by establishing the Ion Channel Working Group (ICWG). This group builds on considerable experience and expertise of its members in the field of ion channel biophysics, pharmacology, and the understanding of translation from in vitro to in vivo models. The ICWG is tasked with bringing together expertise and resources required to deliver best practice recommendations for generating ion channel data needed for in silico human cardiac AP reconstructions of proarrhythmic liabilities. The SPS also supports the CiPA effort in the form of staged funding of the experimental work to be performed. Other committees critical to the success of CiPA include the In Silico Working group (ISWG), under the direction of the FDA, and the Cardiac Stem Cell Working group (SCWG), sponsored by HESI. The ISWG is responsible for the development and validation of the best in silico model of human ventricular electrophysiology for the AP reconstruction of drug effects on the individual ion channel, as determined by the work of the ICWG. The role of the SCWG is to define best practice for experiments using human stem cell–derived cardiomyocytes in an effort to validate the effects observed on ion channels, and/or in silico modeling, and to unmask effects that, for various reasons, were not revealed in either the ion channel or in silico work. Finally, a subcommittee from the joint HESI/CSRC Clinical Translation Working Group selected a series of 28 compounds to be tested in the CiPA paradigm that were categorized into three different risk groups based on their torsadogenic potential (high, intermediate, very low, or none; see Table 1 ) according to published and publically available data and expert opinion7,14,15 (Credible Meds; FDA AERS Database, FDA labeling). The intent in the selection of the compounds is twofold: (1) to provide a battery of compounds for training, testing, and validating the in silico and stem cell CiPA models and (2) to select compounds that cover a wide spectrum of electrophysiological endpoints including the degree of torsadogenic risk, actions on ion channels (with attention to multichannel blockers), varying levels of block at clinical exposure, and inclusion of some compounds with non-hERG TdP risk. The complete list of selected compounds is presented in Table 1 .
List of 28 Comprehensive In Vitro Proarrhythmia Assay (CiPA) compounds and their intended clinical use, categorized into three different risk groups based on their torsadogenic potential.
The Need for a New Paradigm
To appreciate the necessity of modifying the current strategy, one must revisit the evolution of the regulatory guidelines in order to appreciate our current position. Indeed, beginning in the late 1980s, spontaneous case reports of cardiotoxicity related to the use of the nonsedating H1-antihistamine terfenadine began to appear in the literature. Eventually, this drug was shown to cause TdP and sudden death by prolonging cardiac repolarization following inhibition of the delayed rectifier potassium channel, IKr. 16 As similar reports with other noncardiovascular agents began to appear more frequently, the Committee for Proprietary Medicinal Products (CPMP) took action and issued a “Points to Consider” document in 1997 that advocated a series of in vitro and in vivo experiments to assess the risk of QT prolongation (CPMP/986/96). More specifically, the guidance recommended the undertaking of in vitro electrophysiological studies (effects on AP duration) using cardiac tissue that included ion channels corresponding to those contributing to repolarization in human cardiac tissue. Following increasing scrutiny by various regulatory agencies and expert working groups, the International Conference on Harmonization (ICH) issued two imperative guidance documents in 2005 that are still the standard today as safety pharmacology guidelines for the development of new chemical entities: ICH S7B, titled “Non-Clinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals,” describes a nonclinical testing strategy for assessing the potential of a test compound to delay ventricular repolarization, and ICH E14, titled “Clinical Evaluation of the QT/QTc Interval Prolongation and Proarrhythmic Potential for Nonantiarrhythmic Drugs,” requesting that all sponsors submitting new drug applications conduct a thorough QT study to determine if a drug prolongs the heart rate–corrected QT interval (QTc).8,9 Clearly, the introduction and adoption of these guidance documents have proven successful: there has been no withdrawal of marketed drugs for concerns of TdP since they were approved in 2005, while six noncardiac medications were withdrawn from the U.S. market by the FDA between 1995 and 2003, prior to the adoption of these guidelines. Nonetheless, current regulatory concern for any candidate drug to inhibit hERG current and to prolong the QT interval by a magnitude greater than 5 ms leads to extensive evaluation in phase IIb and III trials at significant costs; since 2005, approximately 450 thorough QT/QTc studies have been performed at an estimated cost of approximately 1 billion dollars.17,18 Moreover, the major learning from a decade of clinical studies is that an increase in QTc is a sensitive but not specific endpoint to predict proarrhythmia. 10 Therefore, the current preclinical and clinical strategies are suitable to predict QT prolongation but are inadequate to assess the risk of TdP and need to be revised.
Drawbacks of the Existing Approach and Predominant Focus on the hERG Channel
Since the launch of the ICH guidelines, pharmaceutical companies have established a number of preclinical assays to identify, early in the discovery process, compounds that have the propensity to block the hERG channel.3,5,12 Arguably, the ideal preclinical assay for drug-induced IKr inhibition would involve electrophysiological recording of native IKr in ventricular cardiomyocytes from an appropriate model species. However, the requirement for enzymatic and mechanical myocyte dispersion from intact tissue, together with challenges to effective IKr measurement, make native IKr measurements impractical as a primary safety assay, as inevitably such measurements would be of low throughput. 5 In addition, native IKr is of small amplitude, necessitating its pharmacological isolation from other overlapping currents, further complicating its use as a primary screen. 5 Consequently, for more than 10 years, recombinant hERG channel assays have been employed in preclinical drug safety testing: as early as 2005, hERG assays were used by 93% of surveyed respondents from 119 pharmaceutical companies for preclinical evaluation of drug modification of repolarization in line with ICH S7B. 19 Mammalian cell lines that express hERG are attractive because they permit moderately high-throughput automated patch clamp assays, with measurements of a large-amplitude current compared with native IKr, in the absence of contamination from overlapping conductances.5,20,21 Native IKr may be composed of hERG1a and hERG1b subunits,22,23 and both KCNE1 and KCNE2 subunits can associate with hERG and, potentially, could influence hERG channel pharmacology.24–27 Nonetheless, pharmacological potencies derived from hERG 1a alone expressed in mammalian cells generally approximate well those for native IKr,28,29 and coexpression of hERG 1a and 1b or hERG 1a with KCNE1 or KCNE2 subunits is not routinely needed for screening.
The ICH 7SB guidelines stipulate that radioligand competition binding assays are not a viable alternative to an electrophysiological assay, as they provide no information on a compound’s ability to modify the current itself. 8 However, beyond indicating that IKr/hERG ionic current should be assayed, ICH S7B guidelines are not prescriptive. 8 This has the advantage of permitting flexibility of user approach, both in measurement platform and experimental protocol. However, the potential disadvantage of this flexibility is variability of data due to a lack of standardized approach. The potency of some hERG blockers shows marked sensitivity to stimulus protocol and/or measurement temperature.30–32 The extent to which measurement conditions/protocol influence half-maximal inhibitory effect concentration (IC50) may vary significantly between drugs: one comparative analysis of cisapride and dofetilide using manual patch reported variability of IhERG IC50 for dofetilide between 4 and 46 nM and for cisapride between 7 and 240 nM in published literature derived from mammalian cell lines. 32 This further correlated with greater variability for cisapride than for dofetilide IC50 (7–72 nM versus 4–15 nM) in experiments at a single temperature in a single study, between step, step-ramp, and AP voltage commands. 32 A difficulty that this issue poses for the study of novel chemical entities (NCEs) is that protocol dependence of hERG block is not possible to predict in advance. At present, there is no consensus on a single best protocol for studying IhERG sensitivity to NCEs, although adoption of a standardized approach could reduce variability: a comparative blinded patch-clamp investigation of 12 hERG inhibitors between 2 contract research organizations employing similar measurement methods and standardized solutions, temperature, and protocol showed an IC50 variability between laboratories that was in the order of approximately threefold or less for all but one compound. 33 .
The ICH S7B guidelines recommend that, where possible, full concentration-response relations should be obtained for compounds under examination, except where physicochemical properties limit the maximal concentration that can be tested. 8 Given the hERG channel’s now well-recognized pharmacological promiscuity,5,12,34 this poses problems in the assessment of NCEs: up to ~70% of compounds may interact with hERG at some concentration. 35 Automatically eliminating from further development all drugs that inhibit hERG would be costly and may be unjustified in some cases. Consequently, issues arise as to how “safe” and “dangerous” hERG blockers can be reliably distinguished from one another. Considerable effort has been expended to define a safety index or safety margin that takes into account both potency of an agent against hERG and its effective therapeutic concentration against its intended target (reviewed in refs. 5, 7, and 12). However, hERG safety margins alone may not suffice for decisions as to whether or not to proceed with development or approval of an NCE. First, a sole focus on hERG may miss rare instances in which QT prolongation/TdP could arise from compound effects on ionic currents other than hERG. Second, drug discovery projects may not have available the measured Cmax values necessary for accurate safety margin assessment and may have to rely on Cmax estimates. 12 Third, relating hERG safety margin values to QTc prolongation and risk can be complex. A comparison of hERG block potency with TQT study data for 39 drugs found that a hERG safety margin of 45 optimally linked safety margin to QTc interval prolongation but that QTc prolonging drugs were only five- to sevenfold more likely to exhibit safety margins between 1 and 30 than drugs that do not prolong the QTc interval. 36 .
Additional preclinical in vitro repolarization screens are not currently required, with multiple different approaches used, ranging from single Purkinje fibers to ventricular wedge and perfused heart preparations—each of which has its own strengths and potential disadvantages.3,5 Although such additional tests are not mandatory according to ICH S7B, 8 when used, they are likely to result in variability in type and comprehensiveness of these complementary data.
Functional Effects on Multiple Human Cardiac Ionic Channels
As a first measure, CiPA proposes to evaluate the proarrhythmic risk of compounds by studying the effects on multiple human ventricular ionic channels. Because the study of native currents from primary human cardiac tissue is not a viable approach (due to the same limitations as discussed above in respect of IKr), the core strategy will consist of measuring key ionic currents from recombinant human channels expressed in various heterologous systems. The initial voltage clamp work will be performed manually both at physiologic temperature (~35–37 °C) and room temperature in an effort to establish biophysical and pharmacological characterization of the effects of selected compounds on each current using the gold standard approach. These results will then be used as reference when the work is transitioned to automated high-throughput methods in order to adapt CiPA to the current screening environment of most pharmaceutical companies.
To begin with, it is essential to define which ionic currents play a significant role in conferring proarrhythmic properties to compounds. The duration of the QT interval on the electrocardiogram is defined as the period between the beginning of the QRS complex (depolarization of the ventricles) and the end of the T wave (repolarization of the ventricles; see Fig. 2 ). 37 It is a reflection of ventricular APD, and several distinct ionic channels contribute to defining the morphology and duration of APs, including the rapid inward sodium current (INa) responsible for the rising phase of the AP (phase 0), the late sodium (INa-Late) and L-type calcium channel (ICa-L) in control of the plateau phase (phase 2), and multiple overlapping outward repolarizing potassium currents comprising the transient outward current (Ito; phase 1), the slow (IKs) and rapid (IKr) components of the delayed rectifier potassium channels (phase 3), and the inward rectifier channel (IK1; phase 4), the latter responsible for setting the resting potential of working cardiac myocytes. 38 Based on their fundamental role in defining human APD, and together with the information obtained from a Safety Pharmacology Society survey, the ICWG selected these seven different human ionic channels as initial working material for the CiPA assays. More specifically, as it will be recombinant rather than native channels that are viable for safety testing assays, experiments will focus on the following human recombinant channels expressed in mammalian cell lines: Nav1.5 (rapid INa), toxin-modified Nav1.5 (INa,Late), Cav1.2 (ICa,L), Kv4.3+KChIP2 (Ito), hERG (IKr), KCNQ1+KCNE1 (IKs), and Kir2.1 (IK1). Currents will be studied individually using protocols that will not only assess the potency of block (IC50 determination) but also evaluate kinetics of block that might prove critical in understanding the potential proarrhythmic potential of compounds. 39 Ultimately, only the most informative protocols will be retained as final, and it is likely that the list of seven targeted channels will narrow once their role as proarrhythmic markers are confirmed, or not, based on experimental outcomes. A significant benefit to be expected from this effort will be the establishment of best practices for ion channel studies used to characterize drug effects, such that standardized protocols and methods are developed and adopted in an effort to minimize intra- and interlaboratory variability and allow a more level playing field across the pharmaceutical industry.
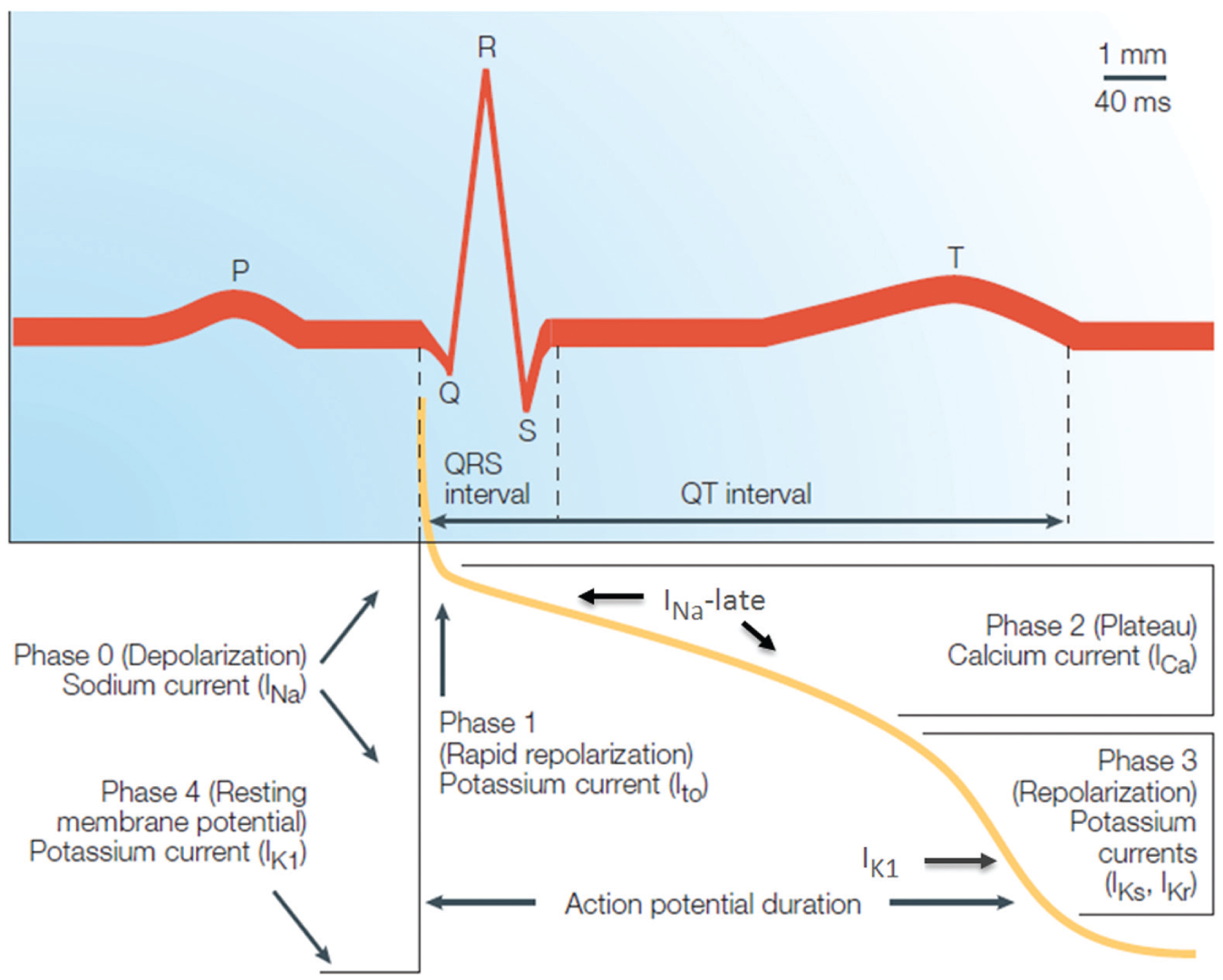
Overview of the different ion channels and their contribution to the morphology and duration of the cardiac action potential (AP). The duration of the QT interval on the electrocardiogram is defined as the duration between the beginning of the QRS complex and the end of the T wave. It is a reflection of ventricular AP duration and represents the time during which the ventricles depolarize and repolarize. Numerous overlapping ionic currents contribute to determining the morphology and duration of the ventricular AP. Adapted with permission from ref. 13: Nat. Rev. Drug Discov.
In Silico Cellular Simulations
The integration of in silico modeling into the CiPA effort offers the potential to provide integrative, cost-effective, and high-throughput solutions to predict drug-induced changes in AP duration by combining the ICWG’s results on individual ion channels. The O’Hara-Rudy (OHR) model of the human ventricle was selected by leading in silico modelers at a consensus meeting in July 2013. The OHR model offers several advantages, including the fact that it is open source (it can be accessed on the Rudy Laboratory research section of the website http://rudylab.wustl.edu); all constants (extracellular ionic concentrations, cell geometry, ionic conductance) and all the initial conditions for state variable and scaling factors have been determined; and, finally, it is fully validated with ionic data described for a human ventricular AP model. 40
The ISWG is focused on evaluating and extending the model so that it can provide assessment of ion pharmacology and clinical risk of TdP. The philosophy behind the use of the model is to use the data generated by the ICWG iteratively to parametrize and validate the performance of the model at each step in the development process while keeping the process as simple as possible by using patch-clamp protocols that are not overly complicated or challenging to apply experimentally. Pilot patch-clamp protocols to parametrize the endogenous IKr current (hERG) are being developed, and at the time of writing, preliminary manual voltage clamp data have been generated for three drugs (dofetilide, cisapride, and verapamil) both at room and physiological temperature. These data will be used to assess the adequacy of the protocol in providing the parameters needed to model dynamic drug block of IKr.
A guiding principle of the in silico effort is to introduce additional complexities into the OHR model only when necessary to increase its predictive accuracy. Initial testing of the model was done using newly generated IC50s for each of the seven targeted channels at physiological temperature for a subset of 12 drugs with well-characterized clinical risk profiles. Comparing the results of these simulations to known clinical responses will help to inform decisions on which candidate channels are needed in the assay and whether the model can be improved. Pilot simulation studies using the OHR model and data for dofetilide suggest that, at least for hERG, IC50s alone may not be adequate in assessing the potential for a drug to prolong the AP and induce EADs. Once the final set of targeted channels and types of pharmacology data needed have been determined and the most promising candidate risk metric(s) identified, the performance of the model and the boundary conditions for using it to predict TdP risk will be defined using newly generated pharmacology data for an independent set of 18 well-characterized drugs.
Stem Cell–Derived Human Myocyte Studies
The primary goal of using human iPSC-derived cardiomyocytes is to identify repolarization effects that are not anticipated from either ion channel studies or in silico reconstruction efforts. Such effects may provide insight on the modulation of cardiac APs by mechanisms that do not directly affect ion channels or may not manifest themselves in the present version of the in silico model. Although iPSC-derived cardiomyocytes offer clear advantages over isolated primary human cardiac cells or cardiac tissue preparations, both in terms of availability and ease of use, numerous aspects of the biology and pharmacology of these cells remain to be determined with certainty (see the Limitations and Challenges section).
In an effort to reach their goal, the SCWG has formed a subgroup under the auspices of HESI. Two technological study platforms were suggested for evaluation, namely, the multielectrode array (MEA) and the voltage-sensing optical (VSO) AP platforms; two-dimensional cell preparations used for the study were prepared according to vendor instructions. A pilot study was initiated in 2014 that consisted of volunteer efforts across 12 work sites and four stem cell providers. This pilot study evaluated the effects of eight blinded compounds selected to test for assay sensitivity to specific ion channel blockers (mexiletine for INa, nifedipine for ICaL, E-4031 for IKr/hERG, and JNJ303 for IKs; across multiple sites and platforms) using changes in either the field potential duration (for MEA) or AP (for VSO) as one primary endpoint. A preliminary comparison of the effects of three drugs across two sites for one stem cell source suggests a good concordance when results were presented as percentage change versus baseline values. Additional experiments and validation work are needed to determine best practices and to reach definitive conclusions on the functional utility of human stem cell–derived cardiomyocytes for determining the proarrhythmic risk of established and future drugs. Toward this goal, a validation study with cardiomyocytes is to commence in the third quarter of 2015.
Impact
If successful, CiPA will have a significant impact on the manner in which cardiac safety assessment is performed in the pharmaceutical industry. Although this is a consortium effort, some general rules around data sharing and use have been developed and compiled under the CiPA Guiding Principles. 18 By moving the evaluation of proarrhythmic risks earlier in the development process, it will allow for the removal of compounds with undesirable effects on cardiac repolarization, alleviating the risk and the costs of unmasking a QT signal of concern in clinical trials.
From a preclinical perspective, the CiPA paradigm will allow standardization of all in vitro ion channel assays to characterize the drug effects on cardiac repolarization. It is also envisioned that it will make available a single, common, and fully validated in silico model to quantify the risk of arrhythmia based on the ion channel data. Finally, it will also establish best practices for the use of stem cell–derived cardiomyocytes, in an effort to confirm the translation between ion channels, in silico modeling, and human cardiac tissue. A concise summary of the similarities and differences between S7B/E14 and CiPA is presented in Table 2 .
Comparison between S7B/E14 and evolving Comprehensive In Vitro Proarrhythmia Assay (CiPA) paradigm.

To a certain extent, CiPA will accomplish what the S7B guidance document has failed to achieve since its inception: standardization of the way preclinical assays are conducted across the pharmaceutical industry in order to remove bias and to provide a reliable and reproducible data set allowing complete assessment of the potential effects of drugs on human cardiac electrophysiology.
Moreover, an early and more complete assessment of proarrhythmic risks, rather than the determination of QT prolongation alone, will reduce the number of false-positive (high-sensitivity) results based on activity at the hERG channel and allow a more accurate determination of true-negative (high-specificity) compounds. As a consequence, it is expected that CiPA will lead to a reduction in the need to conduct TQT studies. Given an individual cost of approximately US $2M per TQT study, CiPA has the potential to make a significant positive impact on the cost of developing new medicines. Moreover, it should lead to an increase in drugs reaching phase 1, as well as improve the accuracy of current or future labeling and allow more drugs to reach the market to treat unmet medical needs. In addition, unlike the approach adopted for the S7B guidance document, CiPA is a consortium effort, allowing sharing of knowledge and expertise to support the project. By doing so, it lends itself to the sharing of resources with the common goal of defining best practice in developing safer drugs and in reevaluating the true cardiac risk of drugs that have been discarded because of their activity on the hERG channel or that are currently marketed but carry a warning for QT prolongation in their label.
Limitations and Challenges
The CiPA initiative is the next logical step following the review and analysis of a decade of data obtained using the ICH-S7B and ICH E14 guidance documents.8,9 It has been touted as “revolutionary,” and to some extent, it is, at the least, a visionary initiative. That said, although it is an attractive proposal, it will require a significant amount of work and likely a few years of testing before it comes to fruition to the point at which regulatory guidelines can be updated definitively. The technology is currently available to perform the ion channel work in a high-throughput environment, albeit most likely at ambient rather than physiological temperature. However, although recording of ionic currents from recombinant channels expressed in various cell lines offers significant advantages over recording endogenous currents in human cardiac myocytes, the underlying properties of these channels may not fully recapitulate that of endogenous channels. For example, when expressed in cell lines, the cardiac calcium channel Cav1.2 displays very slow rates of recovery from inactivation at room temperature and therefore can be recorded only at very slow pacing rates. Given that one important aspect of the CiPA paradigm is to understand not only the potency of compounds but also the kinetics of block, such properties will potentially limit the ability to perform experiments at a more physiologic pacing rate. Therefore, full validation of the ion channel assays for the targets chosen will require time, effort, and funding from sources that may need to go beyond SPS but have not yet been clearly identified. In addition, although the particular ion channels chosen are those collectively most likely to influence APD and arrhythmia susceptibility, other proteins including the Na-Ca exchanger, Na-K pump in the sarcolemmal membrane, and ryanodine receptors in the sarcoplasmic reticulum can contribute directly or (in the case of the RyR) indirectly to electrogenesis; in principle, NCEs might, albeit rarely, influence these processes.
The predictive ability of the ventricular cell model(s) used will depend substantially on the current formulas used, and vigilance will be required to limit the model dependency of the results. Simulations that allow TdP to manifest (rather than simply cellular precursors such as EADs) will require the use of tissue as well as cell models.
Stem cell–derived human cardiomyocytes offer numerous advantages over isolated primary cardiac cells (their use circumvents the use of animals, they can be cultured for extended periods of time allowing for chronic exposure to test compounds, and, potentially most importantly, derive from humans). However, the commercial cells currently available are composed of a mixed phenotype of atrial, nodal, and ventricular cells (often enriched for the latter) that is closer to an immature phenotype and may not fully recapitulate the (electro)physiological properties of adult ventricular cardiac myocytes (e.g., refs. 41–46). Also, there have been some reports in which the pharmacology of standard compounds has proven inconsistent with cardiac cells, raising the question of the reliability of the assay relative to human cardiac tissue.47-50 It should be noted that recordings from stem cell–derived myocytes do not fully recapitulate ECG measurements from the intact heart in situ, although they may reveal the propensity of a compound to delay repolarization and favor EAD generation. In addition, CiPA will not address the issue of compounds that prolong the QT interval via changes in autonomic tone or blood pressure.
Relatively important issues remain to be addressed by the CiPA Steering Committee. For example, at what stage of preclinical drug discovery and development will this new paradigm be applied (lead identification? lead optimization? candidate drug selection?) and at what cost? One can envision, for example, that testing an additional number of ion channels at the lead identification/optimization stage has the propensity to mitigate any potential costs savings consequent to the elimination of TQT studies. Another important issue is whether testing will need to be conducted in compliance with good laboratory practice or good laboratory science. Thus, there may be a significant impact on costs and timelines, depending on the stage at which this new paradigm is implemented. These and other significant issues will need to be addressed before CiPA is ready to be fully implemented.
Although the initial timelines that were proposed to complete the in vitro series of tests for this effort were very ambitious (i.e., revision of S7B by June 2016), in reality the CiPA effort is likely to go through multiple iterations before it reaches a point of applicability. The issues addressed are extremely complex and will require the international consensus of protocols and data from all three core assays, as well as the consensus of regulatory bodies across the globe, in order to be accepted internationally. It is an evolving initiative with evolving workflows that will require scientific, intellectual, and practical contributions from multiple parties and, in consequence, will require time to be successful.
It is evident that the time has come to consider new, more comprehensive ways of preclinical testing for proarrhythmic risk of evolving drugs. The hope is that this new CiPA approach will prevent inappropriate compound attrition due to hERG liability, provide a complete assessment of proarrhythmic risk, reduce animal and clinical work, rescue drugs labeled with cardiac warnings based on small degrees of QT prolongation, and help bring complete and standardized submission packages to regulatory agencies. This will enable new drugs to address unmet medical needs and to reach patients more quickly.
Footnotes
Acknowledgements
The authors would like to thank Drs. Hugo Vargas, Derek Leishman, and Sian Ratcliffe for carefully reviewing the manuscript and providing valued suggestions.
Declaration of Conflicting Interests
This publication reflects the views of the authors and should not be construed to represent the FDA’s views or policies. The authors declare that there are no conflicts of interest.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.