
Abstract
Duchenne muscular dystrophy (DMD) is a genetic, lethal, muscle disorder caused by the loss of the muscle protein, dystrophin, leading to progressive loss of muscle fibers and muscle weakness. Drug discovery efforts targeting DMD have used two main approaches: (1) the restoration of dystrophin expression or the expression of a compensatory protein, and (2) the mitigation of downstream pathological mechanisms, including dysregulated calcium homeostasis, oxidative stress, inflammation, fibrosis, and muscle ischemia. The aim of this review is to introduce the disease, its pathophysiology, and the available research tools to a drug discovery audience. This review will also detail the most promising therapies that are currently being tested in clinical trials or in advanced preclinical models.
Introduction
Duchenne muscular dystrophy (DMD) is a devastating muscle-wasting disorder. It is caused by mutations or deletions in the X-linked dystrophin gene, resulting in the lack of dystrophin protein, or the expression of severely dysfunctional, truncated forms of the dystrophin protein (see Refs. 1–3 for selected reviews). DMD is the most common dystrophic disorder, occurring in, approximately, 1 of every 3500–5000 males born.1,4,5 DMD is typically diagnosed at 5 years of age as deficits in motor function development become apparent. 2 Muscle functions continue to deteriorate with age, and loss of ambulation occurs around 10–12 years of age. By their late teens, most patients also exhibit significant respiratory dysfunction, frequently necessitating ventilation support. Decrease in respiratory capacity is also caused by scoliosis, which often requires surgical intervention. 2 The progressive development of cardiomyopathy is another serious manifestation of DMD and, with the improvements in the management of the respiratory dysfunction, is now considered the leading cause of death in DMD. 6 Cognitive dysfunction also is observed in some DMD patients. 2 Sadly, even with state-of-the-art medical care, most DMD patients do not survive beyond their third decade.
Unfortunately, DMD is only one of many determinant muscle-wasting genetic disorders. Of special interest for this review is Becker muscular dystrophy (BMD), which is caused by a partial loss of functional dystrophin mutations, leading to a milder skeletal muscle dysfunction but severe cardiomyopathy, developing at a later age. The limb-girdle muscular dystrophies (LGMDs) are a group of genetic disorders caused by mutations in several different genes (as discussed in this article). DMD, BMD, and most LGMDs share a common mechanism, the destabilization of the sarcolemma (the myocyte plasma membrane). Due to the mechanical stress caused by muscle contraction, myocytes are uniquely sensitive to perturbations in the mechanical stability of the sarcolemma, and many of the genetic disorders that cause muscle wasting have minimal effects on other organs.
The lack of an effective therapy for DMD along with recent interest in high-value therapies for orphan disorders 7 have triggered a significant effort in both industry and academia to identify disease-modifying treatments for DMD. This review will summarize the strategies used in DMD drug discovery and the most promising directions that are currently being pursued.
The Molecular Basis of DMD
The Dystrophin Gene and Protein
The dystrophin gene is the largest described gene in the human genome. It spans 2.3×106 base pairs (bp), containing 79 exons, resulting in a mature mRNA of 14,000 bp that encodes for a 427 kDa protein. 8 The functions of the various domains of dystrophin have been extensively characterized (reviewed in Refs. 1,3,9). The N-terminus contains the actin-binding domain, followed by the rod-like domain which occupies most of dystrophin, consisting of 24 spectrin repeats. The rod-like domain provides structural elasticity to dystrophin and also contributes to actin binding. The C-terminus consists of the cysteine-rich domain and the C-terminal domain. These domains bind several different proteins, including β-dystroglycan, the syntrophins, neuronal nitric oxide synthase (nNOS), and dystrobrevin ( Fig. 1 ).
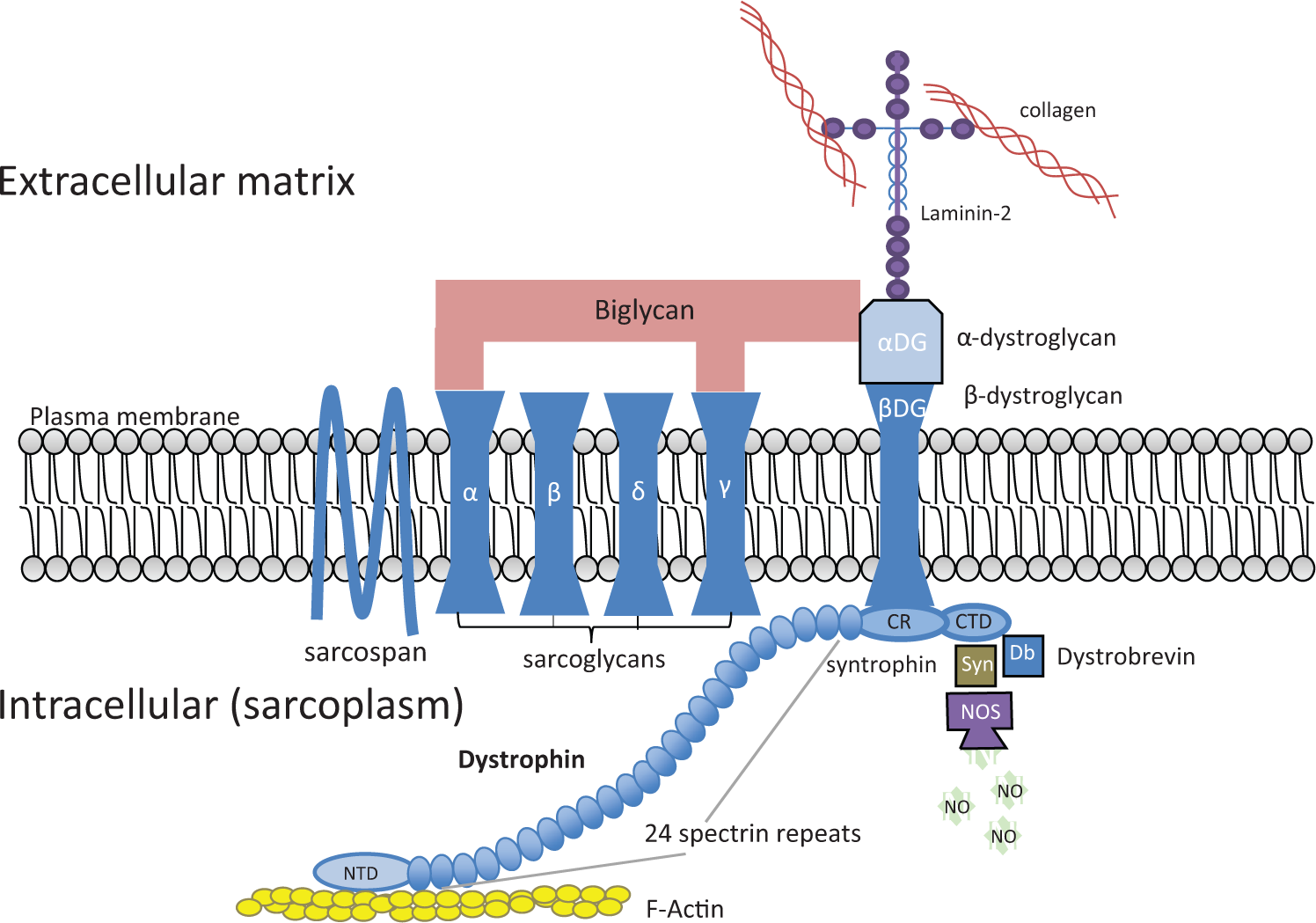
The DGC complex. CR, cysteine-rich domain; CTD, C-terminal domain; DGC, dystrophin–glycoprotein complex; NTD, N-terminal domain.
About two-thirds of the dystrophin mutations leading to DMD are large deletions. These deletions typically cause a translational frame shift resulting in premature translation termination and unstable transcript and protein.1,3 Approximately 10% of the DMD-causing mutations are duplications. 10 Large deletions and duplications tend to cluster in two “hotspot” areas: exons 2–20 and exons 44–53. The rest of the mutations are small deletions or point mutations, affecting dystrophin expression primarily through the introduction of a premature stop codon or by causing aberrant splicing.1,3 Dystrophin function can be partially maintained in the presence of fairly large deletions, especially at the spectrin repeats domain. In fact, an in-frame deletion as large as 46% of the dystrophin protein, found in a BMD patient, allows partial maintenance of dystrophin function. 11 The observation that large deletions in the spectrin-repeats domain lead to only partial loss of function is the basis for some of the most promising therapies that are currently being investigated (discussed further in this article).
The Dystrophin–Glycoprotein Complex
Dystrophin is a component of the dystrophin–glycoprotein complex (DGC; Fig. 1 ; and see Refs. 1,3,9). This complex provides a link between the cytoskeleton and the extracellular matrix, and functions as a “shock absorber” that allows the sarcolemma to maintain structural integrity as mechanical stress is exerted during muscle contraction. The dystrophin protein that provides the link to the cytoskeletal actin filament and other cytoplasmic proteins is tethered to the sarcolemma by β-dystroglycan. β-dystroglycan is associated with the highly glycosylated α-dystroglycan, which links directly to the extracellular matrix (ECM) via laminin-2. 12 α-dystroglycan also binds biglycan, a small extracellular glycoprotein. Biglycan provides a link to the sarcoglycan subcomplex of the DGC, consisting of transmembrane proteins α-, β-, γ-, and δ-sarcoglycans and sarcospan, by binding to α- and γ-sarcoglycans. 9 The importance of the structural integrity of the DGC complex is evident from the muscular dystrophies associated with mutations in DGC complex components. Whereas mutations in dystrophin lead to DMD and BMD, mutations in the four sarcoglycans lead to LGMD2, which bears significant similarities to DMD.3,9 Mutations in the dystroglycans are not commonly associated with muscular dystrophy. However, knockout of the dystroglycans is embryonically lethal in mice, and defects in glycosylation of the dystroglycans, which are thought to weaken the interaction with the extracellular matrix, lead to congenital muscular dystrophy (CMD; see Refs. 3,9). Many other muscular dystrophies are caused by mutations in ECM proteins, additional sarcolemma-associated proteins, as well as proteins involved in sarcolemma repair, further underscoring the importance of sarcolemma integrity for muscle preservation (see Refs. 1,3,9 for more details).
Pathological Mechanisms in DMD
The loss of dystrophin leads to a cascade of pathological events that ultimately cause muscle fiber degeneration. With time, the degenerative process overwhelms the capacity of the muscle to regenerate, and muscle fibers are gradually replaced by collagen, fibroblasts, and fat deposits. As the number of functional muscle fibers decreases, muscle contractility declines. Muscle inflammation stimulated by the necrotic, degenerating fibers also contributes to the development of muscle weakness (see Refs. 3,9,13 for detailed reviews).
Perturbation in Calcium Homeostasis
The sarcolemma of dystrophic muscle fibers was shown to be leaky due to increased sensitivity to contraction-mediated membrane rupture. 14 Because extracellular calcium concentration is four orders of magnitude higher than the intracellular concentration, loss of sarcolemma integrity causes an elevation in intracellular calcium concentration.15–17 Although passive calcium entry via microlesions or activation of nonselective ion channels (e.g., the transient receptor potential channels; see Refs. 13 ) provides the initial trigger, calcium-induced calcium release (CICR) from the sarcoplasmic reticulum (SR) via the ryanodine receptor (RyR) is an important contributor to calcium cytotoxicity.15,18,19 High-cytosolic calcium activates several pathological pathways. The activation of calcium-dependent proteases, calpains, is believed to be involved in the development of DMD pathology by promoting the degradation of muscle proteins. 20 Excess of cytosolic calcium also leads to mitochondrial dysfunction observed in muscular dystrophy, due to the active import of calcium from the cytosol. 9 The pro-inflammatory nuclear factor-κB (NF-κB) pathway is another pathological pathway that has been reported to be activated by the increase in cytosolic calcium concentration.15,21
Oxidative Stress
Oxidative damage is thought to play an important role in the pathophysiology of DMD. Increase in lipid and protein oxidation has been observed in dystrophic muscle.22,23 Levels of NAD(P)H oxidase, a key enzyme mediating the formation of reactive oxygen species (ROS), were found to be elevated in dystrophic skeletal muscle and heart.24,25 Infiltrating neutrophils and macrophages can further contribute to the formation of ROS in the dystrophic muscle. 26 The lack of dystrophin causes the redistribution of nNOS, which is associated with the DGC in normal muscle fibers, to the cytosol, with a concomitant decrease in activity. This mislocalization might contribute to oxidative stress in dystrophic muscle. 9
Inflammation
The extensive muscle fiber degeneration in dystrophic muscle activates an inflammatory response that further contributes to the dystrophic phenotype. The dystrophic muscle is infiltrated by T cells, macrophages, and neutrophils. Elevated levels of the pro-inflammatory cytokines tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), and interleukin-1 (IL-1) are present in dystrophic muscles. 13 TNF-α and IFN-γ are also produced by regenerating muscle fibers in addition to the infiltrating M1 macrophages. 13 As indicated in this article, the NF-κB pro-inflammatory pathway is also activated as a result of the increase in cytosolic calcium concentration. CD8+ T-cell-mediated cytotoxicity and macrophage-mediated cell lysis lead to muscle fiber loss in a vicious feedforward cycle that further enhances the damage caused by muscle fiber degeneration. 13
Fibrosis
The loss of muscle fibers is accompanied by accumulation of fibroblasts and increased deposition of ECM proteins such as collagen and fibronectin, with a resulting reduction of muscle contractility. Fibrosis disrupts muscle function by depriving nutrient supply and by acting as a physical barrier for muscle regeneration. TGF-β is an important mediator of fibrosis, initiating signaling via TGF-β receptors’ autophosphorylation, and in turn leading to the phosphorylation of the smad2/3 proteins. 13 TGF-β enhances synthesis of ECM proteins, and it promotes the conversion of myoblasts to fibroblasts and the differentiation of fibroblasts.27,28
Protease Activation
In addition to calpains that are activated by the elevated cytosolic calcium levels, other proteases also were implicated in contributing to muscle damage in the dystrophic muscle.13,29 Some reports suggest an increase in the expression of the 26S proteasome in dystrophic muscle. This increased expression may help the degradation of oxidized, unfolded proteins, but may also promote the degradation of components of the DGC complex, which is destabilized due to the lack of dystrophin. 29 Matrix metalloproteases (MMPs), namely MMP2 and MMP9, were found to be elevated in dystrophic muscle with levels correlating with disease severity.13,29 Increased MMP activity further compromises the already weakened link between the sarcolemma and the ECM by targeting both β-dystroglycan, a component of the DGC, and the basement membrane protein, collagen IV. MMP9 can also contribute to pathophysiology by activating the pro-inflammatory NF-κB pathway.13,29
Drug Discovery Tools
DMD is a well-defined genetic disorder with a relatively well-characterized natural history. Nevertheless, translating the understanding of the molecular mechanisms underlying DMD to a rational drug discovery strategy and a well-designed drug-screening tree has proved challenging. One of the issues has been the large size of dystrophin, 427 kDa, which prevents restoration of expression using common viral vectors. Another obstacle originates from the role of dystrophin as a scaffold protein that connects the cytoskeleton to the ECM but lacks any known enzymatic or signaling activity that can be quantitated using biochemical or cell-based assays. In spite of these difficulties, significant progress has been made in the development and characterization of drug discovery tools for DMD. Several in vitro cellular systems that allow drug screening as well as in-depth mechanism-of-action studies are now available (see the “In Vitro Tools” section). Well-characterized in vivo disease models for the qualification of in vitro screening leads for advancement into clinical testing are also in place (see the “In Vivo Disease Models” section).
In Vivo Disease Models
Mouse models
The most common in vivo model, used for DMD research, is the mdx mouse. 30 The mdx mouse carries a spontaneous mutation in exon 23 of the dystrophin gene, which completely abolishes dystrophin expression. The mdx mouse is commercially available and has been extensively characterized (see Refs. 31,32 for comprehensive reviews). Detailed standard operating procedures for assessments of muscle functions and pathology in the mdx mouse are available at the TREAT-NMD website (http://www.treat-nmd.eu/research/preclinical/dmd-sops/). Although the mdx mouse harbors a genetic defect that is identical to that of human DMD patients, there are substantial phenotypic differences between the mdx mouse and humans afflicted with DMD. In humans, DMD is characterized by progressive decline in muscle function leading to early death. In contrast, the mdx mouse suffers from an early degenerative crisis at approximately 3 weeks of age that, by 8–12 weeks of age, stabilizes into a chronic condition of mild muscle dysfunction and only a marginal shortening of life span. 33 It was initially hypothesized that utrophin, a dystrophin homolog that is only minimally expressed in mature muscle fibers, can compensate for the dystrophin deficiency in mice.34,35 Later studies have suggested that the milder phenotype of the mdx mouse is the result of more efficient muscle regeneration due to a large reservoir of muscle stem cells (satellite cells).36,37 In spite of the milder phenotype, clear muscle dysfunction and pathology can still be observed in the mdx mouse, and long-term studies can be performed in this model because the life span is close to normal. It is also a common practice to exacerbate the mild phenotype of the mdx mouse by forced treadmill running.38,39
Other dystrophin-deficient mouse models have also been developed. A mouse strain with a targeted deletion of exon 52 has been reported by Araki et al. 40 Of special interest are mouse models that offer a more severe, human-like phenotype. One such model is the mdx mouse lacking the RNA component of telomerase, 37 which shows a diminished muscle-regenerative capacity. Another model is a mouse carrying the mdx mutation in the DBA/2 rather than the C57BL/10 background. The DBA/2 strain has reduced regenerative capacity, yielding a more severe phenotype. 36 A much more severe dystrophic phenotype is observed in the dystrophin/utrophin double-knockout mouse.34,35 The dystrophin/utrophin double-knockout mouse has a much shorter life span of 20 weeks or less,34,35 limiting its utility to short-term studies only.
Other animal models
Animal models using larger mammals as well as nonmammalian vertebrates and invertebrates are also available.41,42 The most widely used large-mammal model is the golden retriever muscular dystrophy (GRMD) model. Like the mdx mouse, the GRMD dog is a result of a spontaneous mutation in the dystrophin gene and exhibits extensive muscle degeneration and necrosis from birth. Respiratory and cardiac dysfunction are observed at a young age, and, in general, the GRMD model resembles human DMD more closely than the mdx mouse model.41,42 However, the high cost of this dog model and the significant interanimal variability in disease manifestation 43 limited the use of this model. Two zebrafish strains with dystrophin mutations were identified.41,44 The zebrafish models offer a cheap and simple pharmacological screening tool because treatment effect can be scored using survival or birefringence microscopy. 44 The Caenorhabditis elegans Dys-1 mutant 41 may offer benefits as a cheap and rapid drug-screening tool and for genetic studies.
In vivo and ex vivo readouts
The progression of muscular dystrophy can be assessed by monitoring in vivo activity, muscle strength, as well as cardiac and respiratory functions. Muscle function can be tested more accurately ex vivo, eliminating confounding behavioral contributions during test performance. Finally, histopathological analysis, as well as analyses of isolated muscle fiber function, can provide valuable mechanistic information.
Early studies in the mdx mouse model have focused on skeletal muscle histopathology.45,46 Following the dystrophic phase, both degenerating and regenerating muscle fibers can be identified. The latter display centrally located nuclei in contrast to normal mature fibers, in which the nuclei are located adjacent to the sarcolemma. As a result of the continuous degeneration and regeneration, muscle fiber diameter is much more variable in dystrophic mice as compared to wild-type mice. Fibrosis is not readily apparent in skeletal muscle from mdx mice. However, fibrosis can be detected and quantitated in the diaphragm, the most affected muscle in the mdx mouse, using collagen-staining dyes such as Masson’s trichrome.31,32 Fibrosis is also apparent in heart sections, albeit at an older age (e.g., Refs. 47,48). An alternative approach to assess muscle pathology is the use of dyes, such as Evans Blue, which penetrates only dystrophic fibers with compromised sarcolemma integrity.31,38
Isolated muscles allow the measurement of muscle function in an ex vivo setting.31,32,49 Muscles are isolated immediately after sacrifice, attached to a force transducer, and force generated after electric stimulation is recorded. The exterior digitorum longus (EDL), the soleus, and diaphragm strips are most commonly used. A decrease in specific muscle force (total force divided by the muscle cross-sectional area) is observed in dystrophic mice. Another significant deficit of dystrophic muscles is the susceptibility to damage caused by eccentric contraction. This is assessed by force decline during a series of forced eccentric contractions (e.g., Ref. 50 ).
Several different methods were developed to evaluate, in vivo, muscle function in dystrophic mice (reviewed in Refs. 31,32). Grip strength of either the forelimbs or the hind limbs is one of the most widely used methods. Maximal, or average, grip force is recorded during 3–5 consecutive trials using a horizontal bar or grid connected to a force sensor. In most cases, grip strength is normalized to body weight. Another common test is treadmill running capacity, which is considered a measure of overall muscle function.31,39 The total distance run prior to stopping due to exhaustion is recorded, with dystrophic mice exhibiting a reduced capacity to endure prolonged running.
It is usually desirable to measure activity under conditions that minimize animal handling to reduce the impact of stress on test results. This is especially important for the mdx mouse, which exhibits enhanced defensive behavior. 51 In general, increased voluntary activity is considered as an indicator of improved muscle function. The open field test measures voluntary movement of a mouse placed in a box equipped with invisible infrared beams that quantitate movement based on the number of beam crossings. An even less “invasive” measure is voluntary wheel running. 52 This test is performed in the mouse’s home cage, which contains a running wheel. The running wheel sensor records the number of wheel rotations, therefore allowing continuous tracking of voluntary activity in the absence of any animal handling.
In the dog GRMD model, loss-of-ambulation age and gait analysis are used to assess muscle function. 43 Cardiac dysfunction is much more apparent in the GRMD dog 42 and the utrophin/dystrophin double-knockout mouse, 35 as compared to the mdx mouse. Cardiac function is typically evaluated by echocardiography 32 or magnetic resonance imaging.53,54 Rhythm abnormalities that are more readily detectable in the GRMD dog 42 are assessed via electrocardiography. 55 Several investigators have used β-adrenergic agonists to unmask cardiac function deficits in the mdx mouse (e.g., Ref. 54 ).
Impairment in respiratory function is present in dystrophic animals. Noninvasive whole-body plethysmography has been used in mouse models of DMD. Mdx mice have demonstrated decreased tidal volume, minute volume, peak inspiratory flow, and peak expiratory flow, with respiratory deficits being more pronounced in mdx/utrophin+/− mice. 56 The mdx mice also show age-dependent impairment in response to hypercapnia (increased CO2 concentration). 57
In Vitro Tools
Recapitulating the dystrophic phenotype in an in vitro system presents a significant challenge. Muscle damage is caused by mechanical stress applied during muscle contraction, which is difficult to replicate in vitro. Nevertheless, a number of in vitro approaches have been implemented successfully for the exploration of disease mechanisms, for the identification of potential therapeutic targets, and for drug screening.
Primary muscle fibers
Muscle fibers from mdx mice were extensively used to dissect the molecular mechanisms underlying disease pathology. In most cases, small, narrow leg muscles such as the EDL or the flexor digitorum brevis (FDB) are used (e.g., Refs. 16,58). Narrow muscles enable easier penetration of ECM-degrading enzymes that allow the isolation of individual muscle fibers. Early studies found an increase in the activity of mechanosensitive calcium channels in mdx muscle fibers. 58 The resting calcium concentration is also elevated in muscle fibers from mdx mice, with further increase when muscle fibers are isolated from mdx mice following forced exercise to increase muscle damage. 16 A detailed study of muscle fiber morphology in EDL and FDB fibers found age-dependent increases in abnormal muscle fibers. Although an increase in cytoplasmic calcium concentration was not observed, an increase in calcium sparks (local, spontaneous calcium release events) during a hypertonic challenge was observed. 59 An increase in calcium sparks was also observed in permeabilized mdx EDL fibers as a result of leaky RyR, even in the absence of osmotic challenge. 18 The observation of an increased calcium leak from the SR of dystrophic muscle was further confirmed by the use of low-affinity calcium dye to probe SR calcium content. 60 Fibers from interossei muscle were used to demonstrate the restoration of normal calcium homeostasis in mdx mice expressing a minidystrophin construct. 61
Primary cardiomyocytes
Impaired calcium homeostasis also was observed in cardiomyocytes isolated from dystrophic mice.19,62–64 Depleted SR calcium content and high incidence of calcium sparks were attributed to leaky RyR that is oxidized, phosphorylated, and nitrosylated in dystrophic cardiomyocytes. Calcium/calmodulin-dependent kinase II (CaMKII) was implicated as the kinase responsible for the increased RyR phosphorylation in dystrophic cardiomyocytes.62,64 Increased levels of NADPH oxidase-2 (NOX-2; Refs. 63,64) contribute to oxidative stress and SR calcium leak. Inducible NOS (iNOS), overexpressed in dystrophic cardiomyocytes, might also contribute to disease pathology by mediating RyR nitrosylation. 19 Indeed, small-molecule inhibitors of CaMKII and NOX-262–64 as well as a RyR stabilizer 19 normalized calcium homeostasis in dystrophic cardiomyocytes.
Dystrophic myoblasts and myotubes
Myoblasts represent an in vitro model system that allows improved assay flexibility and throughput as compared to terminally differentiated muscle fibers. 65 Myoblasts isolated from neonates continue to divide in the presence of high serum concentration but differentiate in muscle-fiber-like polynucleated myotubes at low serum concentration. 65 Similar to muscle fibers, myotubes prepared from dystrophic mice and DMD patients show increased intracellular calcium concentration.15,66 RyR-mediated calcium leak and elevated iNOS levels, observed in dystrophic muscle fibers and cardiomyocytes, also were found in dystrophic myotubes. 15 Immortalized dystrophic myoblasts were generated from dystrophic H-2Kb-tsA58 transgenic mice expressing a thermolabile SV40 large T antigen under the control of interferon-γ. 67 Differentiation of these immortalized dystrophic myoblasts is initiated by transfer to nonpermissive temperature and omission of interferon-γ. 67 The immortalized dystrophic myoblasts were used to confirm hits identified during a screening campaign for small molecules that enhance antisense oligonucleotide (ASO)-mediated exon skipping (Refs. 68 ; see the rationale in the Dystrophin expression restoration section).
The C2C12 cell line
Target identification efforts and screening campaigns relied on the C2C12 cell line. The C2C12 mouse, myoblast-like cells differentiate into myotubes at low serum concentration. 69 Although C2C12 myoblasts are not dystrophic, they allow a muscle cell environment for targeted screening efforts. O’Leary et al. have reported the use of a stable C2C12 cell line expressing an out-of-frame human dystrophin exon 50-GFP fusion construct to confirm small-molecule hits that were found to increase ASO-mediated exon skipping in HEK293 cells expressing a luciferase reporter construct. 68 C2C12 myoblasts stably expressing a GFP construct fused out of frame to mouse dystrophin exon 23 or human dystrophin exon 50 were used in another screening campaign to identify small-molecule enhancers of ASO-mediated exon skipping. 70 The activity of several guanine analogs identified in this screening campaign was confirmed, in C2C12 myotubes and in vivo, in mdx mice. 70 Dantrolene, whose mechanism of action is believed to involve the targeting of RyR, also was found to enhance ASO-mediated exon skipping in a screening campaign using C2C12 myoblasts stably expressing a GFP construct fused out of frame to human dystrophin exon 50. 71 Dantrolene also was found to increase ASO-mediated exon skipping in vivo and to synergize with subtherapeutic levels of ASO to improve muscle function in mdx mice. 71 C2C12 cells stably expressing luciferase under the control of human utrophin promoter were used to screen a chemical library for molecules that enhance utrophin expression (for rationale see the Expression of compensatory proteins section). 72 Of the 14 hits demonstrating concentration responses, one compound was tested for its effect on utrophin mRNA and protein in native C2C12 cells, and was found to increase both. 72
Dystrophic induced pluripotent stem cells
The use of dystrophic induced pluripotent stem cells (iPSCs) is an emerging approach that allows the study of human dystrophic muscle cells. Methods for the differentiation of iPSCs into skeletal muscle cells have been developed (reviewed in Ref. 73 ), and the efficiency of the differentiation protocols has been improved to enable the use of iPSC-derived dystrophic myotubes for drug discovery. 74 iPSCs from DMD patients also have been differentiated into dystrophic cardiomyocytes, which demonstrated elevated levels of resting cytosolic calcium, 75 as observed in primary dystrophic muscle fibers and cardiomyocytes. The iPSC-derived cardiomyocytes were found to have enhanced apoptosis that was corrected, along with the elevated cytosolic calcium levels, by treatment with the membrane sealant Poloxamer P188. 75 Urine-derived stem cells can also be differentiated into cardiomyocyte, 76 thus facilitating access to human dystrophic cardiomyocytes. Recently, iPSCs from DMD patients were used to evaluate gene therapy using the TALEN and the CRISPR-Cas9 systems with encouraging results. 77
Biomarkers and Treatment Outcome Measures
The compromised structural integrity of the sarcolemma of dystrophic muscle fibers leads to leakage of muscle fiber content into the circulation. The abundant muscle enzyme, creatine kinase (CK), is an important biomarker for the initial diagnosis of DMD.2,4,78 CK activity can be determined easily in plasma samples using a simple colorimetric assay, and it is significantly elevated in DMD. However, elevation in blood CK activity is not specific to DMD, and activity was found to decrease as disease progresses and muscle fibers are replaced with connective tissue and fat deposits. More recently, micro-RNAs (miRNAs) were identified as alternative plasma biomarkers. The muscle-specific miRNAs miR-1, miR-133, and miR-206 were found to be elevated in DMD patients and a mouse disease model of DMD.79,80 It remains to be seen whether miRNAs offer a significant predictive value in a clinical setting.
The severity and rapid progression of DMD may lead one to believe that reliable assessment of treatment effects can be achieved. Findings from recent clinical trials highlight the challenges associated with highly variable disease progression rates among subjects. Moreover, variations in test performance not associated with muscle strength, and the opposing effects of increased strength due to growth and declining muscle strength caused by disease progression in younger patients, further complicate outcome assessment.78,81 The 6-min walk test (6MW), in spite of its limitations, is still used as the primary endpoint in most clinical trials. Other measures of muscle function include the 10-s walk/run, the four-stair climb and descent, and time to stand from supine position. In some cases, a combined score from multiple tests is used to achieve a more comprehensive assessment of function. The North Star Ambulatory Assessment (NSAA; see Ref. 78 ) combines the scores of 17 assessed items. A composite score of four Timed Function Tests (TFTs) was used for assessment of muscle function in the Ataluren trial. 81 Most of the tests described here depend on the mobility of patients, which is lost at approximately 10–12 years of age. Handheld devices are used to measure hand grip strength in both ambulatory and nonambulatory patients (e.g., Ref. 82 ). In older patients who suffer from cardiac dysfunction, electrocardiography and echocardiography can be used to monitor the development of arrhythmia and cardiomyopathy, respectively. 6 Respiratory function measures such as forced vital capacity (FVC), peak expiratory flow (PEF), and maximal inspiratory pressure (MIP) are meaningful endpoints in older patients with significant respiratory dysfunction.78,83
Therapeutic Approaches
Standard of Care
Currently, there is no cure or disease-modifying therapy for DMD, and treatment is largely supportive and antisymptomatic. Glucocorticoids, primarily prednisone and deflazacort, reduce muscle inflammation and improve function. They are the only medications that were shown to delay the loss of ambulation and the decline in respiratory functions.2,84 It is now recommended that, if tolerated, glucocorticoid treatment should continue even after loss of ambulation to help preserve upper-limb muscle functions and delay the development of scoliosis. 2 Unfortunately, the use of glucocorticoids is associated with a significant array of side effects, especially in the DMD pediatric population. Among these side effects are attenuated growth, weight gain, glucose intolerance, mood and behavioral changes, and osteoporosis. At an older age, management of respiratory dysfunction and cardiac dysfunction is required. Mechanical respiratory assistance is common by the age of 20, initially at night but also throughout the day during final disease stages. Spinal fusion surgery is performed to correct scoliosis and improve respiration.2,84 Angiotensin-converting enzyme (ACE) inhibitors are the most commonly prescribed medication for DMD cardiomyopathy.2,6,85 They inhibit the formation of angiotensin II, which stimulates the generation of TGF-β that is implicated in DMD pathophysiology. 6 Angiotensin receptor blockers (ARBs), also used in the treatment of DMD cardiomyopathy, might offer a better side effect profile than ACE inhibitors. 85 The use of β-blockers has been tested6,85 but is less established than ACE inhibitors and ARBs in DMD. In summary, the limited effectiveness and/or substantial side effects profile of the inadequate arsenal of DMD medications necessitate the development of more effective therapies to improve the quality of life and survival of DMD patients.
Therapies in Discovery and Development
In the past few decades, a considerable number of therapeutic approaches for DMD were explored in both academia and industry. Essentially, all treatment modalities (RNA, proteins, and small molecules) are being pursued. In this review, we will not attempt to cover all of them but rather will focus on the most advanced therapies and preclinical approaches with a compelling mechanistic rationale ( Table 1 ).
Experimental Therapies for DMD.
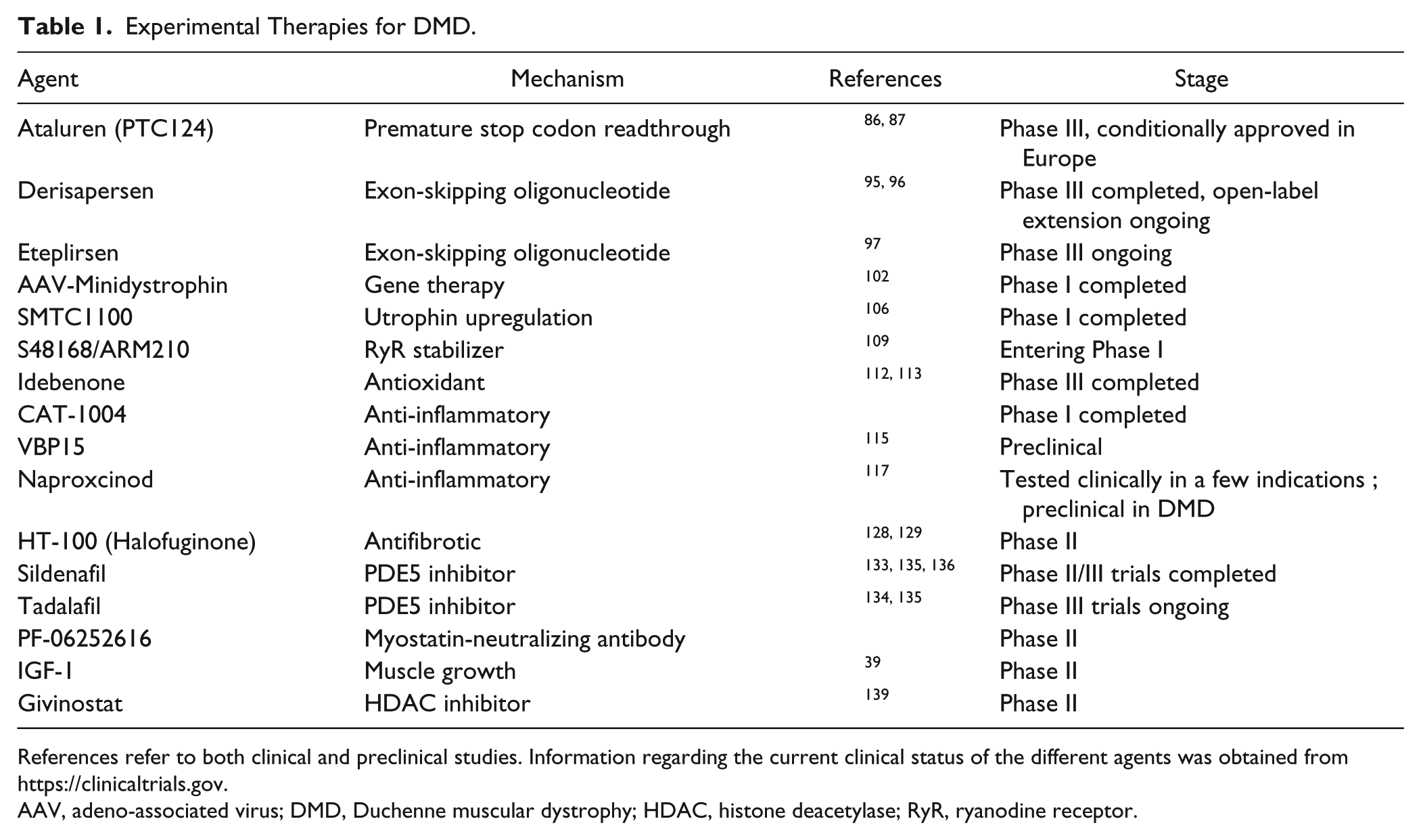
References refer to both clinical and preclinical studies. Information regarding the current clinical status of the different agents was obtained from https://clinicaltrials.gov.
AAV, adeno-associated virus; DMD, Duchenne muscular dystrophy; HDAC, histone deacetylase; RyR, ryanodine receptor.
Dystrophin expression restoration
Among the most exciting developments in DMD therapy are therapies that restore dystrophin expression in DMD patients by overriding the disease-causing mutations. In approximately 13% of patients, loss of dystrophin expression is the result of a premature stop codon. 86 Ataluren, also known as PTC124, was identified and optimized through a screening campaign for small molecules that suppress nonsense mutation and allow the expression of a luciferase reporter carrying a premature stop codon. 87 Ataluren was reported to have selectivity toward readthrough of premature termination with no effect on readthrough of normal stop codons. PTC124 was shown to restore both dystrophin expression and muscle function in the mdx mouse model. 87 In clinical trials, a low dose of Ataluren has decreased the rate of decline in the 6MW test by about 30 m, relative to the placebo control group, following 48 weeks of treatment. A large confirmatory study is ongoing and Ataluren has been conditionally approved for treatment of eligible patients in Europe. 88 It is noteworthy that the high dose of Ataluren did not demonstrate a treatment benefit, 86 and although the mechanism of action of PTC124 was independently confirmed, 89 other investigators have questioned the proposed mechanism of action (e.g., Ref. 90 ).
Another approach for the restoration of dystrophin expression is the use of ASOs that block the splice junction located 5′ to the mutated exon. 91 The idea is to enable the splicing of the affected exon, thus replacing a frameshift nonsense mutation resulting in no dystrophin protein with a dystrophin protein harboring a small, in-frame deletion that retains at least partial functionality, as is the case in BMD. Two different oligonucleotide backbone chemistries, 2′-O-methyl phosphorothioate and phosphordiamidate morpholino (PMO), offering better stability and lower activation of the innate immunity than native RNA, have advanced to clinical testing in DMD patients. 91 Antisense oligonucleotides targeting exon 23 were shown to enable dystrophin expression in the mdx mouse model and improve muscle function and histopathology.92,93 The most advanced exon-skipping ASO target is exon 51, deletion of which can restore the normal reading frame in approximately 13% of DMD patients. 94 The 2′-O-methyl phosphorothioate oligonucleotide, Derisapersen, also known as PRO51, has completed a phase II clinical trial demonstrating a dose-dependent increase in dystrophin expression and a 32-m improvement in the 6 MW test after 25 weeks of treatment.95,96 A larger phase III trial has been completed, but results are not yet officially published. It has been suggested that the PMO oligonucleotide backbone may provide an improved therapeutic window relative to the 2′-O-methyl phosphorothioate backbone. 91 The exon 51–skipping PMO, Eteplirsen, has been tested in 12 patients in a phase II trial, demonstrating increase in dystrophin and improvement in 6MW test performance. 97 A larger phase III study is ongoing.
In considering the potential benefits of the exon-skipping ASOs, it is important to note that the exon 51–skipping ASOs that advance to phase III testing will benefit only a small subset of DMD patients (13%; Ref. 94 ). Additional exon-skipping ASOs targeting other exons are currently in early stages of development and could potentially address as many as 70% of DMD patients. 94 Another important consideration is that the current ASO chemistries do not readily penetrate the heart and will, therefore, not address cardiomyopathy in DMD patients. 98
Expression of compensatory proteins
Gene therapy has been explored both in preclinical disease models as well as in clinical studies in DMD patients. The main obstacles are the large size of dystrophin, the inefficiency of delivery, and immunogenicity of both the viral vector and the dystrophin product. 5 Preclinical studies using a minidystrophin construct, lacking most of the rod domain spectrin-repeats, delivered via an adeno-associated virus (AAV) vector, have shown improvements in both skeletal and cardiac functions in the mdx mouse.99,100 AAV-based vectors were also used to deliver antisense RNA to allow exon skipping and restoration of dystrophin expression.5,101 An exploratory phase I study using AAV delivery of dystrophin via intramuscular injection has demonstrated dystrophin minigene integration. 102 However, dystrophin T-cell immunity was also observed, highlighting a potential complication of therapies that restore dystrophin or minidystrophin expression.102,103
Another approach to compensate for the loss of dystrophin is the upregulation of utrophin, which is structurally similar to dystrophin and maintains many of the dystrophin-binding interactions. 1 In normal mature fibers, utrophin is present only near the neuromuscular junction, with broader distribution observed in developing or regenerating myocytes. 1 Studies in the mdx mouse have demonstrated that utrophin expression can compensate for the lack of dystrophin and ameliorate disease pathology.104,105 SMTC1100 is an optimized, small-molecule activator of utrophin transcription. 106 Treatment of mdx mice with SMTC1100 has enabled an increase in utrophin protein, improvements in in vivo and ex vivo muscle function, as well as normalization of muscle histopathology. 106 SMTC1100 has completed phase I clinical studies.
Mitigation of calcium toxicity
The involvement of dysregulated calcium homeostasis in the pathology of DMD has been well documented (see above). Several studies have examined different strategies to normalize calcium homeostasis in dystrophic models. Overexpression of the sarcoplasmic reticulum calcium ATPase 1 (SERCA1) in the skeletal muscle of dystrophic δ-sarcoglycan null mice decreased resting calcium in isolated myofibers, and improved muscle histopathology and treadmill running endurance. 107 The L-type calcium channel inhibitor Nifedipine also was shown to decrease resting calcium concentration in dystrophic myotubes and improve muscle function in treated mdx mice. 108 Oxidized, nitrosylated, and phosphorylated RyR was shown to mediate an SR calcium leak in dystrophic skeletal muscle fibers and cardiomyocytes.18,19,64 Treatment of mdx mice with S107, a small-molecule RyR stabilizer (termed Rycal generically), normalized calcium homeostasis in skeletal muscle fibers and dystrophic cardiomyocytes.18,19 Furthermore, S107 treatment of mdx mice resulted in improved muscle function, voluntary activity, and muscle histopathology 18 and prevented isoproterenol-triggered arrhythmias. 19 A different Rycal compound, S48168/ARM210, also has been reported recently to demonstrate beneficial effects in mdx mice 109 with clinical testing planned in 2015.
Antioxidants
Oxidative stress is thought to play a role in the pathophysiology of DMD. Administration of the antioxidant, N-acetyl cysteine, to mdx mice was shown to reduce muscle histopathology and protein thiol oxidation. 110 Coenzyme Q10 (CoQ10) is a component of the oxidative phosphorylation electron transport chain and an antioxidant. A small, open-label, CoQ10 clinical trial demonstrated statistically significant improvement in a quantitative muscle testing (QMT) score in DMD patients. 111 Even more encouraging are results obtained with Idebenone, a synthetic analog of CoQ10. In a 9-month preclinical study in the mdx mouse model, Idebenone treatment has improved cardiac diastolic dysfunction and resistance to a challenge with the β-adrenergic agonist Dobutamine, and increased voluntary activity. 112 In a small, double-blind, placebo-controlled, phase II clinical study, Idebenone treatment showed improvement in peak systolic radial strain (not statistically significant) and statistically significant improvement in a measure of respiratory function [peak expiratory flow (PEF)]. 113 A larger phase III trial has been completed and appears to have demonstrated a positive treatment effect on respiratory functions. 114
Anti-inflammatory drugs
Anti-inflammatory glucocorticoids are currently the most effective therapy for delaying the progression of DMD. However, a broad side effect profile limits the utility of glucocorticoids in the treatment of DMD.2,84 There is a substantial effort to identify alternative, efficacious, anti-inflammatory agents with a safer profile.
VBP15 is a glucocorticoid analog that retains potent inhibition of the NF-κB pathway, but lacks the glucocorticoid-responsive element (GRE) transactivation activity that is responsible for some of the side effects observed with glucocorticoid treatments. 115 Indeed, VBP15 was demonstrated to improve muscle function and reduce inflammation without the negative effects of prednisone on body weight, bone growth, and cardiac fibrosis in the mdx mouse. 115 CAT-1004 is a conjugate of salicylate and docosahexaenoic acid that inhibits activated NF-κB, which was implicated in the pathology of DMD. A phase I study has been completed, but preclinical data in DMD disease models have not been published yet. Naproxcinod is an NO-releasing derivative of the nonsteroidal anti-inflammatory drug, naproxen. It has been tested clinically in several different indications in the past but has not been approved yet. 116 Recently, Naproxcinod has been tested in a long-term mdx mouse study demonstrating improvement in both skeletal muscle and cardiac functions. 117 NCX 320, a NO-releasing derivative of ibuprofen, was shown to improve muscle histopathology and voluntary activity in another mouse model of muscular dystrophy, the α-sarcoglycan null mouse. 118
Antifibrotic agents
Muscle fibrosis is one of the hallmarks of muscular dystrophy due to the replacement of muscle fibers by connective tissue as the disease progresses.3,9,13 TGF-β inhibits muscle cell regeneration and differentiation into mature muscle fibers, 27 and promotes myocyte differentiation into fibrotic cells. 28 Several strategies were used to inhibit TGF-β signaling and decrease fibrosis in DMD. Inhibition of TGF-β by a neutralizing antibody improves respiratory function and skeletal muscle function, and decreases hydroxyproline (a marker of fibrosis) muscle content in the mdx mouse model. 119 Blockade of the angiotensin II pathway by either ACE inhibitors or ARBs attenuates TGF-β signaling and improves muscle function and disease pathology in the mdx mouse model.119–121 ACE inhibitors, perindopril and enalapril, have demonstrated efficacy in preserving cardiac function and extending life in DMD patients.122–124 In fact, both ACE inhibitors and ARBs are already in use for the treatment of cardiomyopathy in DMD patients and might even have benefits in presymptomatic patients.6,85 Inhibition of the TGF-β pathway can also be achieved by the broad-specificity tyrosine kinase inhibitor, imatinib, which is an effective treatment for chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors (GISTs). Treatment of mdx mice with imatinib decreased fibrosis in the diaphragm and improved skeletal muscle function.125,126 The plant alkaloid synthetic analog, halofuginone, represents an alternative entry point for the inhibition of TGF-β profibrotic activity. Halofuginone was shown to block TGF-β-mediated Smad3 activation and collagen synthesis. 127 Experiments using the mdx mouse model have demonstrated that halofuginone treatment decreased collagen deposition in skeletal muscle, diaphragm, and heart, and improved skeletal muscle and cardiac functions.128,129 Halofuginone, also known as HT-100, is currently being tested in DMD patients in a phase II trial.
PDE5 inhibitors
The loss of dystrophin leads to disassociation of nNOS from the DGC and loss of activity.3,9 Although the loss of nNOS alone, or nNOS mislocalization, does not lead to a dystrophic phenotype, 130 functional muscle ischemia, presumably as a result of reduced NO production, was implicated in the pathology of DMD. 131 Functional ischemia in DMD can be alleviated by the vasodilatory effect of phosphodiesterase 5 (PDE5) inhibitors. Treatment of mdx mice with the nonselective PDE inhibitor, pentoxifylline, improved skeletal muscle function, decreased oxidative stress, and normalized calcium homeostasis. 132 Treatment of mdx mice with the selective PDE5 inhibitor, sildenafil, improved diaphragm muscle function and decreased fibrosis. 133 Another selective PDE5 inhibitor, tadalafil, was reported to improve muscle histopathology and reduce contraction-induced muscle fiber damage in mdx mice. 134 Both sildenafil and tadalafil were tested in DMD patients and demonstrated to decrease exercise-induced muscle ischemia. 135 However, no beneficial treatment effects of sildenafil on cardiomyopathy were observed in DMD and BMD patients. 136 Several tadalafil trials are still ongoing in DMD patients.
Muscle growth- and regeneration-promoting agents
Enhancement of muscle growth and regeneration is an attractive approach for the treatment of muscular dystrophy. Myostatin, a member of the TGF-β superfamily, is a negative regulator of muscle growth. A myostatin-neutralizing antibody was shown to promote skeletal muscle growth, improve total EDL muscle force (although not specific force), and improve diaphragm muscle histopathology in the mdx mouse model. 137 A myostatin antibody is currently being tested in DMD patients in a phase II trial. Insulin growth factor 1 (IGF-1) is a positive regulator of muscle cell proliferation and differentiation. Treatment of mdx mice with IGF-1 improved muscle function and normalized the electrophysiological properties of EDL muscle fibers. 39 An IGF-1 phase II study has been conducted, but the outcome has not been reported yet. Inhibition of histone deacetylase (HDAC) by trichostatin A (TSA) promotes muscle regeneration and decreased fibrosis and fat deposition in the skeletal muscle of young mdx mice. 138 The HDAC inhibitor, Givinostat, was found to increase muscle growth and improve skeletal muscle histopathology as well as exercise endurance in mdx mice. 139 Givinostat is currently being tested in a phase II clinical trial in DMD patients.
Perspective
The molecular cause for DMD has been known since 1987. 8 Much has been learned about the pathological mechanisms underlying DMD since the discovery of dystrophin. Research tools are in place to support drug discovery efforts, and clinical trials conducted in the past decade have afforded an improved understanding of the natural history of DMD. Nevertheless, currently there is no effective disease-modifying therapy for DMD. A number of treatments that are currently being tested in DMD patients have the potential to offer a breakthrough. It is unlikely, however, that a single therapeutic agent will be sufficient to fully address DMD pathology. Therefore, effective management of DMD will require the understanding of the way different treatment modalities synergize so that an optimal cocktail of therapies can be used at the appropriate stage of the disease.
Footnotes
Acknowledgements
We thank Charu Chaudhry and Ann Rossi for critical review of this manuscript.
Declaration of Conflicting Interests
YB was the senior director of biology at ARMGO Pharma, a company developing S48168/ARM210 and other therapies for DMD. SB declares no potential conflicts of interest.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.