
Abstract
A rapidly accumulating body of work suggests the autophagy pathway is an attractive therapeutic target for neurodegenerative diseases and cancer. To validate autophagy as an anticancer strategy and to assess if systemic inhibition of the pathway will have deleterious effects on normal tissues and physiology, highly selective autophagy inhibitors are needed. While several inducers and inhibitors of autophagy are known, all are nonspecific and none target the enzymes that execute the pathway. A central upstream regulator of the autophagy pathway is the serine/threonine kinase Ulk1 (UNC-51-like kinase-1). Selective molecular probes that function as Ulk1-specific inhibitors are needed to improve our understanding of the autophagy pathway. To identify inhibitors of Ulk1 kinase activity, we developed an HTS-compatible, homogeneous biochemical assay using AlphaScreen technology. This novel assay design uses purified stress-activated Ulk1 and monitors phosphorylation of its full-length native substrate, Atg13. This assay was optimized and validated in a 384-well format by screening the Sigma LOPAC library. Here we report that the Ulk1 AlphaScreen assay is robust and reproducible, with a Z′ factor value of 0.83 ± 0.02 and a signal to background ratio of 20 ± 1.2. Thus, this assay can be used to screen large chemical libraries to discover novel inhibitors of Ulk1.
Introduction
Autophagy is an ancient, cannibalistic (literally “self-eating”) pathway in which building blocks of the cell are recycled via the lysosome, both as a homeostatic mechanism and as a survival strategy during nutrient limitation or times of stress. 1 The pathway is initiated by the formation of isolation membranes (phagophores), which are derived from the outer membrane of the mitochondria and/or the endoplasmic reticulum (ER). These then fuse to form double-membrane (neutral pH) vesicles, coined autophagosomes, that engulf their cargo, which typically includes long-lived proteins, bulk cytoplasmic material, and aged or damaged organelles (e.g., mitochondria). Autophagosomes then fuse with the lysosome, which degrades the delivered cargo to recoup essential building blocks and adenosine triphosphate (ATP) that are necessary for cell survival. 1 Aberrant regulation of autophagy is associated with a variety of human conditions, including neurodegenerative disorders, liver disease, and cancer. 2
In cancer, the role of autophagy is complex, with genetic studies revealing apparent conflicting dual roles, raising doubts as to whether autophagy inhibition is a viable anticancer strategy. Autophagy was shown to act, on one hand, as a tumor suppressor during cancer initiation and, on the other, as an essential mediator of tumor progression.3,4 Recent studies using genetically engineered mouse models of pancreatic and lung cancers coupled with studies silencing autophagy in patient-derived xenografts have ameliorated many of these concerns. These studies suggest an important role for the autophagy pathway in both KRAS- and BRAF-driven tumor progression and reveal impressive preclinical efficacy upon autophagy inhibition across a panel of genetically distinct tumors, thus supporting a rationale for the development of selective autophagy inhibitors as anticancer therapeutics.5–8 Furthermore, many studies note that cancer cells increase flux through the autophagy pathway as a protective mechanism following the treatment with chemotherapeutic agents and radiation therapy. Accordingly, inhibition of the autophagy pathway sensitizes otherwise resistant cells to such agents, implicating inhibition of autophagy as a broad-spectrum anticancer strategy to improve the efficacy of existing therapies. 3
Pharmacologic agents that specifically target the autophagy pathway have yet to be developed. The lysosomotropic agents chloroquine and hydroxychloroquine have been used to impair the autophagy pathway, where their properties as weak bases suppress the degradation of autophagic cargo by lysosomal enzymes. However, these drugs impair all lysosomal activity, and recent clinical trials report dose-limiting toxicity of these agents at concentrations that do not inhibit autophagic flux. 9 Assessing the potential efficacy and safety concerns of systemic inhibition of the autophagy thus requires the discovery of potent and selective inhibitors of key enzymes that execute the pathway. Importantly, concerns regarding whether pharmacologic inhibition of autophagy would lead to toxicity is lessened by the demonstration that systemic ablation of Atg7, the essential E1 enzyme of the pathway, has potent antitumor effects in mutant KRAS-driven lung cancers without significant toxic effects on normal tissue. 8 These studies suggest that an acceptable therapeutic window may be achievable pharmacologically.
A key regulator of the autophagy pathway is a serine/threonine Ulk1 kinase, which is coined Atg1 in yeast. There are three homologs of Atg1 in vertebrates—Ulk1, Ulk2, and Ulk3 (uncoordinated family member [Unc]-51-like kinases 1–3)—yet only Ulk1 is widely expressed. Atg1 kinase activity is required for the induction of autophagy and is inhibited by TOR-directed phosphorylation of both Atg1 and Atg13, which prevents their association. 10 The same principles apply in mammalian cells, where the autophagy pathway is suppressed by PI3K-Akt-mTOR and is activated by AMP kinase (AMPK) and where both Ulk1 and Atg13 are substrates of mTOR and AMPK. 3
Stabilization and activation of Ulk1 require its association with the Hsp90-Cdc37 chaperone complex, and this interaction is necessary for Ulk1-directed phosphorylation of Atg13 on serine-318 (S318). Furthermore, Ulk1 kinase activity and phosphorylation of Atg13 S318 play critical roles in controlling autophagic flux following amino acid deprivation and in the autophagic removal of damaged mitochondria (mitophagy). 11 Based on our characterization of the Ulk1 cellular signaling complex 11 and on the obvious need for specific inhibitors/activators of this pathway, we developed a fully validated high-throughout screening (HTS)–compatible Ulk1 biochemical assay that will enable the discovery of new small-molecule inhibitors of Ulk1. Assay components included purified full-length human Ulk1 and biotinylated Atg13, and by using these, we developed an HTS-compatible Amplified Luminescent Proximity Homogeneous Assay (AlphaScreen). Notably, a pilot screen using the Sigma LOPAC library demonstrated that this Ulk1 inhibitor AlphaScreen assay is both robust and sensitive. Thus, this assay can be used to screen large chemical libraries to discover novel inhibitors targeting Ulk1 and the autophagy pathway.
Materials and Methods
Cell Culture and Reagents
Chemicals including the library of pharmacologically active compounds (Sigma LOPAC1280) were purchased from Sigma Aldrich (St. Louis, MO). The LOPAC library was reformatted at a 2.5-mM concentration in DMSO into 384-well white Optiplates (Greiner Bio-One, Monroe, NC). Human embryonic kidney cells (HEK293T) from American Type Culture Collection (ATCC, Manassas, VA) were maintained at 37 °C in a humidified 5% CO2 atmosphere and cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS), 100 IU/mL penicillin, and 100 µg/mL streptomycin. Mouse monoclonal anti-Flag (FLAG M2, F3165), Atg13 (SAB4200100), and Ulk1 (A7481) antibodies were purchased from Sigma. Phospho-specific Atg13 antibodies (600-401-C49S) were obtained from Rockland (Pottstown, PA). IRDye-labeled antibodies were purchased from LI-COR Biosciences (Lincoln, NE). The AlphaScreen reagents were from PerkinElmer (Waltham, MA).
Immunoblotting
Concentration of protein was measured using the BCA Protein Assay (Pierce, Waltham, MA). Then, 20 µg of protein was added to Laemmli sample buffer and heated to 90 °C for 5 min. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel electrophoresis was performed using NuPAGE 4% to 12% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose using the iBlot system (Invitrogen). Membranes were blocked in Odyssey blocking buffer (LI-COR Biosciences) and incubated with the primary antibody overnight. After repeated washing with TBST (Tris Buffered Saline with Tween 20), blots were incubated with the appropriate IRDye-labeled secondary antibodies (LI-COR Biosciences) for 1 h and visualized using Odyssey infrared imaging (LI-COR Biosciences).
Protein Expression, Purification, and Biotinylation
For purification of full-length Ulk1, an NTAP-Ulk1 expressing vector was expressed in HEK293T cells. Purification was performed as described. 12 Briefly, cells were transfected using calcium phosphate precipitation, and after 24 h, Ulk1 was purified from using the InterPlay TAP Purification Kit according to the manufacturer’s instructions (Agilent Technologies, Santa Clara, CA). Eluted protein was concentrated by filtration (Amicon, EMD Millipore Darmstadt, Germany) to a final concentration of 0.3 mg/mL, and dithiothreitol (DTT) and glycerol were added to a final concentration of 2 mM and 10%, respectively, and stored in aliquots at −80 °C.
For purification of biotinylated N-terminal Bioease-tagged, C-terminal flag tagged full-length human Atg-13, Escherichia coli BL21 (AI) cells were cotransformed with the Avi-Atg13-Flag expression construct and the BirA plasmid (GeneCopoeia, Rockville, MD). A 10-mL overnight culture was used to inoculate 500 mL LB media containing 50 µg/mL ampicillin and 33 µg/mL chloramphenicol. Cultures were grown at 37 °C until the OD600 reached ~0.5. The temperature was adjusted to 30 °C to optimize expression. D-biotin (Supelco, Bellefonte, PA) was added at a final concentration of 50 µM, and expression of Atg-13 and BirA was induced for 4 h with 0.2% L-arabinose and 0.5 mM IPTG, respectively. Bacteria were collected by centrifugation, and proteins were extracted with 15 mL lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100) containing complete protease inhibitors, sonicated five times for 10 s each, incubated on ice for 30 min, and clarified by centrifugation at 4 °C (24,000 × g for 20 min). The protein was purified by anti-FLAG M2 affinity chromatography (Sigma Aldrich). A 1-mL bed volume of Flag-agarose was equilibrated in buffer A (50 mM Tris-HCl, 150 mM NaCl, pH 7.4). The clarified lysate was diluted in buffer A to a concentration of 1 mg/mL and loaded onto the column at 0.25 mL/min. The column was washed with 20 column volumes of buffer A and eluted with buffer B (0.1 M glycine-HCl, pH 3.5). DTT, EGTA, and glycerol were added to the protein sample to a final concentration of 2 mM, 0.1 mM, and 10%, respectively.
Ulk1/Atg13 AlphaScreen Assay
The Ulk1 kinase assay was performed in kinase buffer (KB, 50 mM Tris-HCl [pH 7.5], 20 mM MgCl2, 1 mM DTT, 0.01% Triton X-100 [pH 7.4], DMSO 0.01%, and BSA 0.1 mg/mL) under the following conditions: 5 µL KB containing 10 nM full-length Ulk1 (5 nM final concentration) was mixed with the test compound (100 nL) and incubated at room temperature for 15 min. Next, 2.5 µL KB containing 200 nM Atg13 as well as 2.5 µL KB containing 200 µM ATP (final concentrations 50 nM and 50 µM, respectively) was added. After a 30-min incubation at room temperature, 5 µL detection buffer (DB, 1× AlphaLISA Immunoassay Buffer; PerkinElmer, Waltham, MA) containing anti–phospho-Atg13 antibody and 20 mM EDTA was added to the reaction and incubated for 30 min. Then, 4 µL anti–rabbit acceptor beads (80 µg/mL in DB) and 4 µL streptavidin donor beads (160 µg/mL in DB) were then added. After a 3-h incubation at room temperature, the AlphaScreen signal was measured at a wavelength of 520 to 620 nM using the EnVision (PerkinElmer). Experiments were performed under green light conditions (100 lux) at room temperature in low-volume, AlphaPlate-348 light gray, untreated source plates that were obtained from PerkinElmer (cat. 6005350).
Ulk1/Atg13 ADP-Glo Orthogonal Assay
The Ulk1 kinase assay conditions were exactly as described for the AlphaScreen assay. At the end of the 30-min incubation at room temperature, 10 µL ADP-glo reagent was added to the 10-µL reaction volume and incubated for 40 min. Next, 20 µL Kinase detection reagent was added and incubated for a further 45 min. Luminescence was read after a 1-s integration time on the EnVision plate reader.
Steady-State Kinetics
Initial velocity studies used to determine the steady-state kinetic constants for ATP and NTAP-Ulk1 were conducted in 25 µL kinase buffer containing the following final concentrations: 100 mM HEPES (pH 7.5), 20 mM MgCl2, 2 mM DTT, 1 µCi of [γ-33P]ATP (3000 Ci/mmol) (PerkinElmer), 5 to 100 µM ATP (Sigma Aldrich), and 0.025 to 4 µM NTAP-Ulk1, complemented with phosphatase inhibitor cocktail (Sigma Aldrich) and complete protease inhibitors (Roche, Indianapolis, IN). The reactions were initiated with 5 nM NTAP-Ulk1 (final concentration), and the mixtures were incubated at room temperature for 15 min. Under these conditions, the reaction was linear. The reactions were quenched with 20 µL of 100 mM EDTA. Then, 20 µL of the stopped reaction mixture was spotted in duplicate on an Immobilon-P 96-well plate (Millipore, Billerica, MA). The samples were vacuum-filtered and washed three times with 200 µL of 75 mM phosphoric acid to remove unincorporated [γ-33P]ATP. After a fourth wash with H2O and a final filtration step, 100 µL of Microscint-20 (Packard, PerkinElmer, Waltham, MA) was added to each well, and samples were analyzed on a Packard Topcount liquid scintillation counter. Km determinations using the Ulk1/Atg13 AlphaScreen assay format were performed using assay conditions as described above. Data are the average of three independent experiments. Kinetic data were determined by nonlinear regression analyses.
Data Analysis
Z′ factor values and signal-to-background (S/B) ratios were calculated as described. 13 IC50 values were determined using the Prism software (nonlinear regression and a four-parameter algorithm; GraphPad Software, La Jolla, CA). Data analysis for the LOPAC 1280 compound library was performed using Symyx Assay Explorer version 3.2 (Symyx Technologies, Sunnyvale, CA).
Results
Validation of Signal Detection, Specificity, and Reagent Generation
To confirm selectivity of our signal detection method, we coexpressed full-length Ulk1 along with wild-type (WT) Atg13 or an AtgS318A mutant in HEK-293T cells. As expected, coexpression of Ulk1 and WT Atg13 led to a robust increase in the levels of phospho-Atg13S318 by Western blot, whereas coexpression of Ulk1 with Atg13S318A failed to induce a detectible signal. Thus, this antibody is highly selective for Atg13 phosphorylated at S318 ( Fig. 1A ).
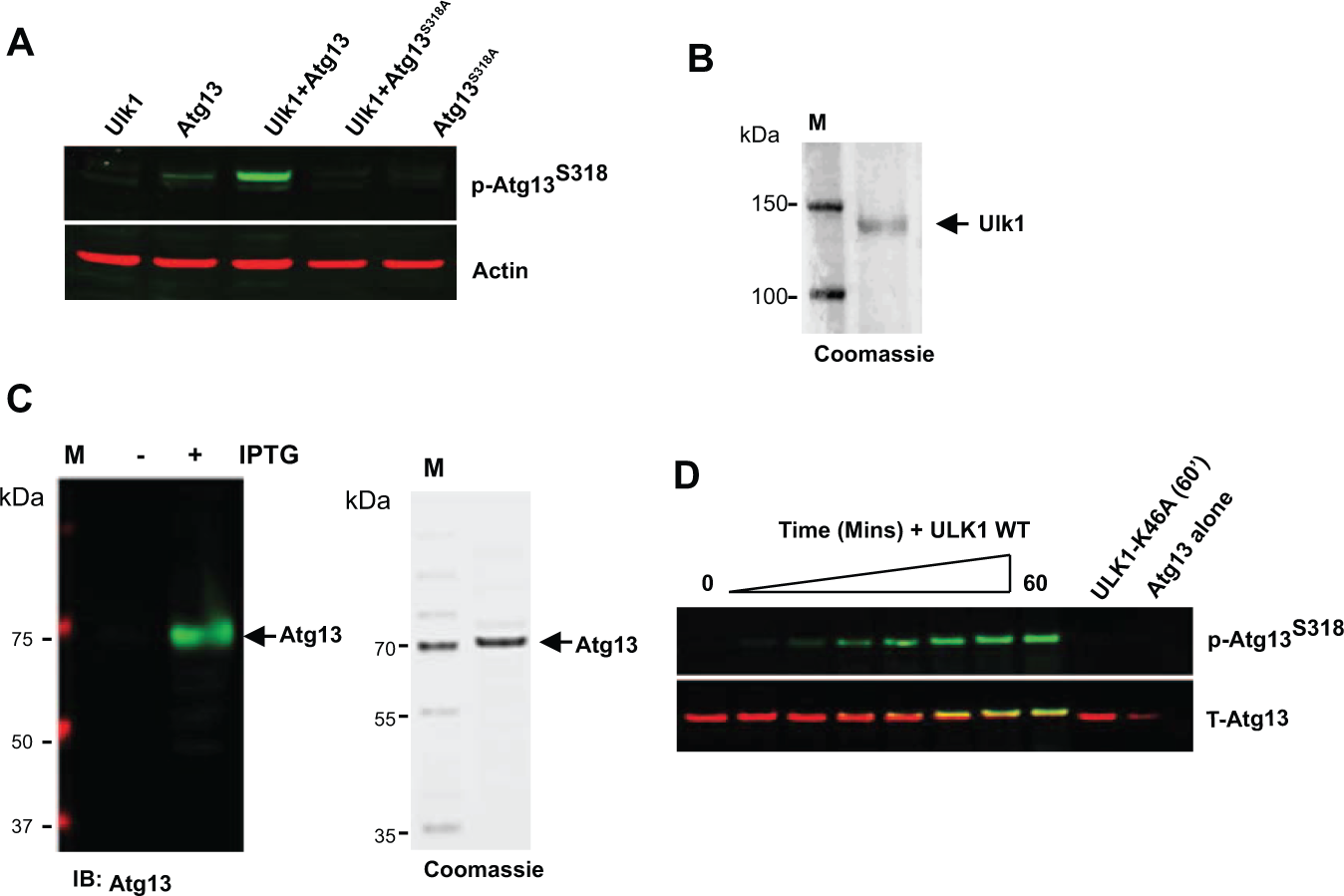
Detection of Ulk1-directed phosphorylation of Atg13-S318 and purification of assay components. (
To generate purified Ulk1 kinase for use in our biochemical assay, we expressed full-length NTAP-Ulk1 in HEK293T cells and purified the enzyme to near homogeneity ( Fig. 1B ). 12 For purification of full-length human Atg13, expression was induced in E. coli along with a biotin ligase (BirA). The recombinant biotinylated Avi-Atg13-FLAG protein was then purified by Flag affinity chromatography. The purity of this protein was confirmed by SDS gel electrophoresis followed by immunoblotting for total Atg13 as well as Coomassie staining ( Fig. 1C ). Furthermore, purified Atg13 was efficiently phosphorylated on S318 following incubation with full-length Ulk1 but not following incubation with the kinase-inactive form of Ulk1 (Ulk1-K46A) ( Fig. 1D ). Therefore, the signal detected is not due to the activity of a contaminating kinase and suggests that the purified Avi-Atg13-FLAG protein is an ideal substrate for detection of Ulk1 kinase activity in vitro.
Ulk1/Atg13 Inhibitor AlphaScreen Assay Development
To test if phosphate incorporation into Atg13 could be detected using AlphaScreen technology, we developed a bead-based, chemiluminescent proximity assay (
Fig. 2A
). Steady-state rate constants for Ulk1 were first determined using a standard radiometric filtration-binding assay, whereby incorporation of radioactive phosphate into Atg13 is measured. Km values for ATP and substrate were determined as 24.1 ± 4.2 and 223 ± 21 µM, respectively (
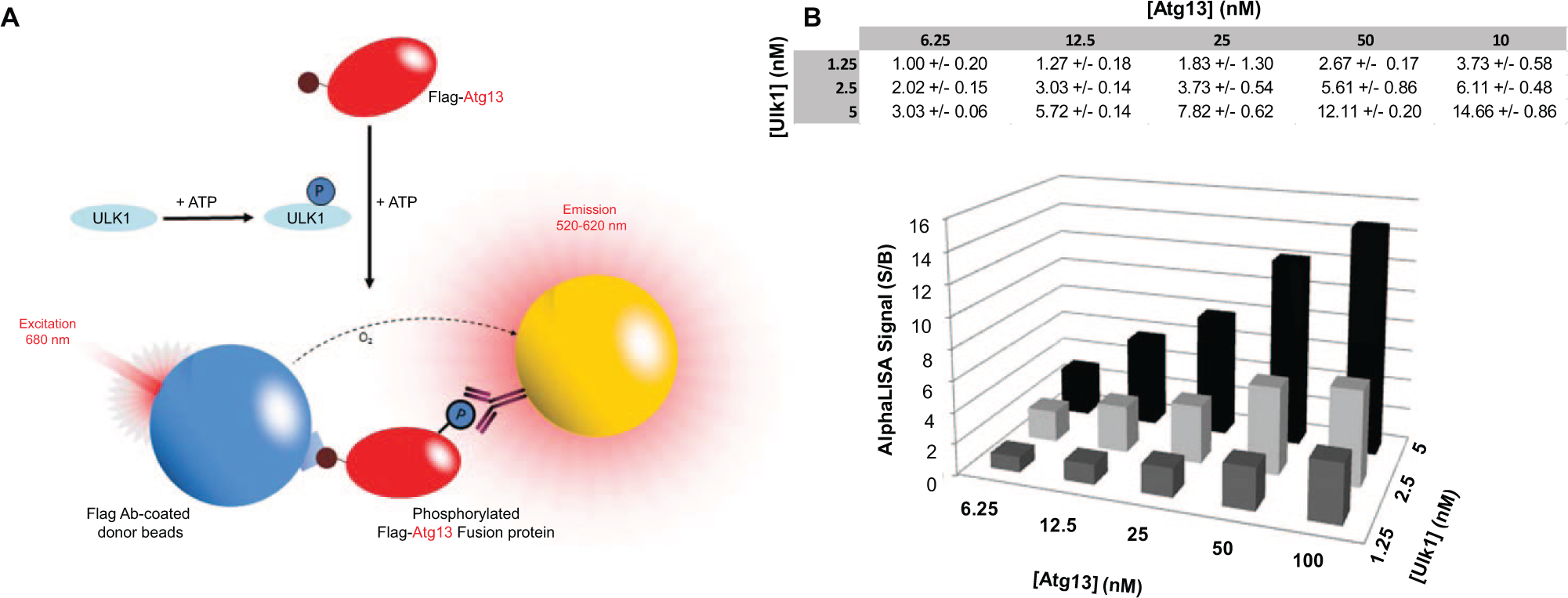
Principles of the Ulk1 AlphaScreen assay. (
Detection and optimization of Atg13 phosphorylation using AlphaScreen assay technology. (
HTS Pilot Screening
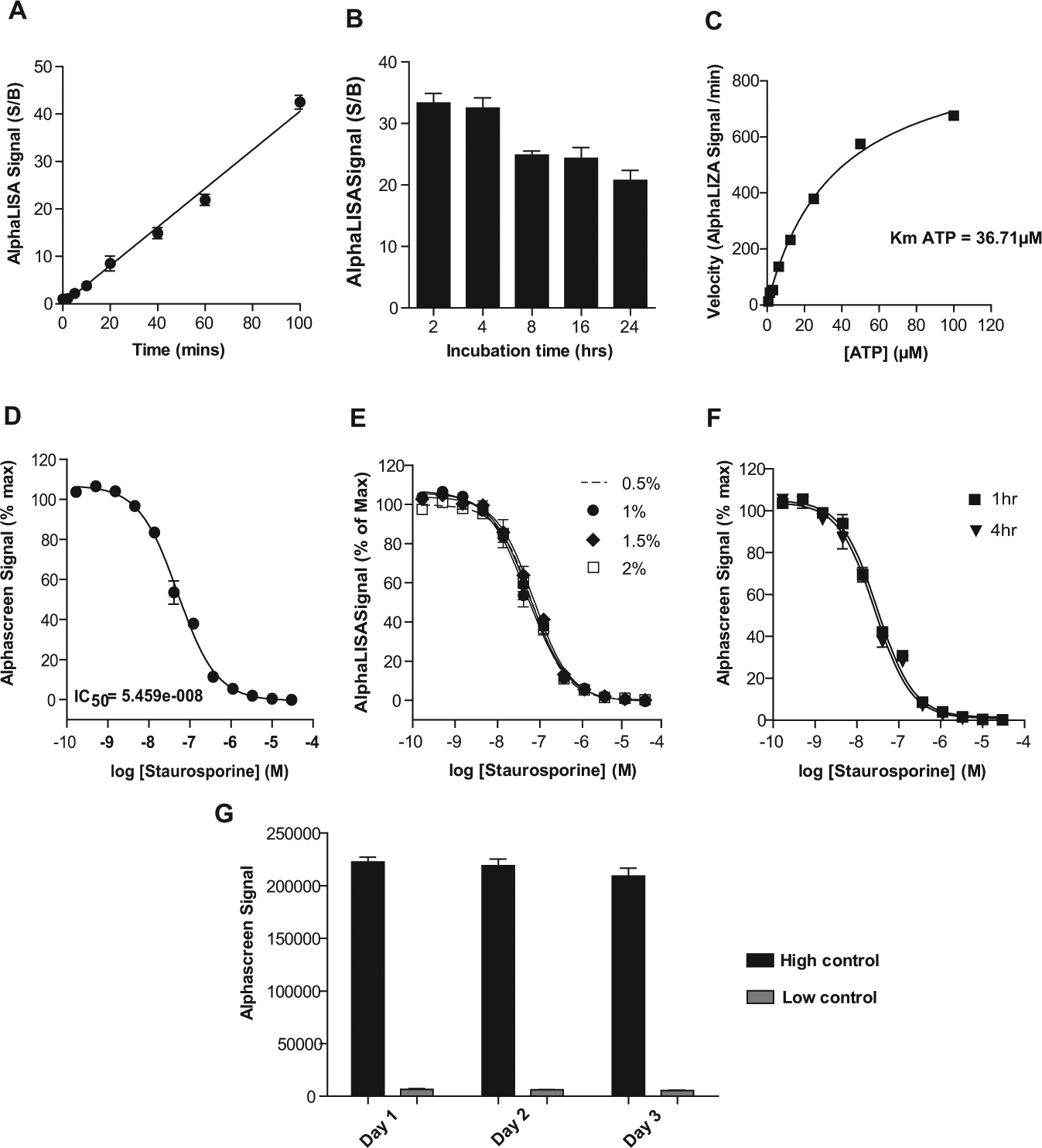
To assess the HTS compatibility of our assay, we first assessed the effects of staurosporine, a nonselective kinase inhibitor, on Ulk1 activity in a 384-well format ( Fig. 3D ). Notably, S/B and Z′ values were ~29-fold and 0.81, respectively (n = 16 high and n = 16 low values), confirming the robustness and reproducibility of this assay. We also examined the effect of DMSO concentration and reagent stability on assay performance. No significant differences in IC50 values of staurosporine were evident in assays having DMSO concentrations ranging from 0.5% to 2% v/v ( Fig. 3E ), which are typical of kinase HTS campaigns. Moreover, master mixes containing substrate, enzyme, and detection reagents were stable when stored at room temperature for at least 4 h, indicating that enzyme activity is maintained within this time frame ( Fig. 3F ). Finally, examination of batch-to-batch reagent stability revealed no significant differences in the performance of the assay when performed over 3 different days. The highly reproducible mean and standard deviations of the AlphaScreen signal are shown for high and low controls ( Fig. 3G ) with S/B ratios of 34, 36, and 38 for days 1, 2, and 3 respectively and with Z′ values of >0.8 in all cases. Thus, batch-to-batch reagent quality is maintained and is suitable for a large compound screening campaign.
To determine the performance of our optimized assay under semi-automated HTS conditions (25 µM compound in 1% DMSO), we screened the Sigma LOPAC (Library of Pharmacologically Active Compounds; 1280 compounds). As a positive control (100% inhibition), we used an IC100 of staurosporine, and DMSO was used as a negative control (0% inhibition). An activity scatterplot of all compounds tested, as well as positive and negative controls, is shown in Figure 4A . To assess plate homogeneity and assay reproducibility, each plate contained high (n = 16) and low (n = 16) signal control wells. Furthermore, Z′ factors for the pilot screen were calculated (0.83 ± 0.02), confirming assay robustness. In addition, the scatterplot of measurements from replicate plates yielded a correlation coefficient of r2 = 0.97, indicating high reproducibility in hit identification ( Fig. 4B ). During miniaturization and optimization to the 1536-well format, measures to maintain uniform plate temperature will be employed to reduce potential edge effects ( Fig. 4A ).
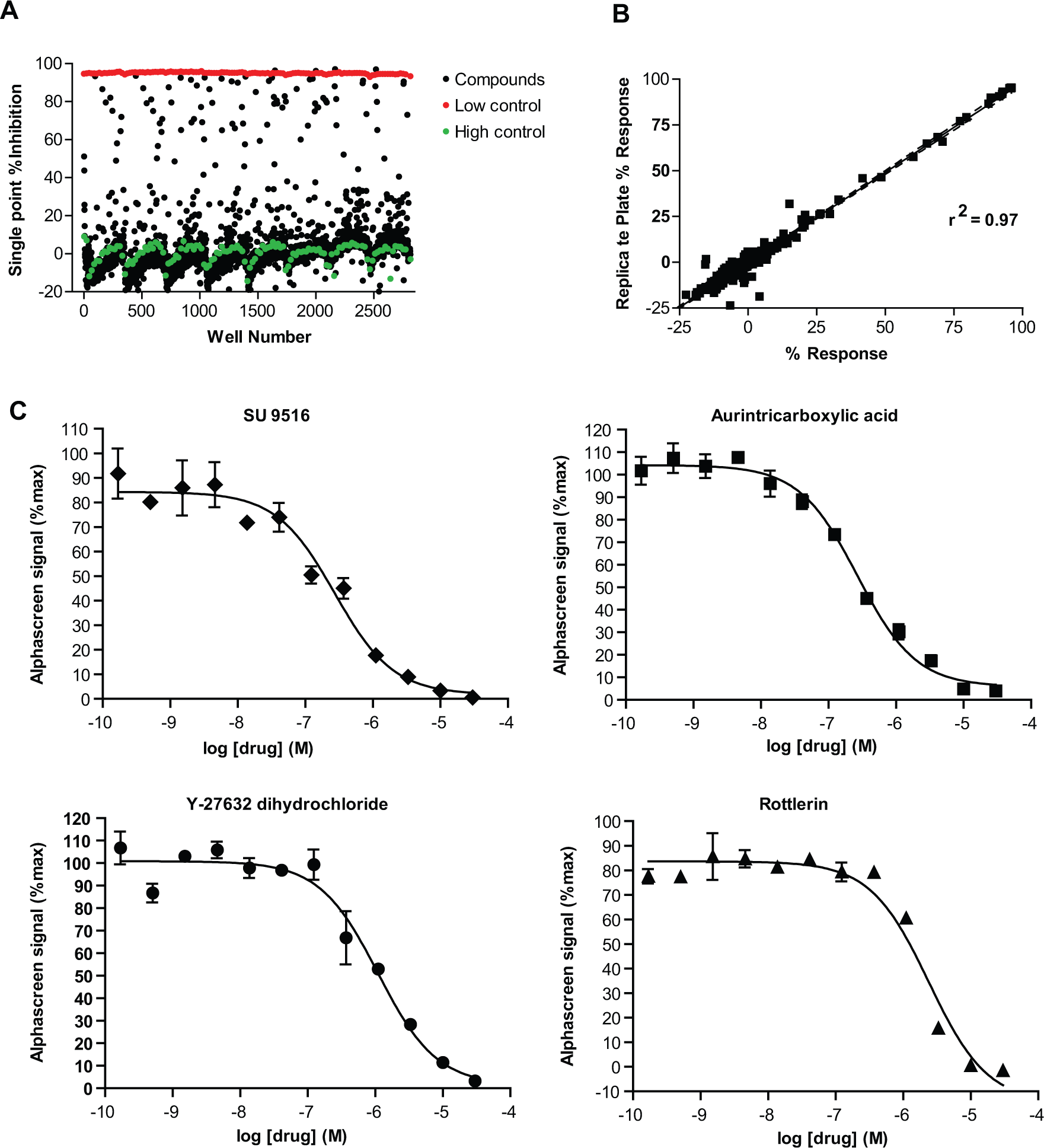
Analysis of the high-throughput LOPAC pilot screen by scatter plot. (
The AlphaScreen signal for each compound was converted into percentage inhibition (%INH) relative to the staurosporine (IC100) positive control. Any compound that gave greater than the assay average plus three times the standard deviation was considered suitable for follow-up. Using this hit cutoff criterion, we identified 39 hits (hit rate of 3%), many of which were known protein kinase inhibitors, consistent with expectations. IC50 values for the top 15 inhibitors were determined (
Comparison of all of our hits with those obtained with a previous screen of the LOPAC library using a similar AlphaScreen kinase assay identified only three common hit compounds (calmidazolium chloride, aurintricarboxylic acid, and reactive blue 2).
14
Of these, only reactive blue 2 inhibited the “true hits” assay AlphaScreen signal by greater than 50%, further indicating that this hit alone is a false positive. Furthermore, using an orthogonal assay that uses an entirely different detection scheme, we confirmed the hits identified from the primary assay (
In summary, we report here the development of a novel HTS-compatible biochemical assay designed to identify small-molecule inhibitors of Ulk1. Our selective assay, which uses full-length active human Ulk1 kinase and its native (unphosphorylated) substrate Atg13, is based on an amplified luminescent proximity homogeneous (AlphaScreen) platform that is an extremely sensitive, nonradioactive, homogeneous “mix-and-read” format. We demonstrate HTS compatibility and robustness in a 384-well format; stability and performance are not affected by DMSO concentrations and conditions commonly used in HTS screens. Finally, a pilot screen of the Sigma LOPAC library using semi-automated conditions produced excellent S/B, Z′ values, and plate-to-plate reproducibility. Collectively, these findings demonstrate that we have developed a robust and reproducible HTS compatible assay suitable for screening large compound libraries for the identification of novel small-molecule inhibitors of Ulk1. While this manuscript was under review, Lazarus et al. 15 published the first structure of Ulk1 with two high-resolution crystal structures of the kinase bound to inhibitors, which aided in generating improved Ulk1 inhibitors. We anticipate that Ulk1 HTS campaigns will increase the diversity of starting compounds, and aided by structure-based design, highly valuable, potent, and selective small-molecule Ulk1 inhibitors will be generated to facilitate the study of autophagy. We are currently miniaturizing our validated 384-well microtiter plate assay into a 1536-well format to enable a lower cost, large-scale HTS campaign.
Footnotes
Acknowledgements
We sincerely thank Ms. Pamela Clark-Spruill for secretarial assistance, Chunying Yang for technical assistance, and the Duckett laboratory for input and editing of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by CA169142 (to J.L.C. and D.R.D.), by the Rendina Family Foundation (D.R.D.), by the NCI Comprehensive Cancer Center (grant P30-CA076292) to the H. Lee Moffitt Cancer Center & Research Institute (J.L.C.), and by funds from the state of Florida to Scripps Florida (D.R.D.) and to the H. Lee Moffitt Cancer Center and Research Institute (J.L.C.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.