
Abstract
Sterile alpha motif and histidine-aspartate domain-containing protein 1 (SAMHD1) is a recently discovered enzyme that plays a central role in nucleotide metabolism and innate immunity. SAMHD1 has deoxyribonucleoside triphosphate (dNTP) triphosphohydrolase activity that depletes the dNTP substrates required for DNA synthesis in cells. The involvement of SAMHD1 in biological processes as varied as viral restriction, endogenous retroelement control, cancer, and modulation of anticancer/antiviral nucleoside drug efficacy makes it a valuable target for the development of small-molecule inhibitors. We report a high-throughput colorimetric assay for SAMHD1 dNTP hydrolase activity that takes advantage of Escherichia coli inorganic pyrophosphatase to convert PPPi to 3 Pi. The assay was validated by screening a library of 2653 clinically used compounds. Fifteen primary hits were obtained (0.57% hit rate); 80% of these were confirmed in a direct secondary assay for dNTP hydrolysis. The zinc salt of the antibiotic cephalosporin C was a potent inhibitor of SAMHD1 with an IC50 of 1.1 ± 0.1 µM, and this inhibition was largely attributable to the presence of zinc. The assay also screened a targeted library of nucleosides and their analogs, revealing that the antiviral drug acycloguanosine (acyclovir) is an inhibitor possessing excellent properties for future fragment-based drug development efforts.
Introduction
Sterile alpha motif and histidine-aspartate domain-containing protein 1 (SAMHD1) is tetrameric deoxyribonucleoside triphosphate (dNTP) triphosphohydrolase that disrupts the synthesis of nonhost nucleic acids. SAMHD1 is composed of two domains: an N-terminal sterile alpha motif (SAM) domain of unknown function and a C-terminal histidine-aspartate (HD) domain with metal-dependent phosphohydrolase activities. Upon activation by dGTP or GTP, the activity of the HD domain catalyzes the hydrolysis of any dNTP to its deoxyribonucleoside (dN) and triphosphate (PPPi). 1 Crystal structures and biochemical analyses have shown that activation by binding of guanine nucleotides to a guanine-specific activator (A1) precedes the binding of dNTPs to a second nonspecific activator (A2) site on each monomer, which in turn drives the formation of the catalytically competent tetramer.2–4 SAMHD1 also has activator-independent RNA binding and exonuclease activities,5,6 although the presence of the nuclease activity has been disputed. 1
SAMHD1 is found across many cell types 7 but is most highly expressed in immune cells of the myeloid lineage, where it acts as a viral restriction factor. 8 The SAMHD1-catalyzed depletion of dNTP pools in these cells restricts infection of diverse retroviruses 9 and DNA viruses 10 by depriving the viral DNA polymerases of dNTP building blocks. SAMHD1-mediated restriction of HIV-1 infection in dendritic cells prevents the activation of CD4+ T cells and, paradoxically, dampens subsequent adaptive immune responses. 11 Although SAMHD1 inhibits the productive infection of myeloid immune cells, the aborted reverse transcripts that accumulate in these cells triggers an inflammatory response and death of bystander CD4+ T cells in a process termed pyroptosis. 12 Inhibition of SAMHD1 would prevent the accumulation of aborted transcripts and would therefore be expected to prevent T-cell depletion in response to HIV infection and possibly slow the progression to AIDS.
SAMHD1 also has essential roles in nucleotide metabolism and homeostasis.7,13 Loss-of-function mutations in SAMHD1 are associated with autoimmune disorders such as the inheritable neuroinflammatory disease Aicardi-Goutières syndrome (AGS) 13 and the inflammatory disease systemic lupus erythematosus (SLE). 14 Both AGS and SLE stem from an immune response to aberrant intracellular nucleic acids that mimic chronic viral infection. 15 Loss-of-function mutations in SAMHD1 16 or epigenetic silencing of the SAMHD1 locus 17 are also associated with the development of various cancers, possibly through increased mutation rates caused by dNTP pool imbalances. 18 SAMHD1 expression also increases the efficacy of nucleoside drugs that are used to treat viral infections and cancer, likely by decreasing the concentration of competing intracellular dNTPs. 19
The involvement of SAMHD1 in the above biological processes motivates inhibitor design for both basic science and potential clinical applications. The large number of nucleotide binding sites (12 per tetramer) and the need for allosteric activation and oligomerization suggest that SAMHD1 would be highly amenable to inhibition by small molecules. Indeed, we have designed a nucleotide analog that inhibits SAMHD1 by the surprising mechanism of destabilizing the catalytically competent tetramer, 20 but the utility of this compound was limited by poor bioavailability. Here we report a cost-effective high-throughput colorimetric screen for SAMHD1 dNTPase activity using Escherichia coli inorganic pyrophosphatase as a coupling enzyme for the detection of inorganic phosphate. This method can be readily applied to screen large chemical libraries for inhibitors of SAMHD1 that could be functional in cell culture. Such inhibitors would be especially valuable for investigating the function of SAMHD1 in primary immune cells that are genetically difficult to manipulate. Specific inhibitors of the dNTPase activity would be particularly useful to determine whether the dNTPase or the RNA exonuclease activity of SAMHD1 is responsible for retroviral restriction and retroelement control as these roles are currently disputed in the literature.6,8
Materials and Methods
Human SAMHD1 Overexpression and Purification
Full-length human SAMHD1 was expressed as a PreScission protease-cleavable His10 fusion in E. coli BL21-DE3 cells and purified by Ni-NTA and cation exchange chromatography as described previously. 4 The protein concentration was calculated by its absorbance at 280 nm using an extinction coefficient of 76,500 M–1 cm–1 (ProtParam, ExPASy). Typical yields were 20 mg of SAMHD1 per liter of bacterial growth with an estimated purity of >95% as determined from sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) with visualization by Coomassie blue staining. The purified protein (100 µΜ) was flash frozen in small aliquots (20 to 50 µL) at -80 °C in storage buffer (50 mM Tris⋅HCl [pH 7.5], 150 mM KCl, 5 mM MgCl2, 1 mM DTT, 20% glycerol). Thawed aliquots for activity assays were stored at -20 °C for a maximum of 3 d before discarding.
Inorganic Pyrophosphatase (PPase) Overexpression and Purification
The inorganic pyrophosphatase gene with its native promoter, Shine-Dalgarno sequence, and terminator was PCR-amplified from E. coli K12 genomic DNA using oligonucleotide primers (forward: 5′ ATT TTA GGA TCC AGA CGA AAA CAA GCG AAG ACA TTC 3′; reverse: 5′ ATT TTA AAG CTT GTG TGT TTA TTT ATC GCG GGC). The PCR product was ligated into the BamHI and HindIII sites of pUC19, and the sequence of the insert was verified by sequencing. The plasmid (pUC19-PPase) is available upon request. E. coli DH5α cells were transformed with pUC19-PPase and grown in LB medium at 37 °C for 15 h. The cells were harvested by centrifugation, and the cell pellets were stored at -80 °C until purification. The cell pellet was resuspended in lysis buffer (50 mM Tris⋅HCl [pH 7.5], 100 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, protease inhibitors [Sigma P2714], and 0.5 mg/mL lysozyme) and rotated at 4 °C for 1 h. The crude lysate was clarified by centrifugation at 30,000 g for 30 min at 4 °C. Nucleic acid was precipitated by the slow dropwise addition of one-half volume of cold 10% streptomycin sulfate on ice. The nucleic acid was removed by centrifugation at 30,000 g for 30 min at 4 °C. The supernatant was adjusted to 20 mM MgCl2 by addition of 2 M MgCl2 stock, then heated in a 70 °C water bath for 30 min. The solution was returned to ice for 30 min, and the precipitated protein was removed by centrifugation at 30,000 g for 15 min at 4 °C. The supernatant (which contains PPase) was warmed to 20 °C and adjusted to 70% saturated ammonium sulfate. The solution was stirred for 30 min, and the protein precipitate (containing PPase) was collected by centrifugation at 30,000 g for 30 min at 20 °C. The pellet was resuspended in a minimal volume of storage buffer (50 mM Tris⋅HCl [pH 8.0], 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 30% glycerol) and dialyzed overnight at 4 °C. The purified protein (900 µM) was aliquoted (500 µL) and stored at -20 °C. The PPase concentration was determined using the Bradford assay with bovine serum albumin as the standard. Typical yields were 200 mg/L pyrophosphatase with >75% purity by SDS-PAGE, which is sufficient for the screening assay. We found that the activity of PPase obtained from this method was identical to commercially available preparations (Sigma I5907).
Enhanced Malachite Green (MG) Assay for SAMHD1 dNTPase Activity
Because SAMHD1 produces PPPi and a dN as products, the PPPi product was converted to inorganic phosphate (Pi) in a coupled reaction with pyrophosphatase before detection colorimetrically using the well-known MG phosphate detection reagent. 21 A working stock of MG solution was prepared by dissolving 0.40 g of MG hydrochloride (Sigma M9636) in a 360 mL solution consisting of 300 mL ddH2O and 60 mL concentrated H2SO4 (JT Baker 9681). Separate ddH2O stocks of 7.5% (w/v) ammonium molybdate tetrahydrate (Fluka 09880) and 11% (v/v) Tween-20 (Sigma P7949) were prepared. The MG detection reagent was prepared fresh daily by mixing 20 mL of the MG solution with 5 mL of ammonium molybdate and 0.4 mL of Tween-20 solution. Standard curves for phosphate detection were prepared in clear polystyrene round-bottom 96-well plates (Costar 3795) by serial dilution of a standard solution (80 µL final well volume; Ricca Chemical Company 5839). Flat-bottom 96-well plates performed similarly and can be substituted if desired. The phosphate standards used the same buffer and quench conditions as the enzymatic reactions (see below). The MG detection reagent was added in a ratio of 1:4 (20 µL of detection reagent to 80 µL of reaction solution). Plates were mixed by rotary shaking at 780 rpm for 30 s and were then incubated at room temperature for 90 to 240 min. The increased incubation time in this enhanced version of the MG assay increases the linear range for Pi detection and the overall signal-to-background of the assay (see the Results section). The absorbance at 650 nm for each well was measured by Multiskan Ascent plate reader (Labsystems).
In the high-throughput screening (HTS) format, SAMHD1 reactions were performed in a total volume of 40 µL (50 mM Tris-HCl [pH 7.5], 50 mM KCl, 5 mM MgCl2, 0.05% Brij-30) using 96-well round-bottom plates. All liquid dispensing was performed using a TomTec Quadra 96-320 liquid handler with reagents aspirated from polypropylene storage plates (Costar 3363) or deep-well assay blocks (Costar 3959). Positive control reactions (no inhibitors) were prepared by aspiration of 10 µL of a mock solution consisting of buffer and 4% DMSO, 20 µL of substrate solution consisting of 0.2 mM dGTP in 2× reaction buffer, and 10 µL enzyme solution consisting of 2 µM SAMHD1 and 20 µM PPase into a single tip with 20 µL air gaps separating each solution. The reagents were dispensed into wells with a 20 µL air blowout and touch off, and the plates were immediately mixed by rotary shaking. The final concentrations of all components in each reaction well are listed in Table 1 . Controls established that the final concentration of 1% DMSO has no effect on the SAMHD1 activity. Reaction quench solutions were prepared by aspiration of 40 µL of 20 mM EDTA and 20 µL of MG detection solution into each tip using a 20 µL air gap separation and then dispensing the quench solutions into the reaction wells using a 20 µL air blowout and touch off. After quenching, the plates were mixed by rotary shaking and the absorbance was measured as above after a 90 min incubation. The phosphate present in each well was determined by comparison with a standard phosphate curve.
Optimized 96-Well High-Throughput Screening Parameters and Assay Statistics. a
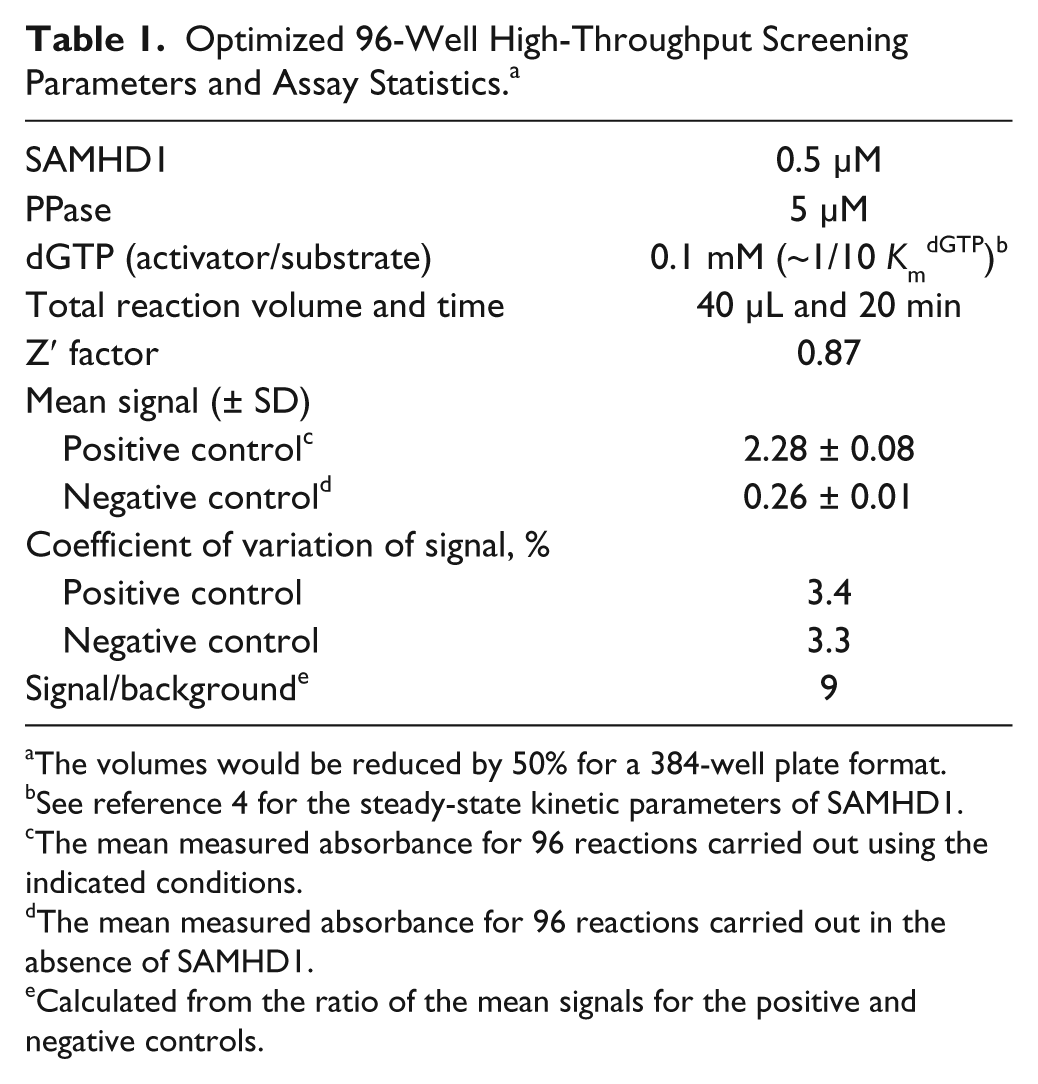
The volumes would be reduced by 50% for a 384-well plate format.
See reference 4 for the steady-state kinetic parameters of SAMHD1.
The mean measured absorbance for 96 reactions carried out using the indicated conditions.
The mean measured absorbance for 96 reactions carried out in the absence of SAMHD1.
Calculated from the ratio of the mean signals for the positive and negative controls.
Pilot Library Screening
The optimized SAMHD1 assay was used to screen a pilot library of 2653 clinically used compounds (a generous gift of Dr. Jun O. Liu). The reactions were performed in reaction buffer with final concentrations of 0.5 µM SAMHD1, 5 µM PPase, 0.1 mM dGTP, 1% DMSO, and 10 µM library compound prepared as described above. After 20 min, reactions were quenched with EDTA and the MG detection reagent, and the absorbance was measured after 90 min color development time as described above. Each screening plate contained 80 compounds and 16 DMSO-only controls. Half of the DMSO-only control wells were used for the SAMHD1 + PPase positive activity controls and the remaining were used for the PPase-only negative activity controls. The percentage SAMHD1 activity that remained in each compound well was calculated from the average of the positive and negative control reactions on that plate (% activity remaining = 100 × (Awell – <Aneg>)/(<Apos> – <Aneg>), where Awell is the absorbance of a well containing a library compound and <Apos> and <Aneg> are average absorbance values for the control wells for the same plate. The Z′ statistical factors for each plate were calculated using eq 1. 22
Thin-Layer Chromatography (TLC) Secondary Assay
Compounds that were identified as hits in the primary enzyme-coupled MG assay were confirmed using a direct TLC assay that separates the dNTP substrate from the nucleoside product. 4 In the secondary assay, SAMHD1 reactions were prepared under the same conditions as in the MG assay, except that PPase was omitted and the dGTP substrate was spiked with trace amounts of tritium-labeled [8- 3 H]dGTP. Small portions (2 µL) were quenched at various times by direct spotting on a C18 reversed-phase TLC plate. The TLC plates were developed and quantified as described previously to obtain a reaction rate for each inhibitor, which was compared with a DMSO control. 4
Dose-Response Curves and IC50 Determinations
Dose-response curves were performed using the MG assay. Master solutions of each compound were prepared from the solids and were serially diluted by twofold into enzyme reactions. The reactions were otherwise carried out exactly as described above. The percentage SAMHD1 activity remaining as a function of inhibitor concentration was fit to eq 2 employing a variable Hill slope parameter
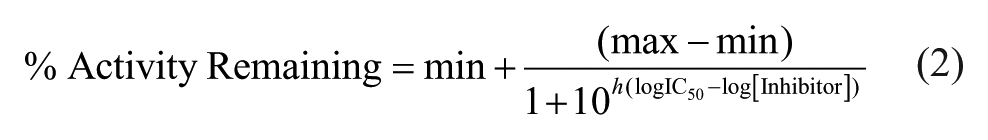
where max and min are the maximal and minimal percentage activity values and h is the Hill slope using the program Prism 6 (GraphPad Software).
Results and Discussion
Enzyme-Coupled MG Assay
The triphosphohydrolase reaction catalyzed by SAMHD1 produces dN and triphosphate products, neither of which is directly amenable to high-throughput detection. However, reliable colorimetric 21 and fluorescent 23 methods do exist for the detection of free inorganic phosphate (Pi). We therefore envisioned an assay in which the production of triphosphate by SAMHD1 was coupled to the detection of Pi by enzymatic PPPi hydrolysis. To be feasible in this application, the coupling enzyme should be easy to purify in high yields and highly specific for the desired PPPi product with little or no activity on the dNTP substrate. We found that inorganic pyrophosphatase (PPase) from E. coli satisfies these requirements. We thus optimized a chromatography-free purification procedure that gives a high yield (200 mg/L) of active PPase of sufficient purity for screening and determined that it has minimal activity on the dGTP substrate of the SAMHD1 reaction (see below).
Optimization of the MG Assay
A simple and economical method for detecting Pi is the change in absorbance of a solution of MG and molybdate upon complexation with phosphate. 21 Although a continuous enzyme-coupled assay for Pi exists, 23 this assay is prohibitive to implement in an HTS format because it requires stoichiometric complexation of phosphate binding protein with Pi. In contrast, the MG assay is easy to implement and requires only inexpensive reagents.
MG has been used to detect phosphate in many applications,
21
but we were initially disappointed in the linear range and signal-to-background obtained using reported methods. Nevertheless, by simply increasing the incubation time for development of the MG complex from 10 to 90 min before making the absorbance measurements, we were able to extend the linear range for phosphate detection from 4 to 12 nmol (
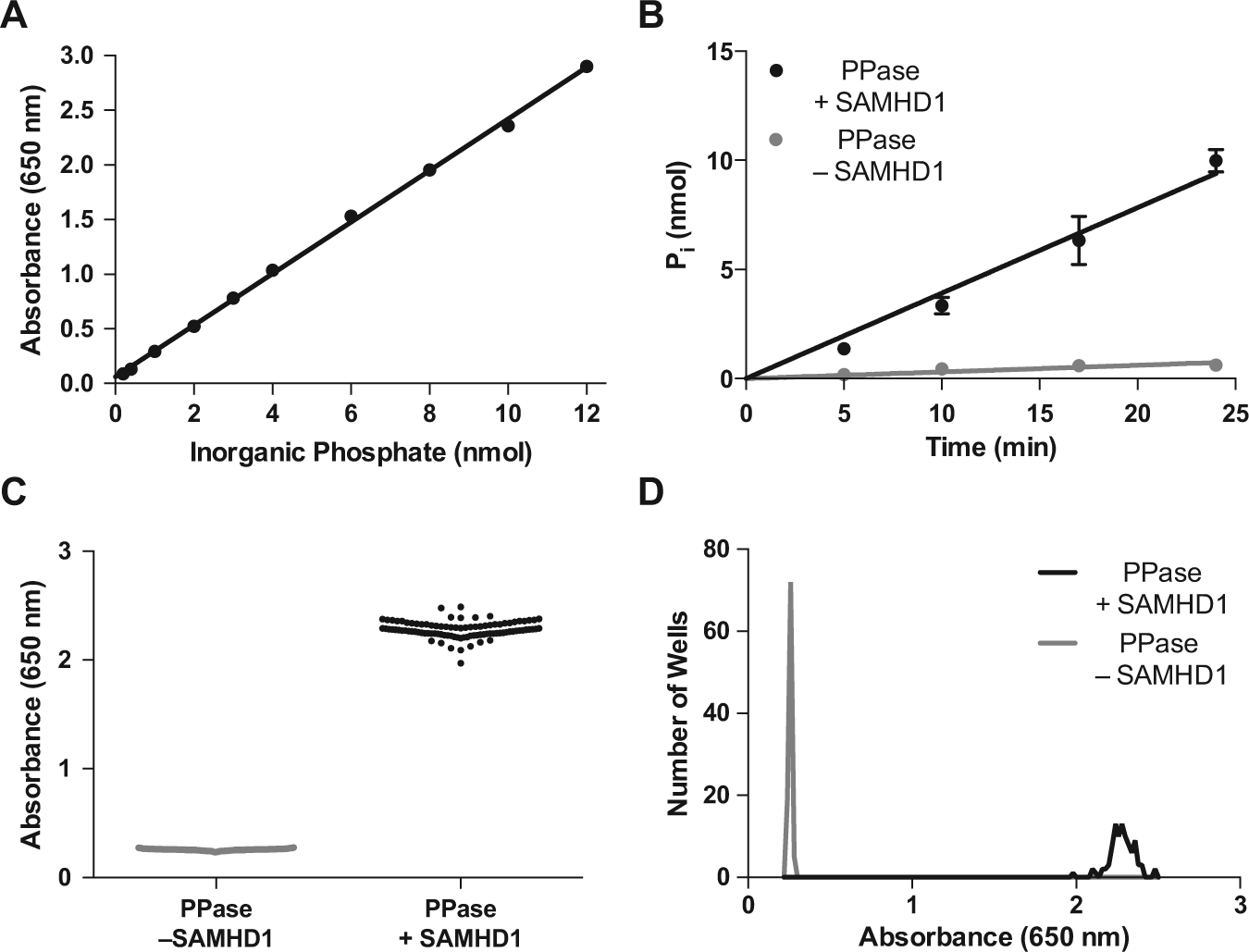
Development of the enzyme-coupled SAMHD1 assay. (
Pyrophosphatase Coupling Reaction
For an enzyme-coupled reaction to be useful, the coupling enzyme must be present in sufficient quantity such that its reaction is fast compared with the reaction of interest. To determine the PPase concentration that was required to meet this condition, a series of experiments were performed in which the SAMHD1 and dGTP substrate concentrations were fixed at the desired concentrations for the HTS and the PPase concentration was varied from 1 to 9 µM. From this analysis, we ascertained that the rate of Pi formation was zero-order in [PPase] when its concentration exceeded about 5 µM (
We note that during our HTS optimization, another group reported an MG-based colorimetric assay for SAMHD1 but did not report its use in an HTS application. 24 Our enhanced MG assay has several advantages over this previous version. First, the reported assay employed yeast exopolyphosphatase to couple the production of PPPi to detection of Pi + PPi. Because our assay uses PPase as the coupling enzyme, PPPi is converted completely to 3Pi, and the color yield per PPPi is threefold greater. This also has significant cost benefits because the enhanced assay uses one-tenth the concentration of the dGTP substrate as compared with the reported assay, and dGTP is the major contributor to the assay cost. Second, bacterial expression of recombinant yeast exopolyphosphatase is only 0.5% the level of pyrophosphatase, and its purification involves more steps and expensive resins. 25 Third, our enhanced assay uses a 10-fold lower concentration of full-length SAMHD1 enzyme as opposed to the N-terminal truncated form used in the published MG assay. We have found that the N-terminal truncation reduces the maximal activity by about 50% as compared with full-length SAMHD1. In general, it is always more desirable to screen the enzyme form that is expressed in cells. 4
Development of the HTS
Based on the time dependence of the SAMHD1 reaction under the desired conditions of the HTS, a 20-min reaction time was selected that corresponded to about 75% reaction. To test the statistics and reproducibility of the enhanced MG assay under these conditions, two 96-well plates of replicate reactions containing either SAMHD1 and PPase (positive control) or PPase alone (negative control) were prepared. The two plate sets showed low well-to-well variation (coefficient of variation = 3.3% and 3.4%), and the mean absorbance increase was 2.02 ± 0.08 between the reactions in the presence and absence of SAMHD1, corresponding to a signal-to-background of nine ( Fig. 1C , D ). The high Z′ = 0.87 and the low coefficients of variation in both the positive and negative control wells indicate that this assay has favorable properties as an HTS. 22 The optimized assay conditions and statistical parameters for the 96-well format are summarized in Table 1 . The expected reaction volumes and cost per well would be halved upon miniaturization to 384-well plates (and even further for a 1536-well format), while the compound throughput would increase approximately fourfold.
Pilot Library Screening
The optimized assay was used to screen a library of 2653 clinical compounds. 26 The screening results approximated a normal distribution with a mean around 95% of the activity of the DMSO-only controls ( Fig. 2A , B ). Setting a hit threshold of 3σ below the mean (<71.5% activity), 29 hits were obtained (1.1% hit rate). A more stringent cutoff of 6σ (<48% activity) yielded 15 hits (0.57% hit rate). The Z′ factors calculated from the eight positive and negative control wells on each plate gave an excellent average Z′ of 0.90 ± 0.04 ( Fig. 2C ). Although this library is relatively small, it has a representative chemical diversity suggesting that the screen can be readily applied to larger libraries.
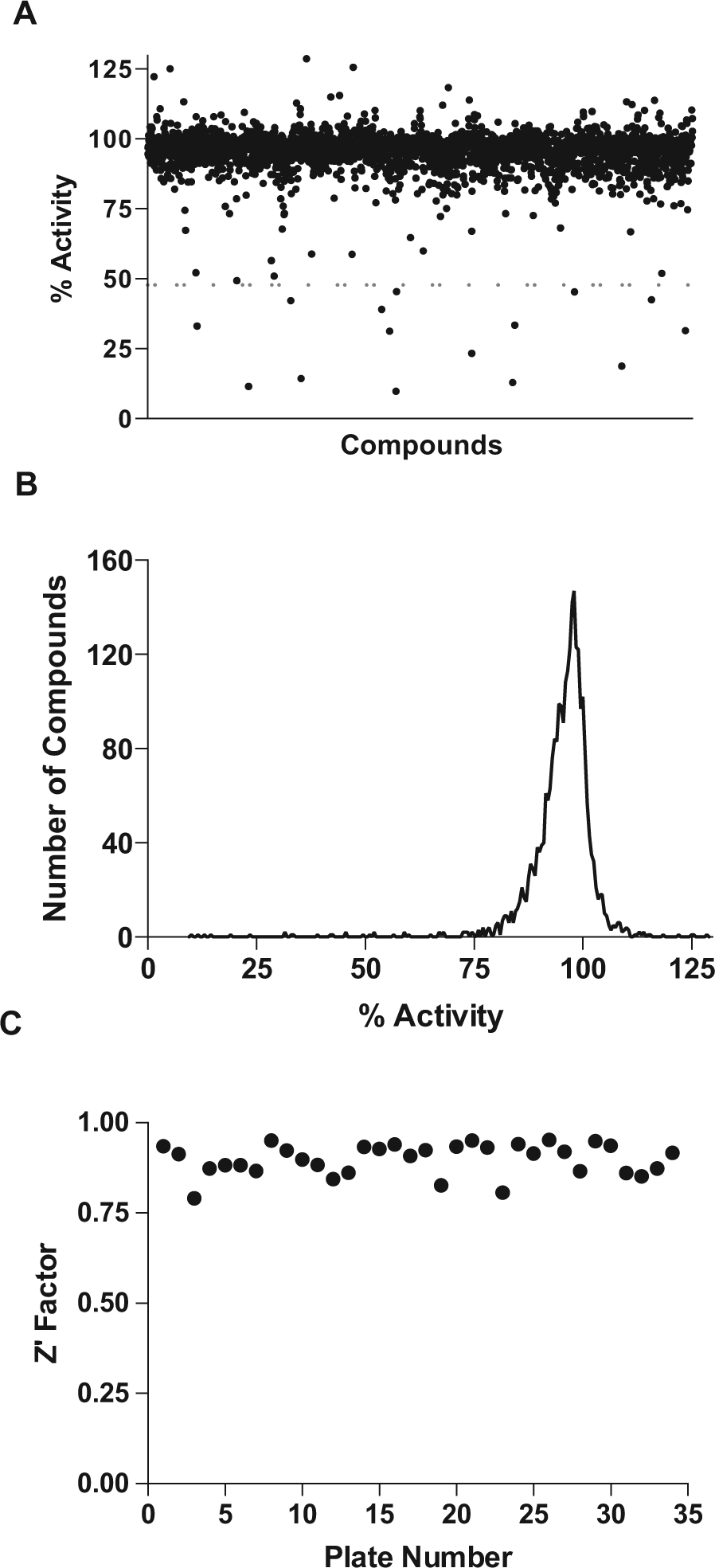
High-throughput screen of the Hopkins library of 2653 clinically used compounds.
26
(
Hit Validation
The 6σ cutoff was used to focus on 15 hits that were detected in the primary screen. The secondary screen uses reversed-phase TLC separation of [
3
H8]-dGTP from the nucleoside product.
4
This is a rapid and economical secondary screen that can be used to make single-time-point activity determinations on hundreds of compounds in several hours using a 12-channel pipettor. Of the 15 compounds tested, 12 showed similar or greater inhibition in this direct assay, corresponding to a validation rate of 80% (
Fig. 3A
). The structures of these hits as well as their activity in the MG primary and TLC secondary screen are shown in
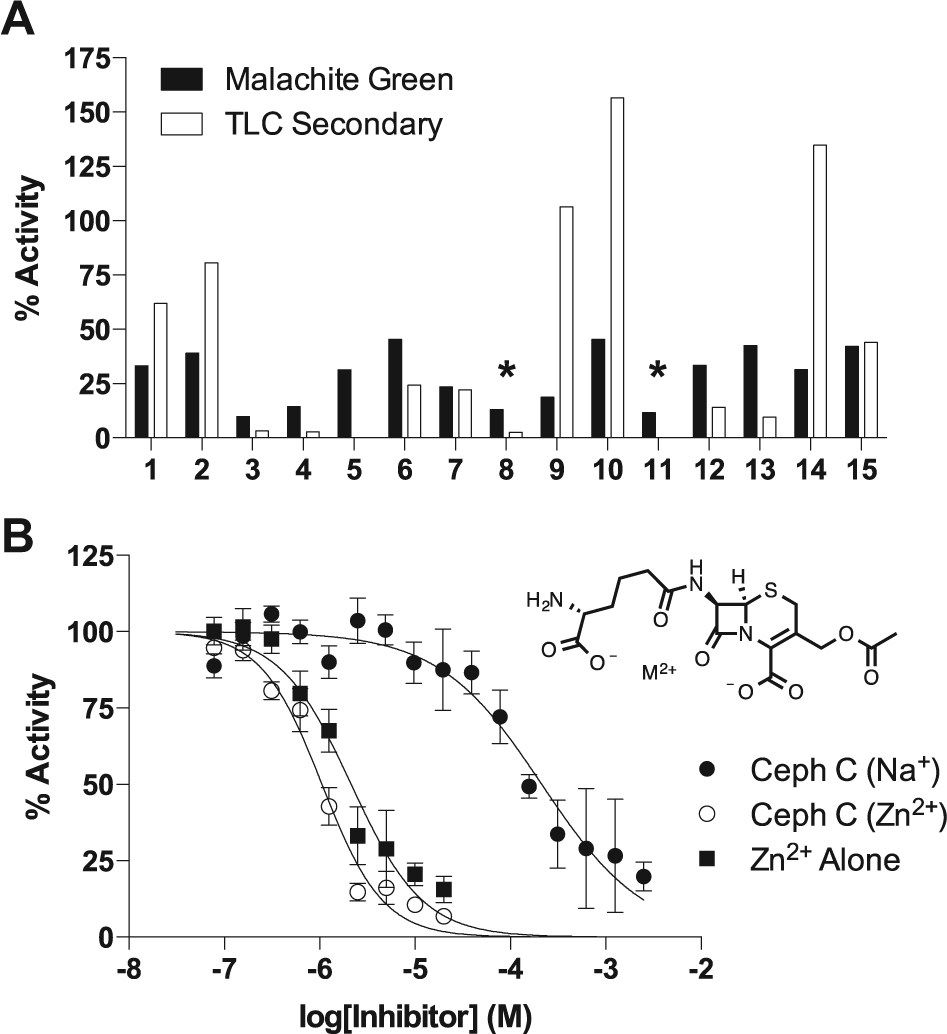
Hit confirmation. (
The Zn2+ salt of Ceph C was obtained as a solid for further analysis. We were unable to procure the sodium salt from any commercial vendor, so it was generated from the Zn2+ salt by ion exchange using Dowex-Na+ resin. Dose-response studies using the two salt forms of Ceph C showed that the Zn2+ salt was a 200-fold more potent inhibitor (IC50CC-Zn = 1.1 ± 0.1 µM, h = 1.4 ± 0.1; IC50CC-Na = 213 ± 30 µM, h = 0.8 ± 0.1; Fig. 3B ).
Some cephalosporin antibiotics are known to form tight complexes with divalent metal ions,
27
including Zn2+, which suggested that the inhibition might result from an E-Zn2+–Ceph C enzyme-metal-bridged complex. However, further investigation showed that Zn2+ alone (as the chloride salt) was also inhibitory (IC50Zn = 2.1 ± 0.2 µM, h = 1.2 ± 0.1;
Fig. 3B
). The inhibition of SAMHD1 by Zn2+ ions even in the presence of 5 mM MgCl2 has not been investigated previously, although crystal structures have been reported that use Zn2+ as a catalytically inert metal.
2
The inhibition by Ceph C-Zn2+ and Zn2+ alone was completely rescued by the addition of approximately stoichiometric amounts of EDTA, which selectively chelates Zn2+ over Mg2+ (
Screening of Nucleosides and Nucleoside Analogs
The unique activation mechanism of SAMDH1, which involves binding of activating nucleotides to two proximal binding pockets on the enzyme surface, makes it an excellent target for fragment tethering-based strategies. In this regard, nucleoside fragments have proved to be useful starting fragments for the development of inhibitors of enzymes involved in nucleotide metabolism. One example was our finding that a uracil substrate fragment could serve as a starting scaffold for development of a potent inhibitor of human dUTP hydrolase (dUTPase) by fragment tethering. 30 Medicinal chemistry efforts by Taiho Pharmaceuticals have built on the uracil fragment to yield a potent inhibitor of dUTPase that is in clinical trials.
To identify a potential inhibitory nucleoside fragment that targets SAMHD1, dose-response curves were obtained for the canonical dNs: deoxythymidine (dThy), deoxycytosine (dCyt), deoxyuridine (dUrd), deoxyadenosine (dAdo), and deoxyguanosine (dGuo; Fig 4A ). Of these, only dGuo showed significant inhibition (IC50 = 488 ± 50 µM, h = 1.0 ± 0.1), probably because of its binding in the guanine nucleoside-specific A1 activator site.2,4 The structural requirements for binding were further explored using several analogs of deoxyguanosine ( Fig. 4B , C ). Several important structure-activity relationships were obtained from these results: (1) the exocyclic amine of guanine is important for binding because dIno, which lacks this amine, is significantly less inhibitory; (2) the conformation and/or steric features of the ribose ring are important because the rGuo and ddGuo analogs show significantly weaker inhibition; and (3) a sugar ring is not critical for inhibition because acGuo (acycloguanosine or acyclovir) inhibits with potency similar to dGuo itself (IC50 = 764 ± 82 µM, h = 1.0 ± 0.1).
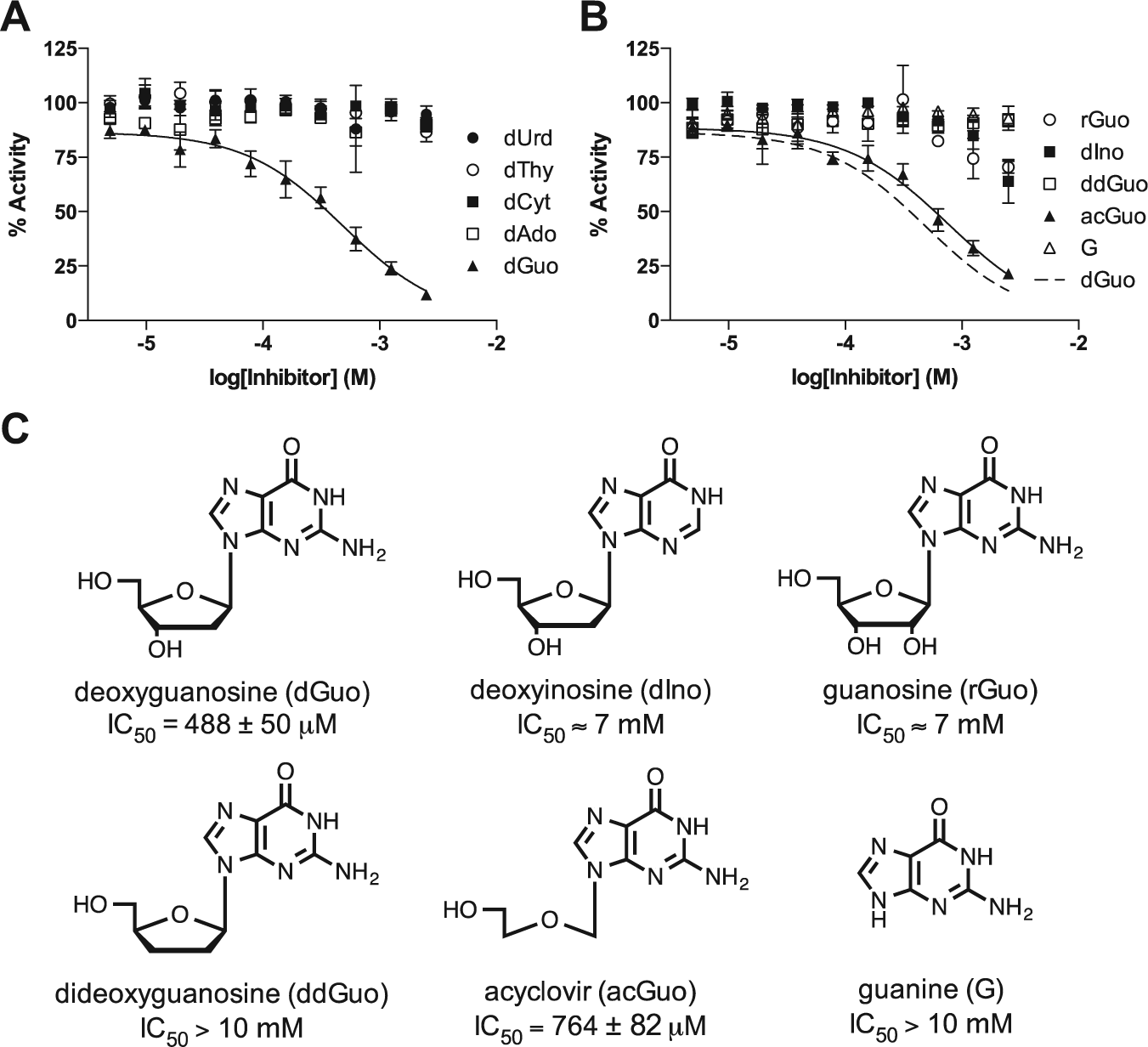
Screening of a targeted library of nucleosides and analogs. (
The identification of acyclovir as an inhibitor is potentially useful for fragment tethering studies because of its small size (MW = 225 g/mol) and the absence of the ribose ring that is prone to ring-opening and anomerization reactions. In addition, this simple nucleoside fragment has a limited number of nucleophilic moieties that would need to be protected for further chemical modification of this scaffold. It is also of note that the triphosphate form of acyclovir is a clinically important antiviral drug, suggesting that other nucleoside prodrugs or their phosphate forms might have off-target activity against SAMHD1. In addition, it is possible that the promiscuous dNTPase activity of SAMHD1 could also degrade the active triphosphate forms of some antiviral and anticancer nucleoside drugs, which has not been thoroughly explored. 19
In summary, we have validated a robust high-throughput assay for screening for inhibitors of SAMHD1. The assay has a large signal-to-background and minimal well-to-well variability and is appropriate for efficient and economical screening of large compound libraries. In addition, the screen is also applicable for rapid screening of targeted libraries, which has already led to the identification of acyclovir as a potential lead compound for fragment-based drug design. This assay should facilitate the discovery and development of cell-permeable inhibitors of SAMHD1 that will be useful for basic science research in the immunology, virology, and cancer fields and may also lead to new therapeutics.
Footnotes
Acknowledgements
The authors would like to thank Dr. Jun O. Liu and Ruojing Li for supplying the Hopkins Drug Library and Erik C. Hansen for helpful discussions about this work.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (NIH) grant T32 GM008763 and NIH grant RO1 GM056834 (J.T.S) and grant 108834-55-RGRL from the Foundation for AIDS Research (J.T.S). K.J.S is the recipient of an American Heart Association Predoctoral Fellowship 14PRE20380664.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.