
Abstract
Therapeutic antibodies have become an established class of drugs for the treatment of a variety of diseases, especially cancer and autoimmune/inflammatory disorders, and a sufficient patent protection is a prerequisite for their successful commercialization. As monoclonal antibodies and their therapeutic potential have been well known for decades, the mere production of yet another therapeutic antibody is in many jurisdictions not considered a patentable invention. In contrast, antibodies with novel structural features and/or improved properties may be patentable. When drafting the claims, care should be taken to obtain a broad patent scope that protects both the antibody of interest and related antibodies having the same functional features, thereby preventing competitors from marketing a functionally equivalent antibody. Furthermore, the application should contain experimental evidence showing the improved properties of the claimed antibody. After the filing of a priority patent application, patent protection should be initiated at least in countries that are of particular commercial importance. Subsequent inventions relating to novel uses, formulations, dosage regimens, and combinations with other treatment modalities should be protected by further patent applications to extend patent term.
Introduction
The interaction with the patent world is often difficult and time-consuming for researchers. Because applications have to be filed before any publication of the scientific results to avoid the danger that the own publication is detrimental for the fate of the patent application, researchers sometimes feel the strong pressure not to communicate with their colleagues on recent scientific results (e.g., on scientific congresses). Furthermore, when a publication is written, often the corresponding patent application has to be prepared simultaneously, with the consequence that the interaction with the patent attorneys may represent a challenge for the researchers.
On the other side, under the current financial situation where funding from third parties is essential for continuation of scientific projects also in the antibody field, a strong patent protection is also essential for the researchers themselves. In fact, monoclonal antibodies have become a mainstream therapeutic modality and represent one of the fastest growing segments in the pharmaceutical industry. Presently, some of the most valuable drugs on the market are antibodies (see, e.g., Rituxan from Roche [Basel, Switzerland], with global sales of $7.9 billion), which shows that therapeutic antibodies have blockbuster potential. Since the first generation of mouse, chimeric, and humanized antibodies received regulatory approval in the late 1990s, so-called next-generation antibodies with structural and functional modifications have been developed. These modifications are aimed at increased efficacy and pharmaceutical properties but also allow the generation of novel intellectual property and a better life cycle management.1,2
It is therefore imperative that researchers and patent attorneys come to an easy and open communication with the goal of both achieving a good publication and a well-written patent application.
Once this patent application is written, it may be filed as a priority application in one country worldwide—for example, as a US provisional application (which is not examined and expires after 1 year) or as a European application. After 1 year at the latest (the so-called priority year), further filings may be performed claiming the priority of the first application. Usually, at this stage, an international (Patent Cooperation Treaty [PCT]) application is filed. After further 18 or 19 months at the latest, this PCT application has then to be converted into national or regional stage applications in those countries or regions where patent protection is required. In many cases, national or regional phases are entered at least in the United States, Canada, Europe (before the European Patent Office), China, Japan, and South Korea. In all elected countries or regions, the national patent offices then examine the application under their rules and come to a decision on the patentability of the claimed invention.
After grant of a patent, it is always possible to challenge this patent in revocation proceedings. Consequently, having obtained broad patent protection is not a guarantee that these claims will survive litigation.
Obtaining and defending patents is a process with different interests of several parties. On one hand, there is the interest of the innovator (inventors and investors) to obtain broad patent protection. On the other hand, there is the interest of the competing parties to prevent broad claims in order to be free in their commercial developments. Third, there is the interest of the public in scientific progress but also to have drugs available at affordable prices. For example, payors in the health care system welcome the arrival of generic drugs on the market. It is the duty of public policy makers to balance these competing interests. The aim of this article is to provide guidance to scientists and inventors on how to obtain broad patent protection for their inventions in the antibody field. As it will be explained below in more detail, it is the policy of the patent offices worldwide to defend the rights of the inventors, their competitors, and the public, which results in the tendency to only grant patents for what the inventors have actually invented and to avoid too broad patent protection.
The Specific Situation of Antibody Patenting
Patenting of antibodies differs from the patenting of other biological substances in some aspects. Before the completion of the Human Genome Project and comparable large-scale sequencing projects of other species, when it was still difficult to identify new (human) proteins, patent protection for antibodies directed to these newly identified proteins was an easy goal to achieve. Usually, the protein itself was then covered by the main claim, and only in subsequent claims, the antibody binding to the protein was claimed. Of note, the discovery of a new protein allowed obtaining claims for antibodies binding to this protein without actually having disclosed any specific antibodies in the patent application.
However, as of today, in view of the fact that sequencing of genomes is usually considered straightforward, the patent offices around the world in many cases consider the identification of a new protein as obvious and not patentable. Furthermore, and even more important, most of the therapeutically interesting proteins are now known. Only in exceptional cases where the identification of the protein has faced considerable difficulties may it still be possible to obtain patent protection for a naturally occurring protein (or at least for a specific epitope). In the United States, it was easier to obtain patent protection for proteins defined by their structure. However, there the situation has become even more complicated in view of recent Supreme Court decisions (e.g., the Myriad or the Prometheus decision), putting the whole concept of the patentability of naturally occurring substances in question.
Consequently, researchers developing an antibody are often faced with the situation that the target of the antibody is either already known or at least not considered inventive by the patent offices, resulting in the fact that the target itself cannot confer patentability to the antibody. Furthermore, patent examiners often argue that since the days of Köhler and Milstein, the production of an antibody against a known protein has to be considered a mere routine process, which means that an antibody per se without having specific, superior properties in comparison to other antibodies is normally not considered inventive.
This, however, does not mean that claims directed to antibodies are not allowable anymore. In decision T 645/01, issued in 2003, the competent Technical Boards of Appeal of the European Patent Office decided that a claim directed to a specific antibody binding a known target may be inventive if this antibody exhibits specific properties that already existing antibodies do not have. Such specific properties may include a higher affinity or specificity, the binding to another epitope, a higher or new pharmacological effectivity, or fewer side effects compared with the other, already existing antibodies.
This view has subsequently been confirmed in various decisions of the Boards of Appeal of the European Patent Office and is shared by the most important patent offices worldwide, including the US Patent and Trademark Office. Consequently, even today, it should not be problematic to obtain patent protection for a specific antibody with improved or different properties in comparison to already known antibodies, provided that it was somehow surprising that such an antibody could be obtained or that the production of the antibody was not based on a mere routine process.
Since the new and surprising properties are considered linked to the specific sequence of the antibody, often the patent protection obtained is restricted to the specific antibody identified during the research project. The antibody is then characterized structurally both in the claims and in the patent specification either by specifically naming and claiming the sequence of the antibody or at least the sequence of the six complementarity determining regions (CDRs) of the antibody. Alternatively, it is also possible to deposit the hybridoma producing the antibody at specific depositary institutions, such as the American Type Culture Collection (ATCC), and directing the patent claims to an antibody produced by said hybridoma. The latter option has been used less frequently in the past years because it is today common knowledge how to produce a given antibody once its sequence or the sequences of its CDRs are known.
However, the scope of protection conferred by such patent claims is limited since it is at least today technologically feasible, by using modern antibody libraries, to produce an equivalent antibody that has the same binding capacity as the given antibody but a completely different amino acid sequence. Furthermore, if the claim covers only the specific antibody or its variable regions and not an antibody with the specific CDRs, even other antibody formats containing the identical CDRs would not fall under this claim. Consequently, the main objective of such a claim is to prevent generic companies to come to the market with a biosimilar having the same amino acid sequence as the original antibody, which may still be commercially very valuable.
How to Obtain Broad Patent Protection for Antibodies
A possible way out of this dilemma is to try to obtain a claim wording wherein the claimed antibody is defined by its functional properties. Such functional parameters include the affinity, specificity, or therapeutic activity of the antibody. Furthermore, it may be possible to direct the claims to antibodies recognizing the same epitope as the identified antibody. Such claims protect not only the specific antibody identified during the research project but also other antibodies with another sequence and provide, therefore, a much broader patent protection.
Claims directed to antibodies with specific biological properties without a reference to the antibody sequence in the claims have been granted (e.g., to AbbVie [formally Abbott, Chicago, IL] both in the United States and in Europe in a patent family involving US 6,914,128 and its European counterpart EP 2168984 B1). In essence, the claims were (and are) directed to an anti–interleukin-12 antibody with a given koff and a given IC50. In a prominent US litigation, the issued claims have been considered invalid due to a lack of written description, meaning essentially that the claims were considered too broad. In the opposition proceedings against the European patent, the patent was very recently revoked due to lack of enablement (i.e., the opposition division came to the conclusion that the claims are not enabled over their breadth). Therefore, although the legal basis and practice are different in the United States and Europe, the outcome of the cases was the same—namely, a revocation of the patent, which shows that for the patenting of antibodies, comparable results (albeit on a different legal basis) may be expected in the United States and in Europe.
Sometimes, applicants also combine structural and functional features in the claims. For example, in some cases, claims have been granted that contain, in addition to CDR sequences, functional properties of a given antibody. Such a claim would cover not only the specific antibody but also synthetic antibodies where the CDRs have been grafted into another antibody format, like single-chain antibodies, provided that said antibody derivatives have said functional properties. Such claims are especially important in US cases, where structural features in the claims are considered even more important by the examiners than in Europe.
An Illustrative Example
As an example, let’s assume that a research team has found that protein X is specific for neuroblastoma cells. Furthermore, an antibody has been identified that recognizes protein X on the surface of neuroblastoma cells and inhibits the growth of said cells but not of other control cells, with an IC50 in the range of 10−9 M. These properties provide the antibody with an interesting therapeutic potential. The antibody has been sequenced, and its CDRs are known. Furthermore, such a high-potency antibody against protein X has never been identified so far. In such a case, the following independent patent claims may be available:
1. Antibody, comprising the following CDR sequences: . . . .
In this claim, the sequences should be identified either by the sequence itself or by reference to a sequence listing.
2. Antibody, recognizing the same epitope as the antibody of claim 1
3. Antibody, recognizing the same epitope as the antibody of claim 1, wherein the epitope has the following sequence: . . . .
4. Antibody against protein X, being capable of inhibiting the growth of neuroblastoma cells with an IC50 lower than 10−9 M
Claim 1 would be the easiest to obtain. Claim 2 may be more complicated and would require as much guidance as possible in the specification as to the properties (and possibly sequence) of the epitope and how to produce any antibody recognizing this specific epitope. Often, it may be necessary to include the sequence of the epitope into the claim, as has been done in claim 3. Claims corresponding to claim 3 have been considered patentable also in the United States (see US 13/499,429). Generally, for obtaining such a claim, it may be advisable to have several examples of such antibodies in the application to meet the requirement that the application is enabled over its whole scope (an European requirement) or that it shows a representative number of species falling under the claim (as required in the United States).
Obtaining (and defending) claim 4 may be a real challenge as explained above with respect to the AbbVie cases, given the fact that protein X is already known and the production of high-affinity antibodies is generally known in the art, because usually one would assume that each high-affinity, anti–X antibody has such an IC50. However, there may be specific circumstances where this is not the case. To obtain such a claim, it will then be important to show that this activity is not restricted to the sequence of the identified antibody, which again may be challenging.
Even in cases where broad functional claims are obtained, these may be challenged and potentially rendered invalid in later revocation or infringement proceedings. In view of this, it is advisable to not only rely on the broad claims but also always have a claim directed to the sequence of the antibody, either in a separate (divisional or continuation) application or as an independent claim in the same patent.
Fortunately, in our example, the inventors have made another invention—namely, that neuroblastoma can be (specifically) treated with the help of anti–X antibodies. This enables the following further claim (European style):
5. Anti–X antibody for use in a method for the treatment of neuroblastoma
or (US style)
5. A method of treating neuroblastoma, comprising administering to a patient in need thereof an anti–X antibody in an amount sufficient to treat neuroblastoma
Most likely, this claim 5 is patentable, if the application contains data to show that every anti–X antibody has this activity. This claim covers the use of any antibody for said specific use, including antibodies that will only be developed in the future. Consequently, if the treatment of neuroblastoma is a commercially important option for anti–X antibodies or even the most important one, such a claim would be commercially valuable. Depending on how easy it is to obtain other anti–X antibodies, this use claim may become more commercially important than the (limited) antibody claim itself because it would allow to block fast-follower products produced by competitors or imitators.
If the invention does not reside in the antibody itself but in a specific antibody–drug conjugate, the situation is different. Here, the patentability-conferring feature would be the conjugate, and depending on the circumstances, there may be no need to specifically define the antibody in the claims. Of course, then the claims have to be directed to the antibody-conjugate and not to the antibody alone.
Timing of Filing of the First Application
In view of the discussions above, it is important to focus, right from the beginning of any antibody research project, on the essential parameters that could confer superiority over the antibodies known in the art and, at the same time, could confer patentability to claims being broader than claims directed to a specific antibody itself. The timing of the filing of the application will then depend on two considerations:
In nearly all countries, there is the so-called first-to-file-doctrine. This doctrine means that the applicant who files the application first is awarded a patent. Consequently, in highly disputed projects where many companies and research teams try to identify a therapeutically valuable antibody, the filing of the first patent application should not be delayed.
On the other side, for being patentable, it is necessary, at least for European patents, that all features and properties of the antibody that should form the basis for arguing patentability should be found in the specification as filed. For example, if it is intended to argue patentability on the basis of improved antitumor properties of a given antibody, care should be taken that these improved antitumor properties are indeed described in the patent specification not only theoretically but with the help of scientific results. In essence, it is necessary that the specification makes the intended medical use plausible (see T 1329/04). Here, the situation is different in the United States, where it is much easier to file new experimental data during examination proceedings.
Furthermore, a patent specification must enable the claimed subject matter over the whole scope of the claim—that is, it is required that the skilled person is capable of producing (nearly) all antibodies falling under the claim. At the European Patent Office, this practice, for example, is described in the decision T 791/00 and has been further discussed in the decision T 601/05. Although based on different other legal considerations, the same is to be expected in all jurisdictions worldwide. To meet these requirements, as many examples as possible should be included in the specification, and it should be explained as detailed as possible how to obtain all antibodies falling under the claim. These examples should contain the scientific results showing the superior effects of the claimed antibody (e.g., the higher affinity or specificity, the binding to another epitope, the higher or new pharmacological effectivity, or fewer side effects compared with other, already existing antibodies). In this context, it will be important to include also comparative data for the known existing antibodies. Of course, it takes time to generate such results, and the time necessary to perform these experiments may very well be a reason for delaying the filing of a patent application. Normally, a useful guidance is the perception of the scientist making the experiments. If she or he believes that the results are credible, then an examiner also should come to the same conclusion. In this context, it is important to note that clinical data are usually not required for conferring patentability. In the case law, it is acknowledged that patent applications have to be filed at the beginning of the life cycle of a medical product when clinical studies have not yet been performed, and depending on the individual subject matter, in vitro data or animal studies are in most cases considered sufficient.
In view of this, it is required to have, from the start of the project, the patent strategy in mind and to thoroughly plan the experiments. Depending on the available resources as well as on the scientific strategy of the project, it may be advisable to file a first patent application as soon as possible and then, as explained below, to follow up within the priority year with further patent applications containing further scientific information.
In essence, it does not make any difference where the first patent application is filed. Many applicants decide to file their first application either in the United States (because it is cheaper) or with the European Patent Office, in the latter case to obtain a search report from the patent office within 6 to 9 months. This search report provides information on the patentability of the claimed product, the antibody, and, therefore, helps in making further informed decisions on the patent strategy.
Subsequent Filing Strategy for the First Invention
After the first filing, the applicant has 1 year, the so-called priority year, to make decisions where to internationally file the application. Furthermore, he or she has the option to further work on the invention and to further improve it (e.g., by generating additional supporting data like animal studies). For example, the antibody may be further characterized, and further surprising properties may be identified. These improvements may then be included into the subsequent applications filed after 1 year, as explained below. Consequently, the year after the filing of the patent application may be especially important for the final shape and success of the application.
At the end of the priority year, it is possible to file directly a subsequent patent application (containing optionally the improvements as explained above) claiming priority of the first application in all countries worldwide where it is commercially advisable to file patent applications. Alternatively, it is also possible, and often preferred, to file an international patent application prior to the expiration of these 12 months. This international patent application costs usually about 5 to 10 thousand Euros or US dollars and has the purpose to delay the decision where to file the subsequent patent applications. In essence, an international patent application is nothing more than a place holder; the international patent application stays alive for 18 or 19 months, and after these 18 or 19 months (depending on the countries where the PCT application should be continued), it is necessary to enter the regional or national stages of this application (i.e., to file the application in the individual countries where there is a commercial interest in the invention). Consequently, by payment of the above-mentioned amount of money, it is possible to delay the decision where to file for about 18 months. This allows finding investors for supporting the entry of the national or regional stages of the international application because, and depending on the content and especially the length of the application, costs in the range of 7000 to 20,000 Euros may arise for each country where the individual application is entered.
A further advantage of the filing of a PCT application is that this application is examined by a patent office, and a search report together with a written opinion setting out the examiner’s view on patentability is provided, which again may facilitate the decision whether and where to enter the national or regional stages.
After the entry of the individual regional or national stages, the international patent application is handled as a national or regional patent application and examined under the national and regional standards. For example, in the regional phase before the European Patent Office, the examiner will issue two or three examination reports. In response to these reports, the applicant has the opportunity to amend the claims and/or the specification. Especially, the applicant may include further features into the claim to distinguish the claimed invention from the prior art and/or to characterize in more detail the invention, which may be required depending on the national law. These amendments have to be based on the text of the application, because the applicant is not allowed to improve his or her invention based on knowledge obtained only after filing the application. At the end, hopefully an agreement is reached with the examiner, with the consequence that a patent is issued.
Once a patent is granted in a given country, the patent is normally enforceable with the consequence that any product brought to the market by a third party falling under the claims can be taken off the market with the help of a lawsuit initiated by the patentee. The costs for such lawsuits vary considerably depending either on the country where they are initiated or on the value of the protected product.
Filing of Subsequent Applications for Further Inventions
Apart from the very important decision when to file the first application, there is another reason why right from the beginning, a comprehensive patent strategy is required. The patent term (i.e., the lifetime of a patent) is 20 years after the filing date and 21 years after the priority date. Up to 5.5 years of additional protection are available in Europe by applying for a Supplementary Protection Certificate (SPC) if a market authorization is obtained for a product falling under the claims of a granted patent. Similar provisions for patent term extensions exist in the United States and other countries. The requirements and scope of these extensions differs from country to country, and in many cases, the additional scope of protection is smaller than that of the original patent and may be restricted to the marketed product. Usually, it takes many years until a market authorization is obtained, with the consequence that there are only a few remaining years of patent protection to recoup the investment and make a profit.
Therefore, after the filing of a first patent application, further more specific patent applications should be envisaged and filed, which also cover the intended commercial product. For example, the subsequent application could specify in more detail how the given product, e.g. the antibody, is produced and optimized. Furthermore, also combination therapies or specific formulations or modes of administration could be claimed (of course only if inventive) to obtain longer patent protection. For antibody inventions, the development of an antibody with an (even) higher affinity or specificity may also represent a subsequent invention that could form the basis of a further application.
In this context, one specific, time-sensitive issue has to be considered. Any patent application will be published after 18 months. Any further application filed before said publication (e.g., within 18 months after the first filing) needs only to be novel but not inventive over the first application. Consequently, in this subsequent application, novel but not noninventive features conferring patentability can be added to claims. Such features depend on the individual nature of the antibody and may include another pharmaceutical use of the antibody, a specific formulation, or a more specified dosage regimen. This subsequent application will have a longer lifetime than the first application, and its subject matter is likely to be patentable if the first application is patentable and, of course, nothing has been published in the meantime that is detrimental to patentability of the added features. Consequently, even if such an improvement is made within the above discussed priority year, it may be worthwhile not to include it in the subsequent application (usually a PCT application) filed after 1 year but to initiate a new patent family to extend the time of patent protection.
Gazyva Case Study
Gazyva (obinutuzumab), Roche’s successor to Rituxan (rituximab), represents the first glycoengineered and Fc-engineered monoclonal antibody and was approved by the Food and Drug Administration (FDA) in November 2013 under the breakthrough therapy designation for the treatment of patients with chronic lymphocytic leukemia (CLL) who have not received treatment.3,4 Gazyva is a fully humanized monoclonal antibody directed against CD20 on B cells. However, Gazyva recognizes a different epitope on CD20 compared with Rituxan. Epitope characterization and crystal structures of antibody-antigen complexes revealed that the extracellular loops harbor two groups of epitopes, one recognized by the majority of anti-CD20 monoclonal antibodies (type I mAbs, including rituximab), which induce movement of antibody-CD20 complexes into lipid rafts. Another group of monoclonal antibodies (type II mAbs, including obinutuzumab) does not direct CD20 complexes into membrane rafts. 5 This property contributes different functional features to Gazyva with a non-caspase-dependent cell death and the lack of complement-dependent cytotoxicity. Gazyva demonstrated clinical superiority compared with Rituxan in a recent phase III study. 4
Gazyva Patent Strategy
The patent strategy for this product represents an illustrative example about how patent protection for a commercially important antibody can be obtained. As shown in Table 1 , a sequence of patent applications was filed to cover Gazyva. The first application, a US provisional application, already had been filed in 2003 and, therefore, 10 years before launching the product. After 1 year, an international application (PCT application) was filed, which was then published in 2005 as WO2005/044859. After 30 or 31 months, respectively, national or regional stage applications were filed in several countries, including, for example, Europe, the United States, Canada, Australia, China, and Japan. The corresponding European patent (EP1692182B1) was granted in April 2010, and no opposition was filed against this patent. Consequently, at least in Europe, a patent covering the product was obtained before the product was brought to the market.
Patent Applications Covering Obinutuzumab (Gazyva, GA101).
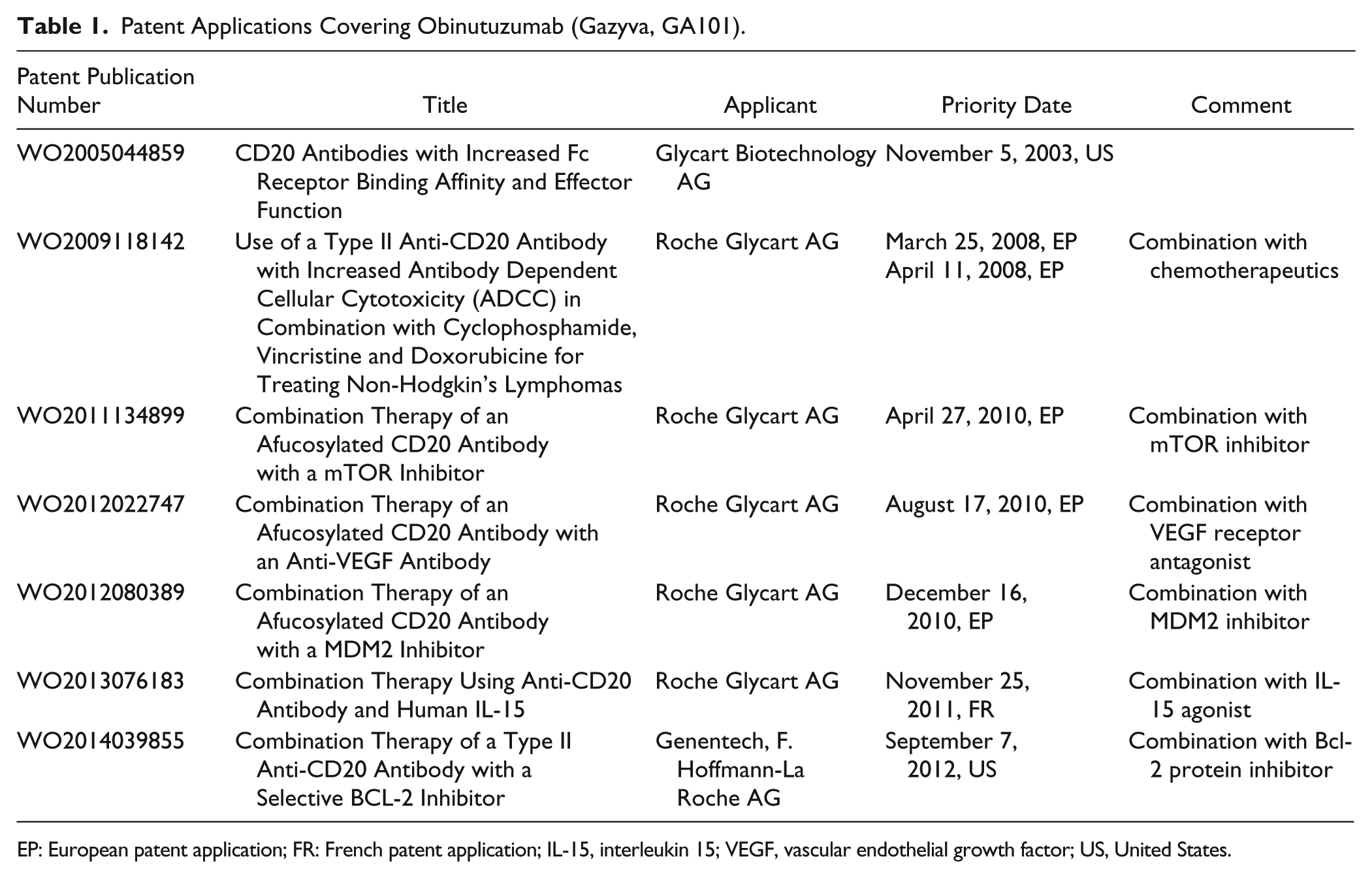
EP: European patent application; FR: French patent application; IL-15, interleukin 15; VEGF, vascular endothelial growth factor; US, United States.
It is interesting to note how the claim language has been revised during European prosecution. Claim 1 of the PCT application was directed to nucleotide sequences encoding the CDRs of the antibody. This claim contained as features only said sequences per se: An isolated polynucleotide comprising a sequence selected from the group consisting of SEQ ID NO. 5, SEQ ID NO. 6, and SEQ ID NO. 7; and a sequence selected from the group consisting of SEQ ID NO. 21, SEQ ID NO. 22, and SEQ ID NO. 23; and sequence ID NO. 24.
Consequently, this claim contained only a few features specifying the variable regions of the antibody without specifying any functional properties. Furthermore, it was not directed to the antibody itself but to nucleotide sequences encoding it. During European prosecution, the examiner inter alia raised the objection that not all essential technical information was contained in the claims and requested the introduction of the framework sequences. Finally, the main claim was amended to read as follows: A humanized, glycoengineered Type II anti-CD20 antibody having increased ADCC as a result of said glycoengineering and having increased ability to induce apoptosis of target cells following said humanizing, wherein said antibody comprises a heavy chain variable region comprising complementarity determining regions (CDRs) of the murine B-Ly1 antibody, wherein: the heavy chain CDR1 is SEQ ID NO. 16; the heavy chain CDR2 is SEQ ID NO. 26; and the heavy chain CDR3 is SEQ ID NO. 28; wherein the heavy chain variable region framework regions (FRs) FR1, FR2, and FR3 of said antibody are human FR sequences encoded by the VH1_10 human germ-line sequence and the heavy chain variable region FR4 of said antibody is a human FR sequence encoded by the JH4 human germ-line sequence, and wherein said antibody further comprises a light chain variable region comprising CDRs of the murine B-Ly1 antibody, wherein: d. the light chain CDR1 is SEQ ID NO. 18; e. the light chain CDR2 is SEQ ID NO. 19; and f. the light chain CDR3 is SEQ ID NO. 20, wherein the light chain variable region FRs FR1, FR2, and FR3 of said antibody are human FR sequences encoded by the VK_2_40 human germ-line sequence and the light chain variable region FR4 of said antibody is a human FR sequence encoded by the JK4 human germ-line sequence.
Consequently, now the claim contains the CDRs of the antibody, a reference to the specific framework sequences as well as several functional features. There was a lengthy discussion between the applicant and the European Patent Office, which included several office actions and amendments of the claims. At the end, the applicant could convince the examiner that with these numerous amendments, the claims are patentable. In particular, the introduction of the antibody sequences resulted in the fact that not only the above formal objection of lack of essential features was overcome but also novelty and inventive step were acknowledged. As to inventive step, the examiner came to the conclusion that the claimed antibody shows an increased ADCC capacity (i.e., an improved property), which was not known in the art.
The patentee so far has filed four divisional applications to cover other, broader subject matter.
Interestingly, the corresponding US application (US 11/889,975) recently issued a claim being directed to the specific sequences of the heavy chain and light chain. Consequently, also the US claims have been directed to the antibody sequence but are still narrower than the European claims. It remains to be seen whether further continuations will be filed.
Parallel patents have also been granted in Japan and Australia, while the Chinese patent application is still pending (according to the ESPACENET database).
However, this application was not the only one that was filed to cover Gazyva. In addition, as can be seen from Table 1 , in 2008, a further (in this case European) application was filed claiming a combination therapy of anti-CD20 antibodies with various chemotherapeutic agents. Also in this case, a follow-up PCT application (WO2009/118142) was filed, and this application inter alia entered the regional stage before the European Patent Office. At least in Europe, this application has not yet been granted. In 2010 and 2011, further patent applications were filed covering further combinations therapies, extending a possible patent protection until 2031 or even later.
Further information on these patent families can be obtained from the ESPACENET database (www.espacenet.com).
In conclusion, therapeutic antibodies are of high potential commercial value. However, it is often difficult to obtain a broad patent protection for a given antibody that also covers comparable antibodies from competitors. Therefore, it is required to establish a good patent strategy right from the beginning of the project. This involves a careful characterization of the biochemical, pharmaceutical, and pharmacological properties of the antibody, preferably in comparison with known antibodies.
Footnotes
Acknowledgements
The author thanks Ulrich Kruse for his helpful input when preparing this manuscript.
Disclaimer
The discussion above reflects solely the personal view of the author and does not constitute legal advice or the views of Isenbruck Bösl Hörschler LLP. For any specific legal question, a lawyer or a patent attorney should be consulted, and no liability is given for any of the information provided in this article.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.