
Abstract
β-Arrestin, a signal adaptor protein, mediates intracellular signal transductions through protein-protein interactions by bringing two or more proteins in proximity. Extracellular signal-regulated kinase (ERK), a protein kinase in the family of mitogen-activated protein kinases (MAPKs), is involved in various receptor signal pathways. Interaction of ERK with β-arrestin or formation of ERK/β-arrestin signal complex occurs in response to activation of a variety of cell surface receptors. The ERK/β-arrestin signal complex may be a common transducer to converge a variety of extracellular stimuli to similar downstream intracellular signaling pathways. By using a cell-based protein-protein interaction LinkLight assay technology, we demonstrate a direct interaction between ERK and β-arrestin in response to extracellular stimuli, which can be sensitively and quantitatively monitored. Activations of G protein–coupled receptors (GPCRs), receptor tyrosine kinases (RTKs), and cytokine receptors promote formation of the ERK/β-arrestin signal complex. Our data indicate that the ERK/β-arrestin signal complex is a common transducer that participates in a variety of receptor signaling pathways. Furthermore, we demonstrate that receptor antagonists or kinase inhibitors can block the agonist-induced ERK and β-arrestin interaction. Thus, the ERK/β-arrestin interaction assay is useful for screening of new receptor modulators.
Introduction
β-Arrestin is a signal adaptor protein that brings its interacting partners together to form signal complexes. 1 β-Arrestin had been initially considered a G protein signaling terminator as it promotes G protein–coupled receptor (GPCR) desensitization and internalization. Recently, much broader roles of β-arrestin have been recognized. In addition to interacting with GPCRs, β-arrestin has been indicated to participate in signal transductions of various receptors, including receptor tyrosine kinases (RTKs), ion channels, and cytokine receptors. 2 Agonist binding to RTKs, such as epidermal growth factor receptor (EGFR) and fibroblast growth factor receptor (FGFR), leads to activation of β-arrestin–dependent MAPK signaling pathways.3,4 Cytokines, such as tumor necrosis factor α (TNFα) and interleukin (IL)–1β, also activate signaling pathways involving β-arrestins.5,6
ERK, a MAPK kinase family member, is also one of the β-arrestin–mediated signaling components. 7 GPCRs can activate ERK via G protein–dependent pathways and/or a β-arrestin–dependent pathway. G protein–dependent ERK activation results in the translocation of active ERK proteins into the nucleus, whereas the arrestin-dependent ERK activation retains active ERK proteins in the cytosol. The onset of ERK activation and duration of active ERK are also different. G protein–dependent ERK activation is transient and occurs within a few minutes, while β-arrestin–dependent ERK activation is slower in onset and lasts approximately 60 min.8 –10 Activation of GPCRs is traditionally measured by G protein–dependent second messengers, including cAMP, IP3, and intracellular calcium mobilization. G protein–independent signaling pathways for GPCRs have been exploited for drug discovery using β-arrestin–based assays.11,12 Several biased GPCR ligands, which selectively activate one pathway over the others, have been identified using β-arrestin–based assays.13,14 The biased ligands with increased target specificity and reduced off-target effects may have great potential to become next-generation GPCR drugs.
Crosstalk between different types of receptors has been recognized. 15 Shared signal complexes may be the basis of cross-communication between different types of receptors that enable cells to integrate a multitude of signals from the environment. Since both β-arrestins and MAPKs commonly participate in the downstream signaling pathways of different types of receptors, the β-arrestin/ERK signal complex could be one of the signal integration nodes. However, little is known about the regulation of β-arrestin–dependent ERK signaling or the effect of the ERK/β-arrestin interaction on receptor signaling pathways.
Although the association of β-arrestins with various kinases has been described in literature, a robust cell-based assay to assess the ERK/β-arrestin interaction directly is not available. Previously, we have developed a cell-based protein-protein interaction LinkLight assay for assessing GPCR and β-arrestin interaction. 12 Here we have applied the LinkLight technology to determine ERK and β-arrestin interaction. We have found that the technology is capable of capturing transient, phosphorylation-regulated protein-protein interactions and generating stable signals without involving reporter gene transcription and translation. The ability to assess transient protein-protein interactions in cells is particularly useful since many protein interactions are regulated by phosphorylation and dephosphorylation in response to environmental stimuli. The interaction of ERK/β-arrestin is mediated by activation of GPCRs, RTKs, and cytokine receptors. Receptor antagonists and kinase inhibitors can block the interaction. Our data demonstrate that the ERK/β-arrestin signal complex is a common mediator in signaling pathways of diverse cell surface receptors.
Materials and Methods
Materials
The compounds and chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Tocris Biosciences (Minneapolis, MN, USA).
Cell Line and Cell Culture
U2OS cells were purchased from American Type Culture Collection (Manassas, VA) and were cultured with McCoy’s 5A medium (cat. 16600-082; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) (cat. 26140-079; Gibco) and 1× Pen/Strep (cat. 15070; Gibco). Cells were cultured in 37 °C incubator with 5% CO2. Cell culture medium was replaced every 3 to 4 days and cells were passed at 90% confluence.
Plasmid Construction and Stable Cell Line Generation
Full-length complementary DNA (cDNA) of human ERK2 without stop codon was subcloned in frame with the TEV protease vector described in our previous publication. 12 The β-arrestin-2–permuted luciferase (β-arr-2-pLuc) expression plasmid was also described in a previous publication. 12 Transfection of U2OS cells was performed with Lipofectamine 2000 according to the manufacturer’s instructions (cat. 11668-027; Life Technologies, Carlsbad, CA). Monoclonal cell lines stably expressing both ERK2-TEV and β-arr-2-pLuc fusion protein were selected with 400 µg/mL G418 (cat. 10131-027; Invitrogen/Life Technology, Carlsbad, CA) and 300 µg/mL Hygromycin B (cat. 10687-010; Life Technologies). Multiple stable clones were selected for evaluation.
Luciferase Assay
The LinkLight U2OS cells stably expressing both ERK2-TEV and β-arr-2-pLuc were seeded into a 384-well white plate (Becton Dickinson, Washington, DC, USA) at 20,000 cells per well with 40 µL regular medium. Cells were cultured overnight. The next day, the culture medium was replaced with 20 µL serum-free Dulbecco’s modified Eagle’s medium (DMEM) followed by addition of 5 µL agonists or stimulants. Agonists were added as serial dilutions with a serum-free DMEM from 10-mM DMSO stocks. For antagonist inhibition assays, the medium was replaced with 15 µL serum-free DMEM. Antagonists or inhibitors were added at 5 µL/well and incubated with the cells for 15 min. Then, the stimulant or agonist was added at 5 µL/well and incubated for 90 min. After the incubation, the plate was equalized to room temperature. An equal volume of Bright-Glo (Promega, Madison, WI) or One-Glo (Promega) or BriteLite (PerkinElmer, Waltham, MA) or NeoLite (Perkin Elmer) luciferase detection reagent was added to cells. Maximum luminescence signals were observed around 5 min after adding luciferase detection reagent (following manufacturer’s instructions). The luminescence signals were determined by using luminescence plate readers (Perkin-Elmer’s EnSpire Alpha and TopCount). Alternatively, after a 90-min compound incubation, the culture medium was replaced with 10 µL of luciferase detection reagent and luminescence signals were recorded.
Measurement of FBS Response
The LinkLight U2OS cells were seeded in a white 384-well plate and cultured overnight with medium containing 10% FBS. The cell culture medium was replaced with fresh media containing FBS concentrations in a serial dilution (final concentration of FBS from 20% to 0.01%). After a 90-min incubation, the luciferase detection reagent was added and luminescence signals were determined.
Measurement of GPCR Agonist Responses
The LinkLight U2OS cells were seeded in 384-well plates. After overnight culture, the culture medium was replaced with a serum-free DMEM and a serial dilution of GPCR ligands was added to the cells. After a 90-min incubation, a luciferase detection reagent was added to the cells and luminescent signals were recorded. The antagonist effects were measured after cells were preincubated with the compounds for 15 min followed by stimulation with 1 µM agonists for 90 min before proceeding to the luciferase assay.
Measurement of RTK Responses
The LinkLight U2OS cells were cultured in a white 384-well plate for overnight. The culture medium was then replaced with a serum-free DMEM. Solutions of epidermal growth factor (EGF) or basic fibroblast growth factor (FGF-b) in a serial dilution were added to cells. After a 90-min incubation with the agonists, the luciferase assay was performed. For antagonist and kinase inhibitor assays, cells were preincubated with antagonists or kinase inhibitors for 15 min followed by agonist stimulation for 90 min and then the luciferase assay.
Determination of Cytokine Receptor Response
The LinkLight U2OS cells were cultured in a white 384-well plate overnight. The culture medium was then replaced with 20 µL serum-free DMEM. A serial dilution of 5 µL TNFα or IL-1β was added to the cells for a 90-min incubation, followed by the luciferase assay. For the measurement of response to kinase inhibitor, the cells were treated with staurosporine for 15 min prior to incubation with 25 ng/mL TNFα or 50 ng/mL IL-1β for 90 min. The luciferase assay was then performed.
Data Analysis
Concentration-response curves were generated by using the Prism software (Graphpad Software, San Diego, CA). All values are expressed as the mean ± SD (n = 3).
Results
Cell-Based Assay to Measure the ERK/β-Arrestin Interaction
Upon activation, ERK is phosphorylated and released from the RAF/MEK/ERK signal complex. 16 The phosphorylated ERK binds to other cellular proteins and translocates to new cellular destinations. Binding of ERK to β-arrestin results in cytosolic retention of the ERK/β-arrestin complex, while its association with nuclear importins results in translocation of ERK into the nucleus. To assess the ERK and β-arrestin interaction, we constructed the ERK-TEV expression plasmid by fusing ERK2 with a TEV protease at its C-terminus. U2OS cells were cotransfected with the ERK-TEV plasmid and the β-arrestin-2–linked permuted luciferase (β-arrestin-pLuc) expression plasmid. Upon the ERK/β-arrestin interaction, the proximity of TEV and permuted luciferase leads to cleavage of the permuted luciferase that contains a TEV cleavage site. The cleaved luciferase fragment spontaneously refolds to form an active luciferase, driven by the self-complementation force. Activity of the reconstituted luciferase can be detected by luciferase detection reagents ( Fig. 1A ).
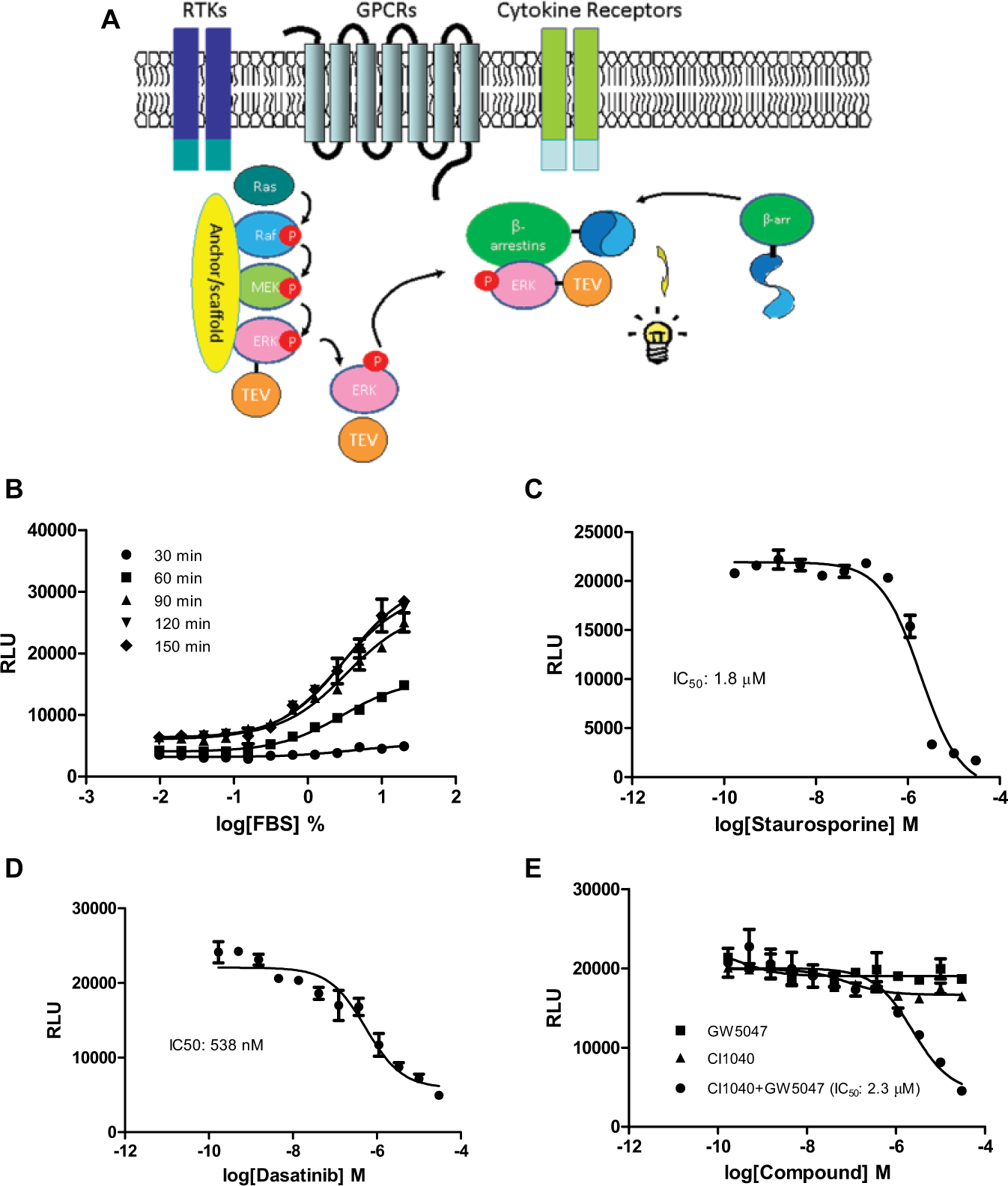
Design of the ERK/β-arrestin interaction LinkLight assay. (
To determine if the ERK/β-arrestin interaction is inducible in the U2OS cell line, we first looked at the response to FBS, which contains a number of growth factors, hormones, and ligands. Many of these factors and hormones bind and activate cell surface receptors. The activated receptors transduce extracellular stimuli through phosphorylation of nonreceptor kinases, such as ERK. We found that treatment of cells coexpressing ERK-TEV and β-arrestin-pLuc with FBS dose-dependently increased luciferase signals (
Fig. 1B
). We also looked at the time course of FBS stimulation. ERK/β-arrestin interaction signals reached a plateau around 120 min. The result indicated an interaction between ERK-TEV and β-arrestin-pLuc or formation of the ERK/β-arrestin signal complex. The data also suggested that in the regular cell culture condition with 10% FBS, ERK could be constitutively phosphorylated or in a dynamic equilibrium between phosphorylation and dephosphorylation states. The U2OS cells transiently expressing only one plasmid (β-arrestin-pLuc) did not show FBS-stimulated signals (
Since cellular signaling pathways involve highly coordinated protein-protein interactions that are regulated by protein phosphorylation and dephosphorylation, we looked at whether the ERK and β-arrestin interaction could be blocked by kinase inhibitors. We found that staurosporine (pan-kinase inhibitor) dose-dependently inhibited ERK/β-arrestin interaction signals ( Fig. 1C ), indicating that the ERK/β-arrestin interaction is mediated by protein phosphorylation. To further investigate which kinase may be involved, we examined the inhibitory effect of some kinase inhibitors. Src kinase upregulates the ERK cascade through direct phosphorylation of RAF and initiates the RAF/MEK/ERK signaling cascade. 17 We found that Src kinase inhibitor dasatinib blocked the FBS-induced ERK/β-arrestin interaction ( Fig. 1D ), suggesting an involvement of Src kinase in ERK activation. However, c-RAF inhibitor GW5074 or MEK kinase inhibitor CI-1040 did not block the ERK/β-arrestin interaction signals ( Fig. 1E ), although RAF and MEK are considered to be immediately upstream of ERK in the RAF/MEK/ERK signaling pathway. Most kinase inhibitors were discovered by cell-free enzymatic assays. However, paradoxical behaviors of these kinase inhibitors in cells were observed.18 –20 To test whether the crosstalk or feedback mechanism is involved in the RAF/MEK/ERK signaling pathway, we treated the cells with a combination of both kinase inhibitors. We found that the combination of GW5074 and CI1040 inhibited the ERK/β-arrestin interaction signals in a dose-dependent manner ( Fig. 1E ). The data indicated that inhibition of either MEK1 or RAF is necessary but not sufficient alone to disrupt the ERK and β-arrestin interaction. The results also suggest that cell-based functional assays may be more physiologically relevant for the identification and validation of kinase inhibitors.
ERK/β-Arrestin Interaction in Response to Activation of GPCRs
Upon activation of GPCRs, GPCRs signal through one or more G protein–mediated signaling pathways. G protein receptor kinases phosphorylate ligand-occupied GPCRs, and the phosphorylation facilitates recruitment of β-arrestins to receptors and promotes receptor internalization. β-Arrestin has also been implicated in linking GPCR-mediated MAPK signaling pathways.
1
To examine if activation of GPCRs promotes the ERK/β-arrestin interaction, we used some of the endogenously expressed GPCRs in U2OS cells. We found that GPCR agonists, including isoproterenol, dopamine, S1P, and adrenaline, dose-dependently stimulated the ERK/β-arrestin interaction signals with EC50 values of 71.9, 140, 337, and 918 nM, respectively (
Fig. 2A
). We also performed homogeneous time-resolved fluorescence (HTRF) cAMP assays with the ERK/β-arrestin LinkLight cells. The results demonstrated that exogenous expression of ERK2-TEV and β-arrestin-pLuc did not affect endogenous GPCR signaling (
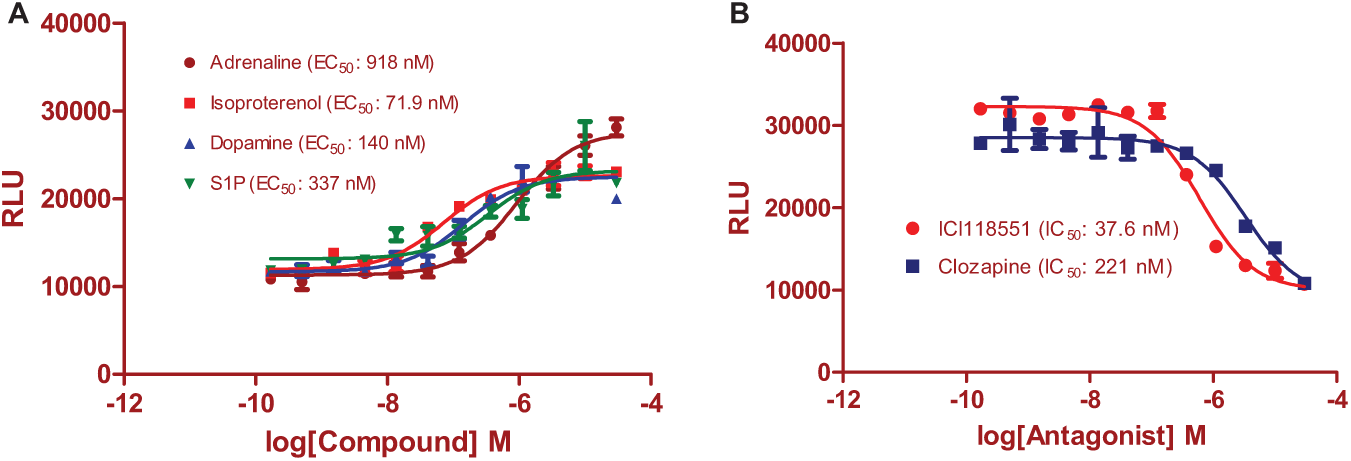
ERK/β-arrestin interaction signals in response to activation of endogenous G protein–coupled receptors (GPCRs) in U2OS cells. (
We then determined whether GPCR antagonists could block the ERK/β-arrestin interaction induced by GPCR agonists. ICI118551 (β-adrenergic receptor-2 antagonist) and clozapine (dopamine receptor antagonist) dose-dependently inhibited the agonist-induced ERK/β-arrestin interaction signals ( Fig. 2B ). The IC50 values for ICI118551 and clozapine were 37.6 and 211 nM, respectively, although they were 3- to 8-fold less potent in comparison to those determined in the classic G protein signaling-based assays.19,21,22
ERK/β-Arrestin Interaction in Response to Activation of RTKs
β-Arrestin has recently been implicated to be part of the downstream MAPK signaling cascades elicited by receptor tyrosine kinases, such as insulin-like growth factor and EGFRs.
2
Binding of ligands to RTKs results in RTK auto-phosphorylation and recruits adaptor proteins to propagate signal transductions, including the MAPK signaling cascade.3,4 To probe the ERK/β-arrestin interaction in response to activations of RTKs, we determined the ERK/β-arrestin interaction signals stimulated by endogenous RTKs expressed in U2OS cells in response to two growth factors, EGF and FGF-b.18,23 We found that both EGF and FGF-b dose-dependently stimulated luciferase signals. The EC50 values for EGF and FGF-b were 3.75 and 3.87 ng/mL, respectively (
Fig. 3A
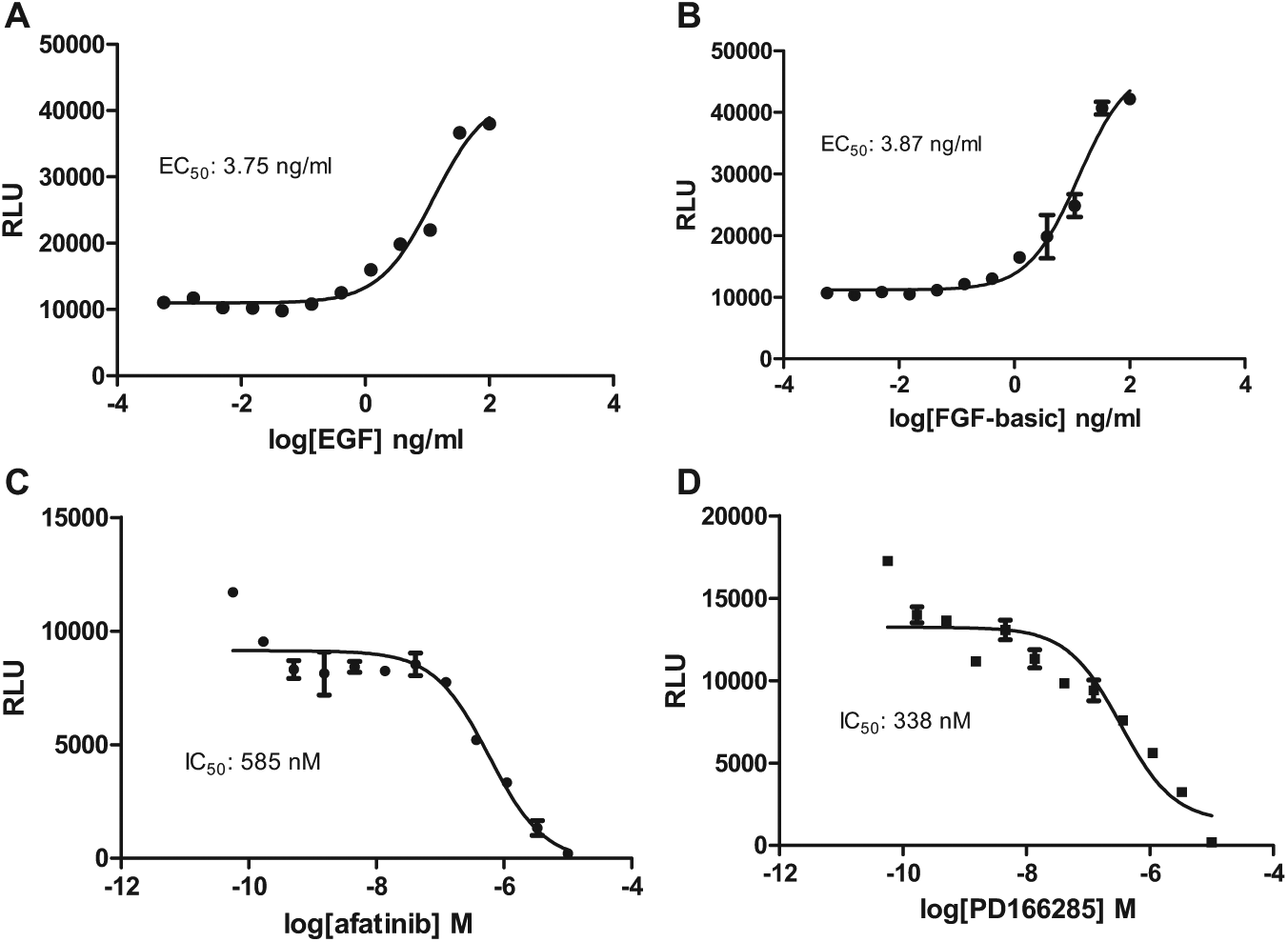
ERK/β-arrestin interaction signals in response to activation of endogenous receptor tyrosine kinases (RTKs) in U2OS cells. (
We then investigated whether specific RTK inhibitors could inhibit the agonist-induced ERK2 and β-arrestin-2 interaction signals. We found that afatinib (EGFR inhibitor) and PD166285 (FGFR inhibitor) blocked EGF and FGF-b induced ERK/β-arrestin interaction signals (
Fig. 3C
ERK/β-Arrestin Interaction in Response to Activation of Cytokine Receptors
TNFα is a master cytokine that mediates inflammatory responses and innate immunity. 20 TNFα has been implicated in disease pathogenesis, including cancer, sepsis, rheumatoid arthritis, diabetes, and inflammatory bowel disease. Major pathways activated by TNFα include caspases, nuclear factor (NF)–κB, and MAP kinases. The early phase of TNFα signaling induces expression of inflammatory cytokines, including TNFα, which results in secondary cytokine expression and secretion. Secondary cytokines contribute to most biological functions of TNFα. Functional involvement of MAP kinases has been indicated in both upstream (TNFα-producing cells) and downstream (TNFα-acting cells) TNFα signaling. 25
To examine the role of the ERK/β-arrestin signal complex in cytokine receptor signaling, we applied TNFα to the U2OS cells that express endogenous TNFα receptor 1 (TNFR1). TNFα induced robust luciferase signals ( Fig. 4A ) with an EC50 of 5.65 ng/mL, indicating the participation of the ERK/β-arrestin signal complex in TNFR1 signaling. We also found that another cytokine, IL-1β, stimulated ERK and β-arrestin interaction ( Fig. 4B ) with an EC50 of 2.8 ng/mL that is compatible to the reported value. 26 U2OS cells are known to respond to IL-1 stimulation.
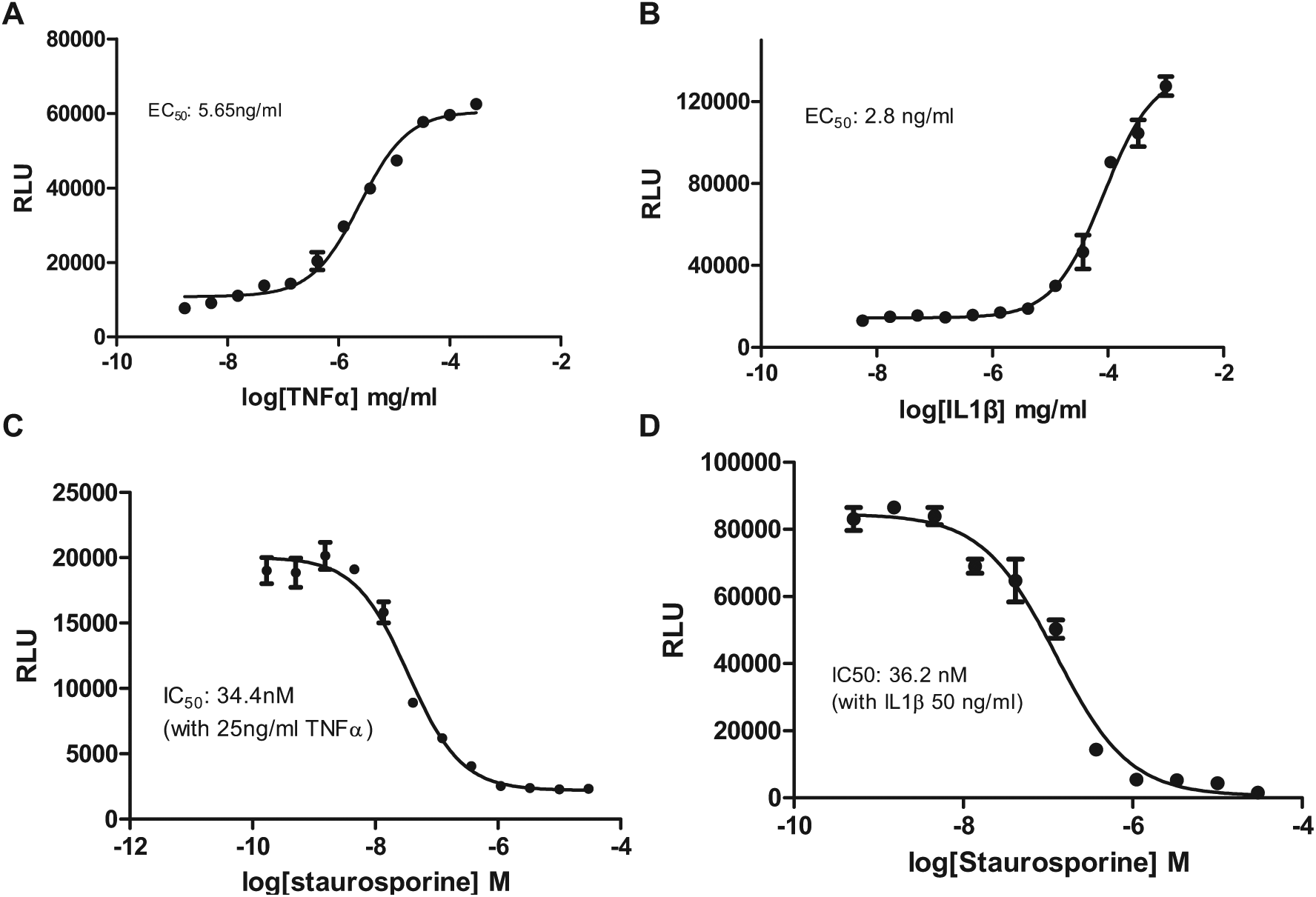
ERK/β-arrestin interaction signals in response to activation of endogenous cytokine receptors in U2OS cells. (
Since the small-molecule TNFR1 and IL1R inhibitors are currently unavailable, we used staurosporine (pan-kinase inhibitor) to block TNFα- and IL-1β–induced ERK/β-arrestin interaction signals. We found that staurosporine dose-dependently inhibited the ERK/β-arrestin interaction signals in response to TNFα or IL-1β stimulation (
Fig. 4C
Discussion
We established a cell-based assay that can quantitatively assess the interaction or formation of ERK/β-arrestin signal complex. This assay has been employed to investigate the relationship of the signal complex in response to activation of diverse cell surface receptors. We demonstrate that binding of a GPCR, cytokine, or RTK agonist to its corresponding cell surface receptor induces the interaction of ERK and β-arrestin or formation of the ERK/β-arrestin signal complex. The interaction is agonist concentration dependent and can be blocked by a specific receptor antagonist or kinase inhibitor. Thus, the ERK/β-arrestin signal complex functions as a convergence point for diverse cell surface receptor signal transduction.
In the past decade, β-arrestins have been found to play much broader roles in cell biology than previously thought. The β-arrestins appear to be important mediators of a broad range of cellular activities such as cell proliferation, differentiation, migration, and apoptosis. Growing evidence indicates that β-arrestins interact with nonreceptor kinases to form signaling complexes for signal propagation and transduction. Activation of diverse cell surface receptors can lead to intracellular kinase activation. One of the major cellular signaling mediators activated by diverse types of cell surface receptors is ERK. The ERK protein mediates multiple signaling pathways, which transduces extracellular signals to intracellular signaling pathways by phosphorylating a large number of protein substrates located in both the cytosol and nucleus.
Many cellular signal transducers are shared by diverse signaling pathways. 27 For example, the signal adaptors are shared by diverse biological inputs. The shared pathways enable cells to integrate signals of diverse external stimuli to the similar cellular responses. These signal adaptors are regulated by highly coordinated phosphorylation-mediated protein-protein interactions. Protein phosphorylation induces protein conformational changes, enables phosphorylated proteins to interact with other proteins, influences protein stability, translocates protein to different subcellular locations, and defines downstream signaling cascades. Accordingly, MAPKs, including ERK, participate in cellular signal transductions elicited by diverse cell surface receptors. 28 The ERK/β-arrestin signal complex could be one of the central cross-communication integrating nodes.
The mechanisms of how β-arrestins participate in the formation of individual MAPK module/β-arrestin complexes may vary among different types of receptors. Thus, GPCRs, RTKs, and cytokine receptors are intimately involved in multilayer forms of crosstalk that influence a variety of cellular signaling processes. We hypothesized that the formation of the ERK/β-arrestin signal complex could be a common signal integration node in signaling pathways of diverse cell surface receptors. By using the ERK/β-arrestin interaction as a functional signal readout, we demonstrated that the ERK/β-arrestin signal complex is formed after activation of endogenous GPCRs, RTKs, and cytokine receptors in U2OS cells. We also demonstrated that GPCR antagonists, specific RTK inhibitors, and kinase inhibitors blocked the ERK/β-arrestin interaction signals induced by the agonists. Thus, our data indicate that different types of receptors, including GPCRs, RTKs, and cytokine receptors, transduce signals at least partially through the ERK/β-arrestin signal complex.
By showing that the ERK/β-arrestin signal complex is a common transducer in multiple receptor signaling pathways, a question of signaling specificity has arisen. β-Arrestin binds to two or more cellular proteins in a signaling cascade and brings them in close proximity. This not only facilitates signal propagation and amplification but also insulates a specific signaling pathway from other signals. Multiple nonreceptor kinases, including PI3K, AKT, JNK, p38, and SRC, have also been implicated to interact with β-arrestins. 29 In addition, extracellular stimuli may have unequal impacts on the formation of various kinase/β-arrestin signal complexes, and their roles in signal transduction need to be investigated further. Signaling specificity can also be achieved by involvement of discrete protein-protein interactions, which may have different responsive time courses. Specific subcellular locations of interacting proteins may also define signaling specificity.
Defining mechanisms for receptor signaling pathways is essential for understanding many aspects of cell biology, cancer biology, and immunology, as well as for effectively targeting signaling pathways for drug discovery. Kinase inhibitors are traditionally discovered by biochemical enzymatic assays, binding assays, and antibody-based protein phosphorylation assays. However, functions of kinases in cells are carried out not only by their intrinsic enzymatic activity but also by cellular protein-protein interaction networks. In cells, kinase and phosphatase activities are dynamically regulated in response to environmental stimuli, which may be very different from those in cell-free enzymatic activity assays. The cellular feedback and crosstalk mechanisms could compromise effects of kinase inhibitors discovered from cell-free biochemical assays.
Our data indicated that the RAF and MEK kinase inhibitor used individually did not block the ERK/β-arrestin interaction. However, the interaction was inhibited by a combination of RAF and MEK inhibitors. This could be explained by the crosstalk and feedback mechanisms in the RAF/MEK/ERK signaling cascade. The result also suggests that the cell-based kinase signaling assay could be a better alternative for the identification of kinase inhibitors. However, the precise mechanism of this phenomenon is still to be identified.
Receptors, including GPCRs, cytokine receptors, and RTKs, account for more than half of confirmed drug targets. Assessing receptor activity by measurements of its functions, such as signaling pathways, is commonly used in the drug discovery process. The traditional RTK kinase assays have used antibody-based or reporter-gene based assays to monitor the signaling pathway indirectly. Instead, our assay directly measures the ERK/β-arrestin interaction, which provides more precise spatiotemporal information. We have demonstrated that the ERK/β-arrestin interaction can be induced by activation of diverse GPCRs, cytokine receptors, and RTKs. Selective small-molecule antagonists modulating the ERK/β-arrestin interaction could be discovered using this cell-based assay through blockade of the agonist response of either an endogenous receptor or an exogenously expressing receptor. Targeting GPCR’s ERK/β-arrestin signaling pathway for drug discovery may identify a new generation of drug candidates that may have better selectivity with fewer adverse effects. RTKs function as key regulators of cell growth, proliferation, and survival. The uncontrolled RTK signaling that leads to cancer has provided the rationale for anti-RTK drug development. Currently, small-molecule RTK antagonists are rarely available. Screening of a small-molecule compound library via an RTK-stimulated ERK/β-arrestin interaction assay may result in the discovery of new functional RTK inhibitors. To identify specificity of hits from screening using this assay, a secondary assay is needed to further demonstrate specificity of hits within the interested signaling pathway.
In addition, the assay time for assessment of the ERK/β-arrestin interaction is relatively short, and the assay signals are independent of reporter gene transcription and translation. The feature is particularly useful for cytokine receptor signaling pathways in which the signal transduction is primarily dependent on the transient protein-protein interactions. Unlike RTKs, cytokine receptors such as TNFα receptors have no intrinsic enzymatic activity. Currently, TNFα receptor activity is measured by secreted secondary cytokines or a reporter gene assay driven by the TNFα response element.
30
However, the biphasic nature of TNFα signaling complicates results obtained from those assays. Our assay directly measures the upstream ERK/β-arrestin interaction that has obvious advantages over those antibody-based and reporter gene–based assays for the measurement of cytokine receptor activity. The cells treated with cytokines produced much more robust signals than did the cells treated with GPCR and RTK agonists. Potentially, other signaling mechanisms may participate. In addition, the signal-to-background (S/B) ratios and Z′ factors of ERK/β-arrestin LinkLight assays were determined for four agonists. The S/B ratio was 3.66-fold and the Z′ factor was 0.69 for 10 µM dopamine, the S/B ratio was 3.78-fold and the Z′ factor was 0.82 for 10 µM adrenaline, the S/B ratio was 8.99-fold and the Z′ factor was 0.78 for 0.1 µg/mL TNFα, and the S/B ratio was 14.5-fold and the Z′ factor was 0.81 for 0.1 µg/mL IL-1β (
Footnotes
Acknowledgements
We thank Dr. Paul S. Wright from Sanofi for suggestions and carefully reading the manuscript and Mr. Paul Shinn from the National Center for Advancing Translational Sciences for preparing compound plates. We also thank Nicolas Pierre at cisbio for providing the HTRF cAMP kit.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: H.E. has the commercial interests of the patented LinkLight technology. The other authors have declared that no competing interests exist.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the NIH/NIMH SBIR grant (R43MH101903) awarded to H.E. and was supported by the Intramural Research Programs of Therapeutics for Rare and Neglected Diseases at the National Center for Advancing Translational Sciences.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.