
Abstract
Metabolomics-based studies are proving of great utility in the analysis of modes of action (MOAs) and resistance mechanisms of drugs in parasitic protozoa. They have helped to determine the MOA of eflornithine, half of the gold standard combination therapy in use against human African trypanosomiasis (HAT), as well as the mechanism of resistance to this drug. In Leishmania, metabolomics has also given insight into the MOA of miltefosine, an alkylphospholipid. Several studies on antimony resistance in Leishmania have been conducted, analyzing the metabolic content of resistant lines, offering clues as to the MOA of this class of drugs. A study of chloroquine resistance in Plasmodium falciparum combined metabolomics techniques with other genetic and proteomic techniques to offer new insight into the role of the PfCRT protein. The MOA and mechanism of resistance to a group of halogenated pyrimidines in Trypanosoma brucei have also recently been elucidated. Effective as metabolomics techniques are, care must be taken in the design and implementation of these experiments, to ensure the resulting data are meaningful. This review outlines the steps required to conduct a metabolomics experiment as well as provide an overview of metabolomics-based drug research in protozoa to date.
Introduction
Metabolomics encompasses the study of small biochemicals within defined biological samples. The study of metabolic changes within a cell population after drug perturbation is promising to be of great use in the analysis of how antimicrobials work. Metabolomics tools have been around for several years and have been fundamental in the discovery of new pathways1–4 and the validation and refinement of known pathways.4–6 Pathway annotation based on genomic reconstructions overlaid onto well-characterized pathways from model organisms offers a simple way to predict an organism’s metabolic potential but inevitably misses organism-specific features. Nutrient requirements, for example, differ in parasitic protozoa compared with their yeast and bacterial counterparts. Parasitic life cycles also complicate pathway annotations, as different life cycle stages may exist in several distinct hosts. The life cycle problem offers a major hurdle for metabolic reconstruction based on genome data alone, as major metabolic changes are evident as parasites move between life cycle stages, requiring different gene sets to be turned on and off.7–9 Transcriptomics goes some way to provide life cycle specificity, but environmental nutrients also shape the metabolome. Our ability to make direct measurements on cellular metabolomes from various organisms is now affecting how we can study metabolism in any cell type, including the parasitic protozoa responsible for diseases such as human African trypanosomiasis, the leishmaniases, and malaria.
Drug Research
Drug research in the area of parasitology is notoriously underfunded, and new compounds are rare. Of the limited set of drugs available for use against parasitic diseases, most are old, have unacceptable levels of resistance, and/or are toxic. 10 Knowledge of how these drugs exert their actions is usually lacking. Phenotypic screens have gained popularity in finding new antiparasitic compounds, making use of the extensive pharmaceutical compound libraries. 11 Much of this work is done through not-for-profit partner organizations such as the Drugs for Neglected Diseases Initiative and the Medicines for Malaria Venture. Compounds active in phenotypic screens hold advantages, including clear membrane permeability and a selectivity index against human cells, that can be easily determined as increasing numbers of compounds have become available for screening against less economically interesting diseases.12–16
A disadvantage of phenotype-based drug screens is that the drug target is not known, so toxic impacts and synergistic effects cannot be predicted. This is where metabolomics analysis of drug action becomes advantageous.
Metabolomics Technologies
Metabolomic-based technologies, which measure the cell’s small-molecule (up to approximately 1400 Da) complement, are increasingly being incorporated into the drug-screening pipeline as the modes of action (MOAs) of new compounds, as well as older compounds, are being sought.
The lab-based experiments in the metabolomics workflow are relatively simple ( Fig. 1 ): cells are grown in culture (animals are rarely used because of unacceptable levels of biological variation), drugs are added, and cell extracts are analyzed using mass spectrometry (MS) or nuclear magnetic resonance (NMR), in which the levels of each metabolite can be recorded. To determine true perturbations to the internal metabolite levels, efficient data deconvolution is required. This is currently more challenging than data collection. New data deconvolution programs are emerging, however, which are facilitating this stage of the analysis. The technologies and software used in metabolomics-based drug discovery will be discussed in this review, as will drug MOAs and mechanisms of resistance found in protozoa using these platforms.
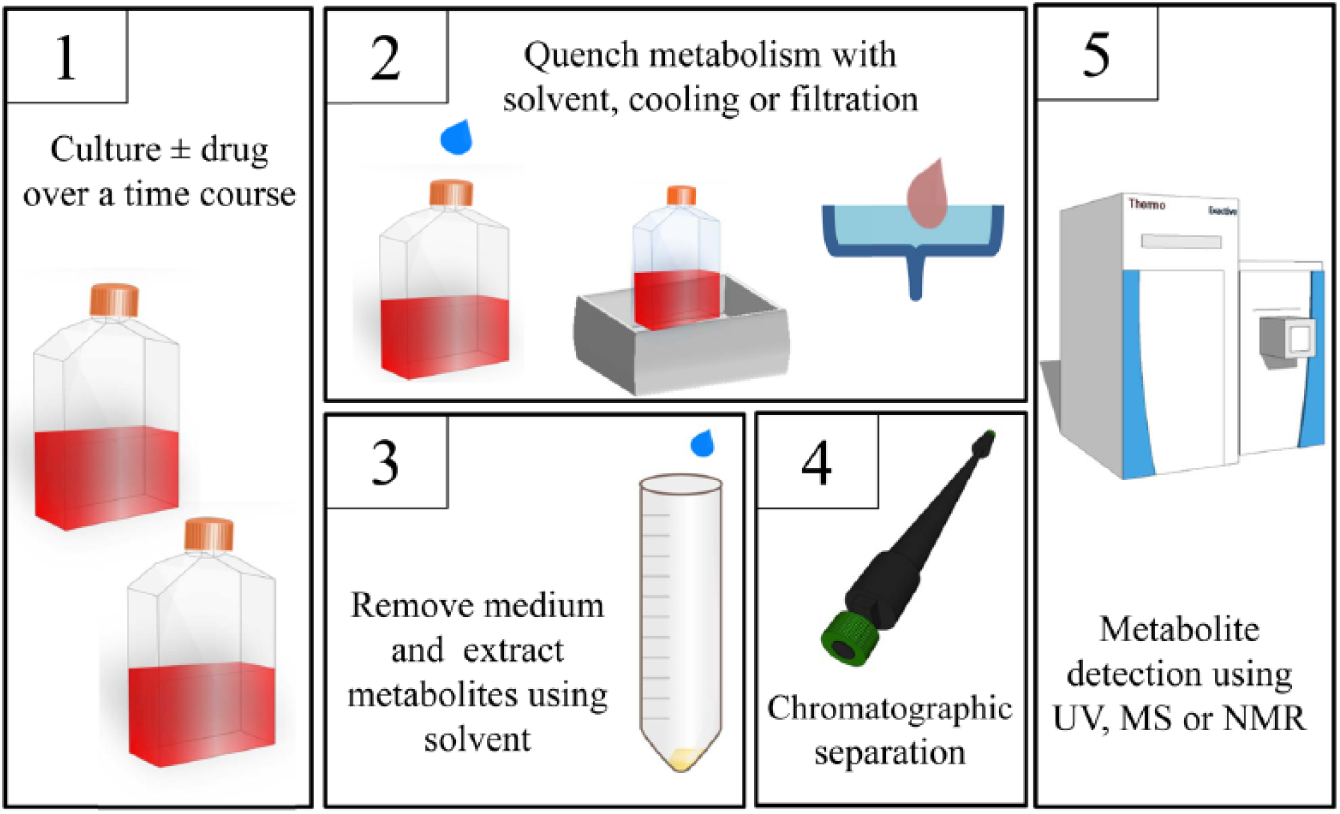
Mode of action metabolomics. Metabolites are grown in the presence and absence of drug. Metabolism is quenched, medium is removed, and metabolites are extracted. Data are collected after chromatographic separation.
Experimental Design
To analyze the MOA of a new active compound in a cell system, it is first necessary to find a cell system that is as biologically relevant as possible. For many parasites, this will involve culturing the cells in a medium that is as close as possible to the host environment. Host environments include cerebrospinal fluid (Trypanosoma brucei), blood (T. brucei and Trypanosoma cruzi trypomastigotes) or an intracellular compartment (Leishmania amastigotes, Plasmodium, and T. cruzi amastigotes). Rich media are often not suitable as they can contain components that mask the effects of the drug or block the action of the drug. 17 As a result, efforts have been made to create minimal media that are as close as possible to the most relevant biological environment.17–20 Metabolomics has recently been used to show that bloodstream-form T. brucei used only glucose, cysteine, glutamine, and aromatic amino acids from the HMI-11 classically used to culture bloodstream-form trypanosomes. 17 A minimal medium comprising these nutrients, salts, buffer, but still requiring serum was devised. The new medium, CMM, provided comparable growth rates to HMI-11 and showed that parasites were much more sensitive to several drugs in a minimal medium where nutrients are not in excess. 17 These media are also useful for flux analyses in which stable-isotope labeled metabolites are tracked in real time through metabolic pathways. 4
In the absence of a minimal medium, defined media21,22 are used to ensure that knowledge of the source of nutrients can be traced back to the cell under study or the medium in which the cells are grown. For Plasmodium, a cell-free culture system is not yet possible, so the contribution of the red blood cells to the metabolome must be taken into account, and additional controls are required. 23
Drug Perturbations
The drug dose and length of time that a cell culture is treated depend largely on the biological question. Creek et al. 17 used time points up until 48 h at a dose that cause a 20% growth inhibition. Others have used a multiple of the concentration required to inhibit growth by 50% (IC50) 2 or the IC90 and time points up until there has been 50% growth inhibition compared with untreated. 24 Whichever drug concentration and endpoint are chosen, a range of time points up until the endpoint is recommended to find trends in the noisy data sets. This allows researchers to establish the earliest changes to the metabolome, which are most likely to relate to the primary target. It is also recommended to conduct a pilot study and power analysis prior to the full experiment so that sufficient numbers of replicates are taken to ensure biological significance. The power analysis tables used to analyze two-dimensional protein gels offer a convenient prototype for other large data sets such as metabolomics. 25 Usually, six replicates are considered to be minimal when the coefficient of variation (standard deviation of the metabolite intensity/mean) is 0.3. 25 For comparisons over a time series, fewer replicates (usually three) may be used. A control of the time course without drug should also be included to account for cell cycle or density-induced changes to the metabolome.
Quenching Metabolism and Extracting Metabolites
A protocol for the rapid quenching of metabolism is essential to any metabolomics experiment. If metabolism is not quenched rapidly, there may be additional changes to metabolism that relate primarily to perturbations during cell handling and extraction. For example, glucose is essential to bloodstream-form parasites, and the cells commence dying as soon as parasites are removed from their glucose source. 26 If cell metabolism is not quenched before cells are processed further, the products of reactionary metabolism are measured. Quenching of metabolism is usually achieved via rapid cooling, filtration, or the addition of a surplus of cold (usually 4 °C or −48 °C) methanol27,28 ( Fig. 1 ; Table 1 ). Quenching bacteria in cold methanol or by filtration is considered to be standard,27,28 but these methods cannot be used on some protozoa; for example, bloodstream-form T. brucei lyse on filters and exhibit metabolite leakage with cold methanol (unpublished observations). Rapid cooling of parasite suspensions, within their culture flasks in a dry-ice ethanol bath, is considered the best practice for suspended protozoa, followed by an extraction in solvent such as methanol or chloroform:methanol:water (1:3:1). 40 For Plasmodium, the standard currently combines quenching of metabolism and metabolite extraction. Generally, infected red blood cells are quenched with methanol (80%, 90%, or 100% in water) at 4 °C or in a dry-ice ethanol bath. Cells are then sonicated and centrifuged, and the supernatant is taken and dried under an N2 gas stream, before reconstituting in the preferred chromatography solvent.31,32,39
Methods used for the quenching of metabolism, metabolite extraction, and data processing in parasitic protozoa.
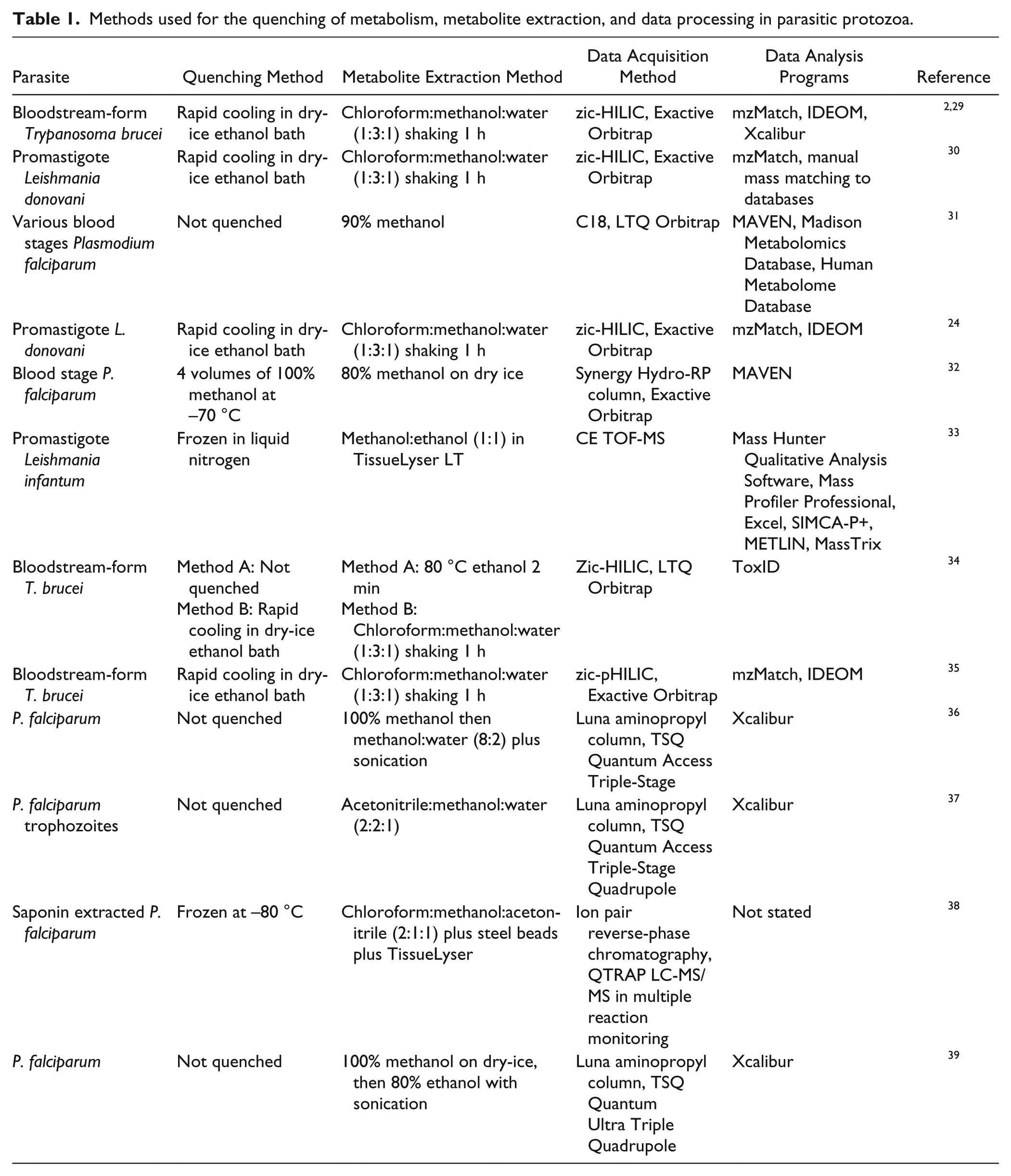
After quenching metabolism, cell types that do not display metabolite leakage can be washed, but washing is not recommended for all cell types. Removing excessive buffer, salts, and other contaminants from the medium is advantageous, as it will improve the life of the chromatography apparatus, especially if separation is based on polarity. Also, any metabolites that are in excess in the medium risk masking smaller changes to the internal metabolome if the medium is not removed. 17
Metabolite extraction generally coincides with the precipitation of membranes, proteins, and other macromolecular polymers from the cells. Metabolite extraction is usually achieved by the addition of a solvent such as methanol or acetonitrile in water for polar metabolites, often with chloroform added to improve the extraction of more hydrophobic metabolites. 40 However, the combination of solvents will depend largely on the column on which the extract is to be run and the metabolite fraction that the user wishes to analyze.
Data Acquisition and Analysis
Many researchers will wish to use a service to generate data from their cell extracts. There are several such services available including Metabolon (www.metabolon.com), Phenomenome (http://www.phenomenome.com), Glasgow Polyomics (www.polyomics.gla.ac.uk), The West Coast Metabolomics Center (metabolomics.ucdavis.edu), The University of Birmingham metabolomics service (http://www.birmingham.ac.uk/facilities/genomics/about/metabolomics), Biocrates Life Sciences (http://www.biocrates.com), and The Metabolomics Innovation Centre (www.metabolomicscentre.ca). These services will vary on cost and the amount of downstream analysis performed.
Metabolite extractions are complex. Usually the complexity of the extract is reduced by separation on a chromatography column into several fractions. 41 Specific metabolites hypothesized to underlie a biological event will dictate to some extent which chromatography apparatus to use. For example, gas chromatography (GC) is usually used for volatile metabolites and C18 or reversed-phase chromatography for lipids. 42 For an untargeted analysis, covering wide areas of cellular metabolism, liquid chromatography (LC) is usually used. Hydrophilic interaction liquid chromatography (HILIC; suitable for separating polar, hydrophilic compounds) and polymeric HILIC (suitable for separating polar, hydrophilic compounds over a large pH range) columns are often used for this type of separation ( Table 1 ), as they optimize separation of hydrophilic compounds. For a review of LC techniques, see Ramautar et al. 43
Mass spectrometers or NMR detectors are the most commonly used instruments for metabolite detection. NMR detects metabolites based on the vibrational energy of bonds. The technique is quantitative and gives information on the structure of the metabolites, which can lead to a definitive identification. NMR is, however, less sensitive than MS although still widely used to detect high-abundance metabolites. NMR is also used in conjunction with MS to get information on the structure of molecules identified by MS as contributing to a biological event but of unknown identity and to quantify these molecules.42,44
MS has advantages over NMR as it is generally able to detect the accurate mass of hundreds to thousands of compounds in one sample and is much more sensitive. 42 The disadvantage of MS is that it takes significantly longer to run (with chromatography) than NMR, so it is lower throughput and lacks quantification unless additional steps (e.g., using labeled standards) are taken.
Targeted approaches such as multiple reaction monitoring (MRM) can also be taken. In MRM, several target ions are isolated and fragmented to obtain metabolite identifications. Targeted approaches such as MRM provide definitive metabolite identifications, whereas untargeted approaches analyze the abundance of every mass peak and attempt to assign metabolite identifications after data acquisition and filtering.
Once raw data have been collected, the peaks representing true metabolites must be picked, aligned, and filtered, and then statistical comparisons must be made. 41 There is currently no consensus on the best way to pick peaks and analyze any differences, and many groups use their own in-house platforms or a combination of tools ( Table 1 ). Most platforms use XCMS, 45 MzMatch 46 (which uses XCMS but includes further downstream data reduction), MZmine, 47 Metalign, 48 or MathDAMP 49 for data reduction and analyzing variation between different treatment groups. Metabolite identification can be achieved using spectral libraries for GC-MS,50,51 but metabolite annotation is a much more difficult task in LC-MS. An exact mass can usually give one or a few formulae, but stereoisomers cannot be distinguished on mass alone, and other orthogonal data are required.44,52 Metabolite annotation is therefore usually performed on the exact mass detected and the retention time or retention index compared with an authentic standard or the fragmentation pattern. The Metabolomics Standards Initiative (MSI) working group outlined the minimum data considered appropriate for a metabolite to be deemed “identified.” 44 The level (between 1 and 4, see below) of identification of each metabolite should be recorded in any published experiment to comply with the MSI. 44
Level 1: identified compounds. For this level of identification, a match of retention time and mass to an authentic standards run in identical experimental conditions is required or mass and fragmentation or mass and stable isotope labeling. These data must be made available to publish the compounds as “identified.” 44
Level 2: putatively annotated compounds. Compounds without chemical reference standards (usually mass and retention time but with no standard to match to), based on physicochemical properties and/or spectral similarity with public/commercial spectral libraries. 44
Level 3: putatively characterized compound classes. Compound matches based on characteristic physicochemical properties of a chemical class of compounds or by spectral similarity to known compounds of a chemical class. 44
Level 4: unknown compounds. Although unidentified or unclassified, these metabolites can still be differentiated and quantified based on spectral data. 44
Several programs are available to aid automatic metabolite annotation or identification. Many groups will again use their own platform. IDEOM 53 uses the mass and retention time of standards to provide metabolite identifications and a retention index to provide annotations. Other programs use the mass alone, searching against metabolite databases such as HMDB, 54 , KEGG, 55 ChemSpider, 56 or METLIN. 57 Stereoisomers can then be distinguished using retention time or fragmentation information. Fragmentation provides data on the structure of a mass and therefore enables a more definitive identification. Such fragmentation analyses are performed using a mass spectrometer with MSn capabilities. 58 To analyze fragmentation data programs such as MAGMa 59 and MetFusion 60 are available, or the user can develop a nonautomated protocol, which may be more useful if the number of features is small.61–63
MOAs and Resistance of Antiprotozoal Drugs
Antitrypanosomal Drugs
Trypanosomes are responsible for a variety of diseases, including human African trypanosomiasis (HAT), nagana in animals, and Chagas disease. The majority of the drugs for HAT, caused by T. brucei subspecies, are toxic or ineffective in the latter stages of the disease. 10 The current recommended treatment for late-stage disease is nifurtimox-eflornithine combination therapy for the gambiense form of the disease or melarsoprol for the rhodesiense form. Nagana, the livestock form of HAT, is responsible for economic losses of US$1.4 billion per year. 64 Drugs for nagana are few, and there is widespread resistance to the main compounds: diminazine and isometamidium. 64 The treatment for Chagas disease, caused by T. cruzi, is benznidazole or nifurtimox. Chemotherapy is effective in the early, acute phase of the disease but not during the intermediate or chronic stages.10,65
Among the first studies using metabolomics to analyze the MOA of an antiprotozoan agent was a study into the MOA of eflornithine, a known ornithine decarboxylase (ODC) inhibitor.2,66,67 After treatment with a sub-IC50 level of eflornithine (20 µM), the substrate for ODC, ornithine, was found to increase significantly (more than sevenfold), and the product of ODC, putrescine, was found to decrease significantly 2 ( Fig. 2 ). The remainder of the metabolome was largely unchanged (other than N-acetylated derivatives of the substrate and product, whose concentrations mirrored those of the parent compounds) at this drug dose. At higher drug doses, greater metabolic changes were observed, including apparent cell leakage of many metabolites at 48 h. 2 The same study analyzed the metabolome during a combination of eflornithine and nifurtimox treatment in an attempt to reflect the treatment in the field. 2 The results highlighted the fact that in vitro eflornithine and nifurtimox are antagonistic, as shown in both the IC50 values in combination by isobologram analysis and each drug’s effects on the metabolome. 2
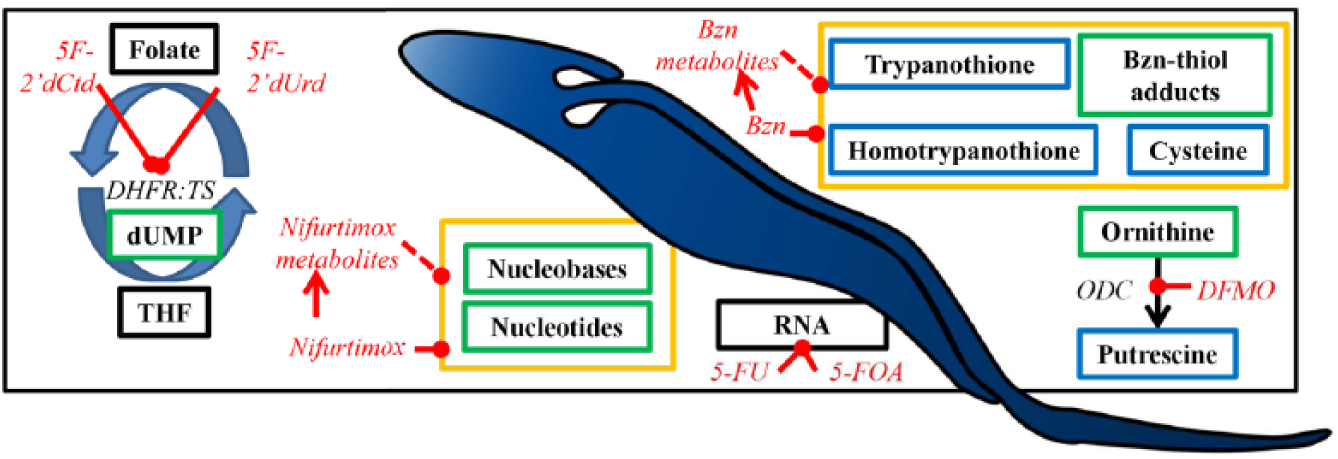
Metabolomics mode of action studies in T. brucei. Enzymes are in black italics, drugs are in red italics, and metabolites are boxed. Blue boxes indicate a decrease in metabolite abundance, and green boxes indicate an increase in metabolite abundance. Black boxes indicate no change in abundance. Yellow boxes indicate metabolite changes for which the inhibited enzyme/process is unknown. DFMO, difluoromethylornithine (eflornithine); ODC, ornithine decarboxylase; 5F-2′dCtd, 5-fluoro-2′deoxycytidine; 5F-2′dUrd, 5-fluoro-2′deoxyuridine; 5-FU, 5-fluorouracil; 5-FOA, 5-fluoroorotate; DHFR:TS, dihydrofolatereductase:thymidylate synthase; Bzn, benznidazole.
The MOA of nifurtimox was also analyzed, and although the primary target of the drug was not discovered, increases in nucleotides and nucleobases were detected, suggesting a degradation of DNA and RNA 2 ( Fig. 2 ). The reduction of nifurtimox to its saturated open chain nitrile derivative 68 was also observed, indicating that this process occurs rapidly (within 1 h at least) after drug treatment. 2 Another nitroheterocyclic trypanocidal drug, benznidazole, is used against T. cruzi, the causative agent of Chagas disease. An untargeted LC-MS–based metabolomics analysis of changes to metabolism in T. cruzi exposed to benznidazole revealed that key cellular thiols (trypanothione, homotrypanothione, and cysteine) were depleted during treatment. Furthermore, numerous metabolites of the drug were observed, as were a multitude of adducts of the drug bound to cellular thiols and other metabolites. 29 Another study on eflornithine looked at the differences between eflornithine-resistant T. brucei compared with drug sensitive T. brucei. 34 There were few differences to the metabolome as a whole, but when challenged with eflornithine, it was found that substantially more eflornithine was taken into sensitive cells (the drug’s mass being readily detectable by MS), indicating a transporter-based mechanism of resistance. 34 This was verified using standard molecular techniques, and an amino acid transporter, AAT6, was shown to import the drug, and its absence was responsible for resistance. 34
The MOAs of novel drugs have also been analyzed using metabolomics. In a recent study, fluoropyrimidine analogues were synthesized, and their action on T. brucei was investigated by LC-MS. This study provided information on the MOA of these analogues, as well as information on the reversibility of the enzymes in pyrimidine biosynthesis, providing evidence that a uridine phosphorylase is expressed in bloodstream-form trypanosomes. 35 5-fluoroorotic acid and 5-fluorouracil appeared to exert their actions through incorporation into RNA ( Fig. 2 ). The action of 5-fluorouracil is therefore different from the action of the drug in humans, where it is incorporated into DNA, leading to double-strand breaks. 35 5-fluoro-2′deoxyuridine and 5-fluoro-2′deoxycytidine appeared to inhibit the bifuntional dihydrofolate reductase-thymidylate synthase enzyme, leading to a large increase in dUMP 35 ( Fig. 2 ). Metabolomics approaches are presently being used to seek MOAs of other known trypanocidal drugs and various new agents that kill the parasites.
Leishmanicides
Leishmania chemotherapy is very much dependent on the infective species and the area of the world where the patient was infected. There is widespread drug resistance to many of the licensed compounds, particularly on the Indian subcontinent, where the more severe form of the disease visceral leishmaniasis persists. 10
Leishmania donovani is the main cause of visceral leishmaniasis in India and has been treated with antimonial compounds for more than 60 y. 69 The MOA of antimony (SbIII) was investigated using a sheathless capillary electrophoresis technique, and increases in L-cystathione, S-adenosylhomocysteine, and glutathione disulfide were recorded 33 ( Fig. 3 ), suggesting an increased response to oxidative stress. Changes in polyamine and thiol levels have previously been noted using high-performance liquid chromatography–based assays,70,71 but it is unclear whether the response is due to the drug itself or a more generalized stress response. Certainly, SbIII has been shown to conjugate to trypanothione (which also contributes to the reduction of the metal from its inactive, SbV form, to the active form SbIII)71,72 to aid expulsion from the parasite,70,73 so the increase in polyamines may also be due to a detoxification mechanism of the parasite, given that spermidine is a component of trypanothione. It is important to note that thiols exist in reduced and disulfide forms, with the reduced forms being important for detoxifying reactive oxygen species (ROS). Susceptible Leishmania treated with SbIII accumulated disulfide forms of glutathione and trypanothione, diminishing the redox capacity of the parasites. 70
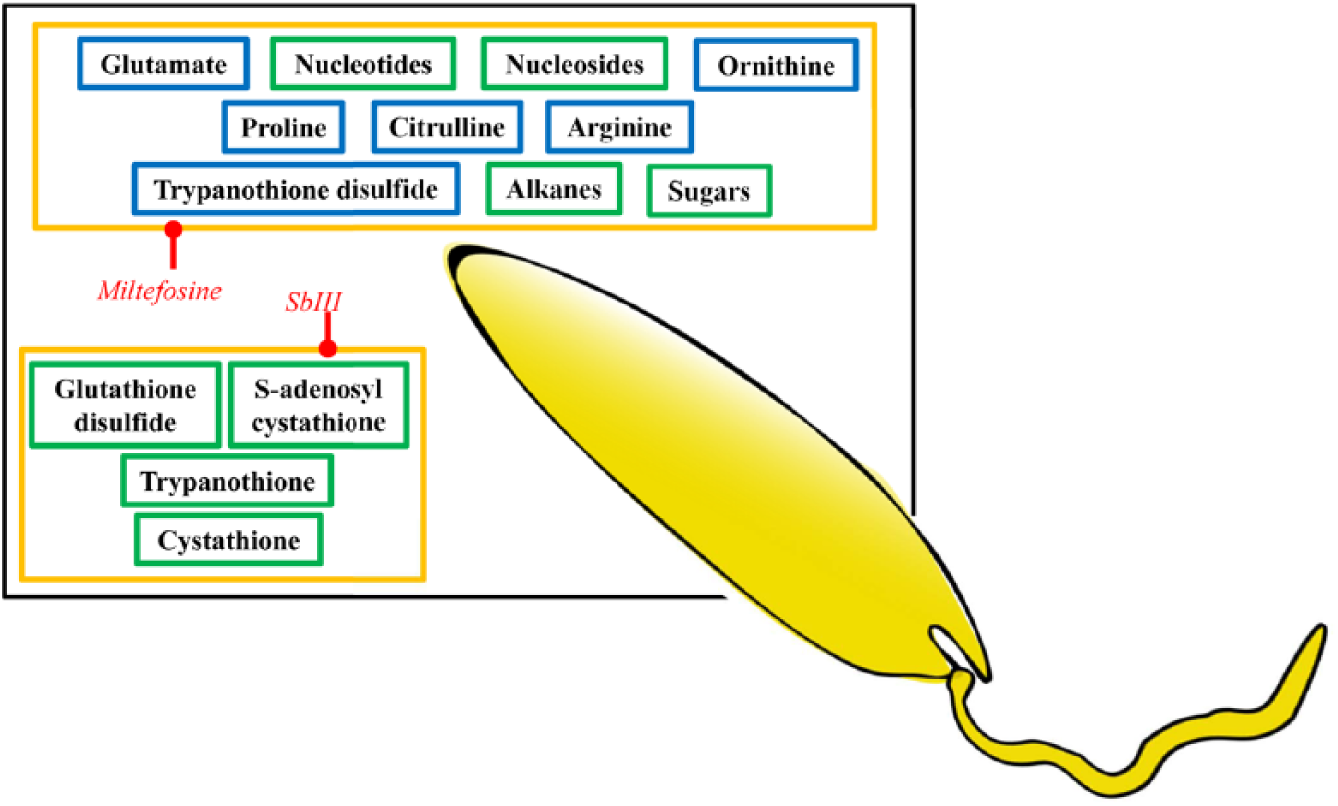
Metabolomics mode of action studies in Leishmania. Drugs are in red italics, and metabolites are boxed. Blue boxes indicate a decrease in metabolite abundance, and green boxes indicate an increase in metabolite abundance. Yellow boxes indicate metabolite changes for which the inhibited enzyme/process is unknown.
It is easier to obtain clinical isolates of Leishmania than T. brucei, and resistance is a very pressing issue, with extremely high rates of antimony resistance in Northern India. 74 As a result of this high incidence of resistance, there are several studies looking at the metabolic differences in clinical resistant isolates. For example, sodium stibogluconate–resistant promastigote isolates were compared with sensitive isolates from the Indian subcontinent, 30 in the presence and absence of drug pressure and during the parasite cell cycle using untargeted Orbitrap mass spectrometry coupled to a zic-HILIC column. 30 The MOA of antimony proved elusive using this technique, as large areas of metabolism were affected. 30 The study did highlight, however, the importance of stage-specific differences to the metabolome as even in insect stages of the parasite, large differences were seen as the parasites moved into a stationary phase of growth. 30 As most studies in Leishmania are performed in promastigote stages, a key question remains as to whether any of the promastigote results will translate to a parasite that exists in humans in a wholly different environment.
The difficulty in growing axenic amastigotes of most Leishmania species means that the majority of studies have been done on cultured promastigote cells. Cultured axenic amastigotes are also not necessarily identical to macrophage amastigotes and reveal altered biochemistry. 75 It is generally accepted that all drug studies that are performed on promastigote cells must also be verified in amastigotes—whether axenic or macrophage dwelling—but a systematic analysis of the changes to parasite biochemistry across the life cycle has not yet been undertaken. This should be a priority for the future if drug studies are to be performed in these very distinct life cycle stages. Life cycle studies should be done on isogenic strains of Leishmania to ensure that a full knowledge of the genetic background is available. The many studies of field strains suffer from a lack of control over genetic differences between outbred strains, which may or may not underlie differences in drug susceptibility or drug action.
Promastigote Leishmania resistant to antimony also appeared to have differences in the fluidity of their membranes. 30 Another study that indicated alterations to the lipid content of Leishmania analyzed metabolic changes in response to miltefosine treatment. 24 A lipidomics analysis, using an Absciex QTrap, revealed no changes to the membrane content of the drug-treated cells, although internal lipid metabolism appeared to have been affected, as evidenced by the increases in alkane fragments 24 ( Fig. 3 ). Sugars and nucleotides were also increased after miltefosine treatment ( Fig. 3 ), suggesting a multifactorial MOA, 24 supported by further work demonstrating a general ROS response. 76 This study also measured the uptake of miltefosine, first using MS and confirmed using a fluorescent miltefosine analog. A transport defect was discovered in the miltefosine-resistant strain, which is a known resistance mechanism.77–79 Interestingly, an antimony-resistant strain, naïve to miltefosine, also had a miltefosine transport defect. 24 Mutations in the miltefosine transporter in the antimony-resistant line raise concerns about the possible development of multidrug-resistant Leishmania.
Antimalarials
The rise of drug resistance in every class of antimalarial 37 is compromising antimalaria therapy, and combination therapies are increasingly preferred. 80 More drugs with novel MOAs are required that will circumvent the current resistance mechanisms.
Atovaquone was discovered more than 20 y ago and is known to disrupt the parasite’s mitochondrial electron transport chain at the bc1 complex. 81 Atovaquone and its quinolone analogue CK-2-68 were shown to inhibit two enzymes in the bc1 complex: NADH:ubiquinone oxidoreductase and cytochrome bc1. Using an MRM-targeted metabolomics approach looking for 35 metabolites, it was shown that following atovaquone-induced membrane depolarization, proton pumping was disrupted but dihydroorotate dehydrogenase activity is also diminished, leading to a depletion in orotate levels 37 ( Fig. 4 ). The lack of orotate then affects pyrimidine synthesis. 37 Despite the fact that atovaquone acts against more than one target, resistance has been relatively simple to select.82,83 Both mutations in the catalytic domain of the bc1 complex 84 and dihydroorotate dehydrogenase overexpression 85 have been identified as mechanisms to reduce sensitivity to atovaquone.
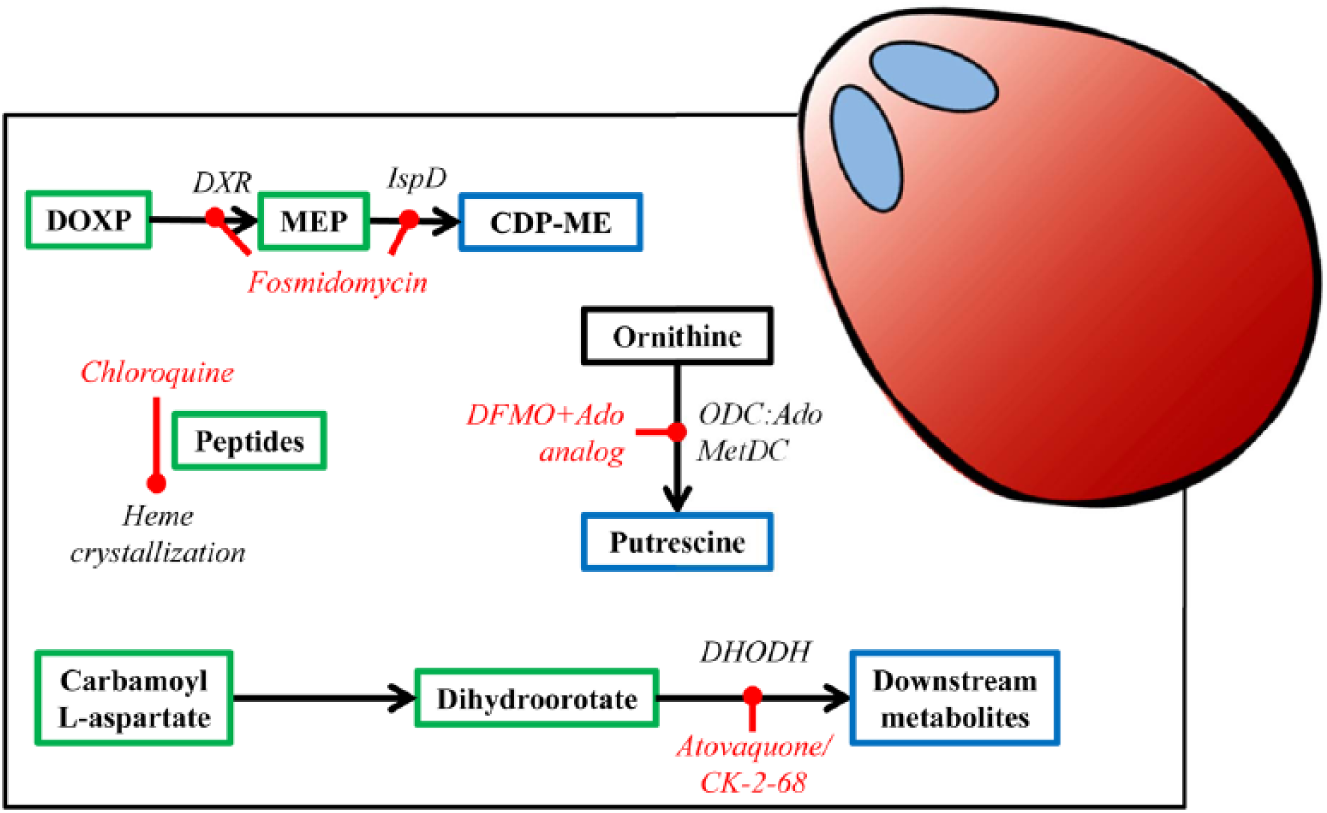
Metabolomics mode of action studies in P. falciparum. Enzymes are in black italics, drugs are in red italics, and metabolites are boxed. Blue boxes indicate a decrease in metabolite abundance, and green boxes indicate an increase in metabolite abundance. Black boxes indicate no change in abundance. DFMO, difluoromethylornithine (eflornithine); ODC, ornithine decarboxylase; DXR, deoxyxylulose 5-phosphate reductoisomerase; IspD, methylerythritol phosphate cytidyltransferase; DOXP, 1-D-deoxyxylulose 5-phosphate; MEP, methylerythritol phosphate; CDP-ME, cytidine diphosphate methylerythritol; DHODH, dihydroorotate dehydrogenase; Ado, adenosine; AdoMetDC, S-adenosyl methionine decarboxylase.
An analysis of six isoprenoid precursor metabolites using MRM revealed a second target of fosmidomycin, a previously known inhibitor of deoxy-xylulose phosphate reductoisomerase (DXR) localized in the apicoplast, an organelle essential to Plasmodium. 38 After fosmidomycin treatment of ring-stage cultures, an increase in methylerythritol phosphate and a decrease in cytidine diphosphate methylerythritol were seen in addition to the changes expected with DXR inhibition 38 ( Fig. 4 ). This suggests that methylerythritol phosphate cytidyltransferase (IspD) is a secondary target for fosmidomycin, which was strengthened by the fact that overexpression of IspD leads to resistance to the compound. 38 It has been shown that P. falciparum grown in excess isopentenyl pyrophosphate (IPP), the end product of isoprenoid biosynthesis, are less susceptible to fosmidomycin treatment. 86 IPP also rescued parasites treated with antibiotics that caused loss of the apicoplast (chloramphenicol, clindamycin, and doxycycline), providing evidence that IPP biosynthesis is the only essential function of the apicoplast. 86
A major area for antiprotozoal development is the inhibition of pathways that help detoxify oxidative stress–related metabolites generated during the periods of rapid growth that these parasites undergo. 87 Two papers have explored inhibitors of these pathways in malaria. In P. falciparum, ODC and S-adenosyl methionine decarboxylase (AdoMetDC) exist as a single, bifunctional enzyme. 88 A combination of the ODC inhibitor, eflornithine and 5′{[(Z)-4-amino-2-butenyl]methylamino}5′deoxyadenosine, an inhibitor of AdoMetDC, was added to late schizonts of P. falciparum, and changes to the metabolome were recorded using MRM of 172 metabolites. 36 Increases in putrescine and spermidine were recorded, but ornithine levels were unchanged due to an increase in ornithine aminotransferase expression, as determined by a global transcript analysis. 36 Global transcript analysis further revealed growth arrest after 19 h posttreatment and an increase in expression of lysine decarboxylase and ornithine aminotransferase as well as a decrease in expression of S-adenosylmethionine synthetase, supporting the metabolite data. 36
A second study aimed to validate glutamate dehydrogenase as a drug target using an untargeted LC-MS approach. 32 Heavy atom–labeled glutamine tracking was used to show that there is no difference in the labeling of the tricarboxylic acid cycle intermediates after knock out of this gene, nor is there an increased sensitivity to oxidative stresses. 32 This leads to the conclusion that, contrary to previous suggestions, glutamate dehydrogenase is not suitable as a drug target. 32 Tracking of heavy labeled metabolites has been used to analyze general metabolism in P. falciparum, probing the possibilities of mitochondrial metabolism89,90 and phospholipid biosynthesis. 91 These types of analysis have uncovered choke points in metabolism, which could serve as future drug targets. Another heavy atom label tracking analysis used LC-MS to track 13C-U-glucose proving that P. falciparum lacks the ability to synthesize nicotinamide adenine dinucleotide (NAD+) de novo and highlighting an essential enzyme in the pathway responsible for producing NAD from exogenous niacin: P. falciparum nicotinate mononucleotide adenylyltransferase. 39
With the rise in antimalarial resistance in many malaria-endemic countries, efforts to understand the resistance mechanisms and how long these mechanisms last when drug pressure is removed from the field are increasing. 92 Chloroquine is the drug for which there is the widespread resistance in the field in P. falciparum and increasingly in other Plasmodium species. 80 The PfCRT transporter, localized to the food vacuole membrane, has long been known as a key determinant of resistance. 93 An analysis of the metabolite levels that are significantly different between drug-sensitive and drug-resistant isolates of P. falciparum found several peptides related to hemoglobin digestion. 31 Efflux of hemoglobin peptides could therefore be an essential role of the PfCRT transporter, and glutathione may be imported concurrently with the efflux of these peptides. 94 The work on chloroquine resistance also provided evidence of a fitness cost associated with resistance, and other work has also suggested a fitness cost to chloroquine resistance, 95 which may promote reemergence of drug sensitivity in areas where there is widespread resistance. 31 It is hoped that once chloroquine compounds are removed from the field, drug sensitivity will reemerge and the compounds will once again become useful as part of a combination therapy.
The Future
As more is known about the normal metabolism of protozoan parasites, particularly through the life cycle stages, perturbations in this metabolism will become easier to analyze. Metabolomics is likely to be a key tool in the analysis of normal and perturbed metabolism and will be used more readily in the analysis of drug MOAs. Currently, untargeted metabolomics approaches reveal numerous unknown metabolites. As these pathways are annotated, a shift to more quantitative, targeted technologies (e.g., using ever more identified metabolites in MRM approaches) can be applied in a bespoke fashion to individual parasite types. Combining metabolomics with other -omic technologies (proteomics, genomics, and transcriptomics) will also help determine resistance mechanisms to these drugs.
Partnerships with pharmaceutical companies are allowing compound libraries to be screened systematically against protozoan parasites, and lead compounds from these phenotypic screens are emerging. The retrospective identification of drug targets can then be used in refining the structure of hits in target-based medicinal chemistry campaigns.
Metabolomics is also being applied to other aspects of pharmaceutical development. For example, metabolic responses predictive of toxicology in human cells can be determined and then sensitive methods used to detect whether such changes can be picked up either in vitro or in fluids derived from treated animals.
An additional tool becoming more readily available is quantitative metabolomics. Instrumentation and data analysis platforms are changing to allow the quantitation of metabolites in a normal analysis pipeline. This may be through NMR technologies or through the use of a spiked sample of universally labeled metabolite extract. 96 Knowing the exact concentrations of metabolites in the various life cycle stages of the parasites will help in the building of in silico models of parasite metabolism that will further be able to predict drug targets and resistance mechanisms.
Integrating metabolomics into the pharmacological pipeline is a priority for the future, and protozoan anti-infectives are at the forefront in this area of research.
Footnotes
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Isabel M. Vincent and Michael P. Barrett both work in collaboration with Glasgow Polyomics.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.