
Abstract
Shikimate dehydrogenase (AroE) is an attractive target for herbicides and antimicrobial agents due to its conserved and essential nature in plants, fungi, and bacteria. Here, we have performed an in vitro screen using a collection of more than 5500 compounds and identified 24 novel inhibitors of AroE from Pseudomonas putida. The IC50 values for the two most potent inhibitors we identified, epigallocatechin gallate (EGCG) and epicatechin gallate (ECG), were 3.0 ± 0.2 µM and 3.7 ± 0.5 µM, respectively. Based on the high level of structural conservation between AroE orthologs, we predicted that the identified compounds would also inhibit AroE enzymes from other organisms. Consistent with this hypothesis, we found that EGCG and ECG inhibit the AroE domain of the bifunctional dehydroquinate dehydratase-shikimate dehydrogenase (DHQ-SDH) from Arabidopsis thaliana with IC50 values of 2.1 ± 0.3 µM and 2.0 ± 0.2 µM, respectively.
Keywords
Introduction
The shikimate pathway produces essential precursors ofthe aromatic amino acids, folate, and vitamins. 1 Whilethe pathway is present in plants and microbes, it is not found in animals. 1 The pathway has received considerable attention due in large part to the identification of one of its enzymes, 5-enolpyruvylshikimate-3-phosphate (EPSP) synthase, as the cellular target of the herbicide, glyphosate (N-(phosphonomethyl) glycine). 2 While a number of studies have sought to identify inhibitors of other enzymes in the pathway,3–9 a relatively small number of compounds have been identified that block the pathway’s fourth reaction, the NADPH-dependent conversion of 3-dehydroshikimate to shikimate.10–12 This reaction is catalyzed by shikimate dehydrogenase (AroE). 1
Here, we have designed an in vitro screen to evaluate the ability of compounds from a combined library of more than 5500 small molecules to inhibit the AroE enzyme from the bacterium, Pseudomonas putida (PpAroE). From our collection of chemicals, we uncovered a group of structurally related polyphenolic molecules with potent inhibitory properties. Two of these compounds, epigallocatechin gallate (EGCG) and epicatechin gallate (ECG), inhibit PpAroE with IC50 values of 3.0 ± 0.2 µM and 3.7 ± 0.5 µM, respectively. Based on the high level of structural conservation observed among AroE orthologs, we speculated that the identified compounds would also be effective inhibitors of other AroE enzymes from diverse organisms. Indeed, we found that EGCG and ECG inhibit the AroE domain ofthe bifunctional Arabidopsis thaliana dehydroquinate dehydratase-shikimate dehydrogenase (DHQ-SDH). EGCG, ECG, and the remaining compounds identified in this study may represent useful chemical scaffolds for the design of novel antibiotics targeting the shikimate pathway in microbes and plants.
Materials and Methods
Protein Expression and Purification
The gene encoding AroE from P. putida KT2440 (PP_0074) was inserted into a modified pET28a vector (Novagen, Gibbstown, NJ) containing a 6XHis-tag/TEV cleavage site, as previously described. 13 Escherichia coli strain BL21 CodonPlus was used for expression of the recombinant protein. Cultures were grown in Luria-Bertani (LB) media with 50 µg/mL kanamycin at 37 °C with shaking until they reached an OD600 of 0.6 to 0.8. Protein expression was induced by the addition of 0.4 mM isopropyl β-D-thiogalactopyranoside (IPTG). Cultures were then allowed to grow with shaking for an additional 4 h at 37 °C. The incubation temperature was lowered to 16 °C overnight, and the following morning, cells were harvested by centrifugation and lysed using a French press. Insoluble cellular materials were removed by centrifugation. Purification of the recombinant protein was performed by nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography over an imidizole gradient (5–300 mM) in buffers containing 50 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 5% glycerol. The purified protein was dialyzed overnight at 4 °C in 10 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 5% glycerol. A. thaliana DHQ-SDH was purified in a similar manner. Preparation of the DHQ-SDH construct is described elsewhere. 14
Inhibitor Screen Design
Screening for inhibitors was performed in a 96-well format by using a POLARstar OPTIMA plate reader (BMG Labtech, Nepean, Canada) to monitor the enzyme-dependent reduction of NADP+ (ϵ = 6220 M−1cm−1) at 340 nm and 25 °C. Each 100-µL reaction contained 50 mM Tris-HCl (pH 8.8; the optimal pH for the enzyme; data not shown), 150 mM NaCl, 60 ng purified recombinant PpAroE, 100 µM shikimate, 2 mM NADP+, and 2.5 µM of a potential inhibitor. Reactions were initiated by the addition of the enzyme. Screening was performed in duplicate using compounds from the TimTec Natural Product Library (~280 compounds; TimTec LLC, Newark, NJ), the Spectrum Collection (~2000 compounds; Microsource Discovery, Groton, CT), and the Library of Active Compounds on Arabidopsis (LATCA; ~3600 compounds; Sean Cutler, University of California, Riverside). The inhibitory activity of these compounds was evaluated by comparing the reaction rate in each well containing a potential inhibitor with a DMSO-treated control. Compounds were classified as inhibitors if they caused ≥25% apparent reduction in turnover rate.
IC50 Determination
The two strongest inhibitors, EGCG and ECG, were reordered from Sigma-Aldrich (St. Louis, MO) and subjected to a detailed analysis. Solutions of both compounds were freshly prepared in DMSO. For determination of IC50 values, reaction progress was monitored at a range of inhibitor concentrations (0–20 µM) in a reaction volume of 1 mL using a Varian Cary 50 Bio UV-visible spectrophotometer (Varian, Mulgrave, Australia). The reaction was initiated by the addition of enzyme (200 ng PpAroE or 450 ng A. thaliana DHQ-SDH) to a reaction mixture containing shikimate and NADP+ at concentrations reflecting their approximate Km values (200 µM shikimate and 50 µM NADP+ for PpAroE 15 ; 700 µM shikimate and 150 µM NADP+ for A. thaliana DHQ-SDH 14 ). IC50 values were determined using the program GraFit (Erithacus Software, East Grinstead, UK) by plotting the relative activity of the enzymes at the different inhibitor concentrations and fitting the resultant profile to equation (1):
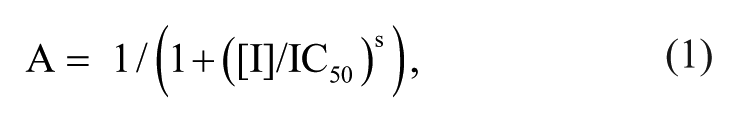
where A is the relative activity remaining in the presence of the inhibitor at concentration, [I], and s is the slope factor.
Structural Analysis of PpAroE
PpAroE crystals were generated using the hanging drop vapor diffusion method. Purified PpAroE (10 mg/mL) and well solution (0.1 M NaHEPES [pH 6.6], 2 M (NH4)2SO4, and 2% PEG400) were mixed in a 1:1 ratio. Diffraction data were collected at the Advanced Photon Source at the Argonne National Laboratory, Chicago. Data were processed and scaled using HKL2000, 16 and the structure of PpAroE was determined by molecular replacement with Haemophilus influenzae AroE (PDB ID: 1P77) using the AutoMR and AutoBuild programs in the Phenix software suite. 17 A summary of data collection and refinement statistics will be published elsewhere. The atomic coordinates and structure factors for AroE can be found in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, New Jersey (http://www.rcsb.org/; ID: 4OMU).
Minimal Inhibitory Concentration (MIC) Determination
A broth microdilution assay was used to evaluate the antibiotic activity of EGCG and ECG against P. putida KT2440. Cells were grown to mid-log phase in LB media before being diluted to a concentration of ~2 × 107 colony-forming units (CFU)/mL. Each well of a 96-well plate containing 100 µL of LB media and EGCG or ECG at concentrations between 0 and 1 mM was inoculated with 5 µL of the diluted cells (~105 CFU). Gentamycin was used as a positive control. The plate was incubated with gentle shaking at 30 °C for 16 h. Following incubation, the optical density in each well was measured at 600 nm and normalized relative to a DMSO-treated control, which was considered to represent 100% bacterial growth.
Results
Inhibitor Identification
We used a high-throughput spectrophotometric screen to identify inhibitors of the reaction catalyzed by PpAroE ( Fig. 1 ). Our combined screening libraries contained more than 5500 natural compounds, drug molecules, and other bioactive chemicals. Each compound was screened at a final concentration of 2.5 µM. Screening was performed in the presence of a saturating concentration of NADP+ (2 mM) in an attempt to avoid identification of compounds that interfere with cofactor binding, as these inhibitors may show broad specificity against other NAD(P)+-using enzymes. Conversely, shikimate was fixed at the nonsaturating concentration of 100 µM during screening to facilitate identification of inhibitors that may act in a competitive manner with respect to the enzyme’s substrate.
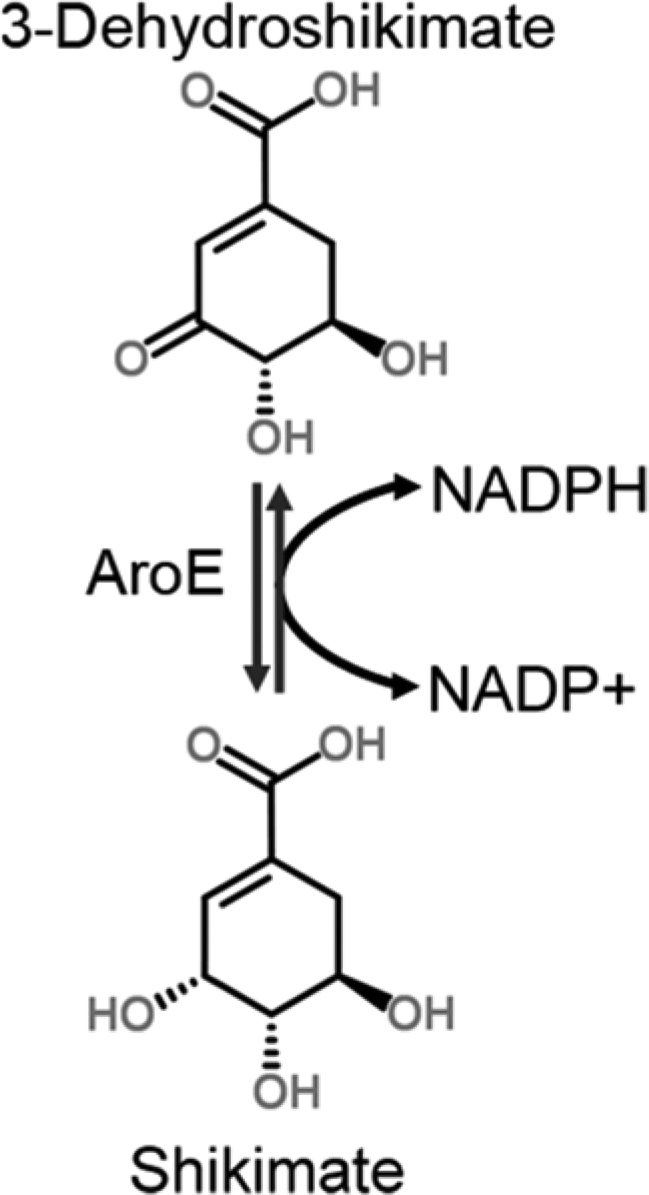
The reaction catalyzed by AroE. The production of NADPH (reverse reaction) was monitored spectrophotometrically in the presence of potential inhibitors.
Using this approach, we identified a total of 24 compounds that caused apparent reductions in the PpAroE-catalyzed reaction rate of at least 25% compared with a DMSO-treated control. The complete collection of inhibitors identified in this screen is shown in Table 1 . Notably, we identified a group of structurally related polyphenolic inhibitors. These compounds share a similar flavanol backbone, and many contain at least one molecule of gallate attached via an ester linkage. Two such compounds, EGCG and ECG, caused >75% inhibition of PpAroE under the screening conditions. Upon further analysis with shikimate and NADP+ held at concentrations reflecting their Km values, 15 we calculated IC50 values of 3.0 ± 0.2 µM and 3.7 ± 0.5 µM for EGCG and ECG, respectively ( Fig. 2 ).
Inhibitors of PpAroE Identified by In Vitro Screening.
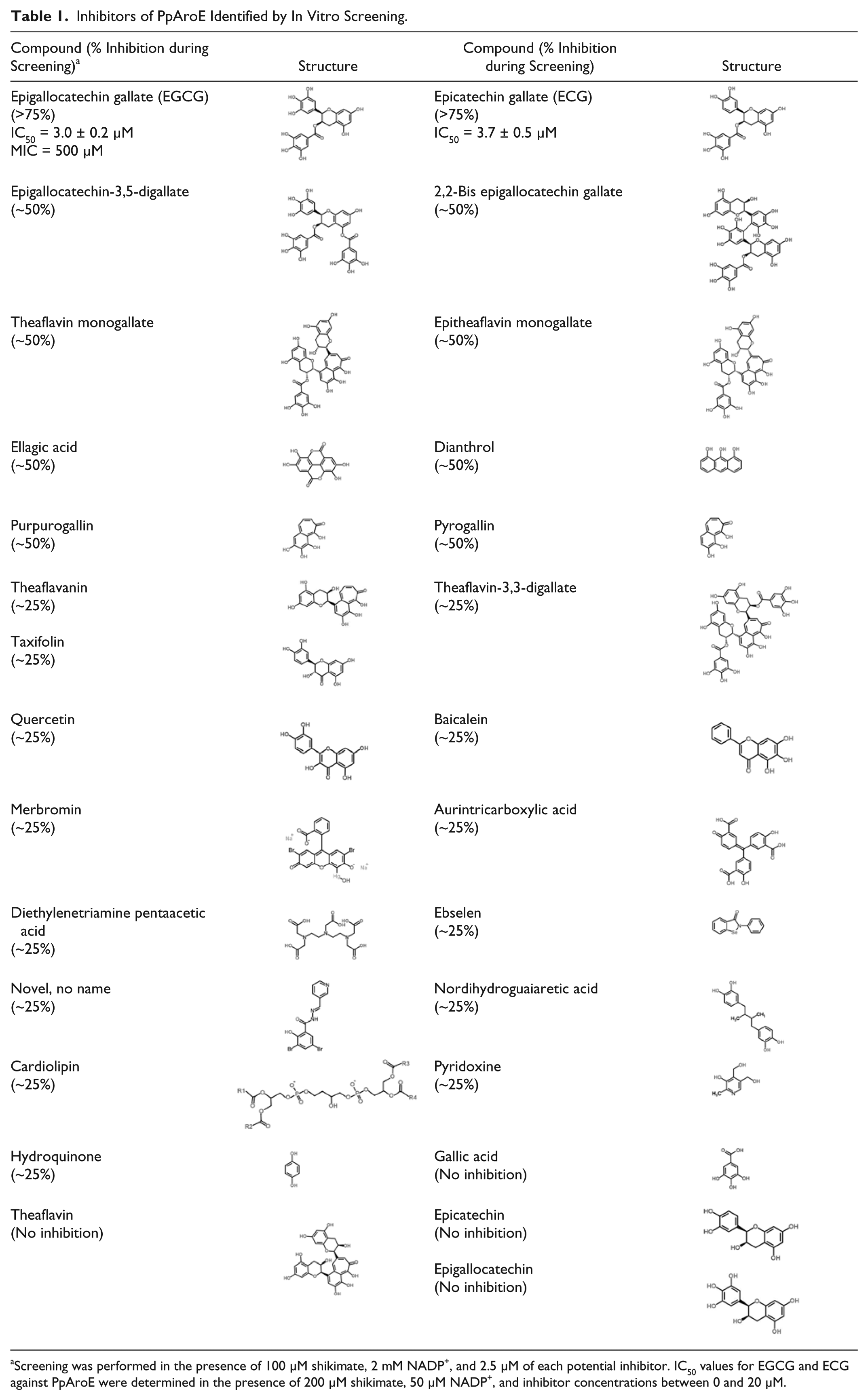
Screening was performed in the presence of 100 µM shikimate, 2 mM NADP+, and 2.5 µM of each potential inhibitor. IC50 values for EGCG and ECG against PpAroE were determined in the presence of 200 µM shikimate, 50 µM NADP+, and inhibitor concentrations between 0 and 20 µM.
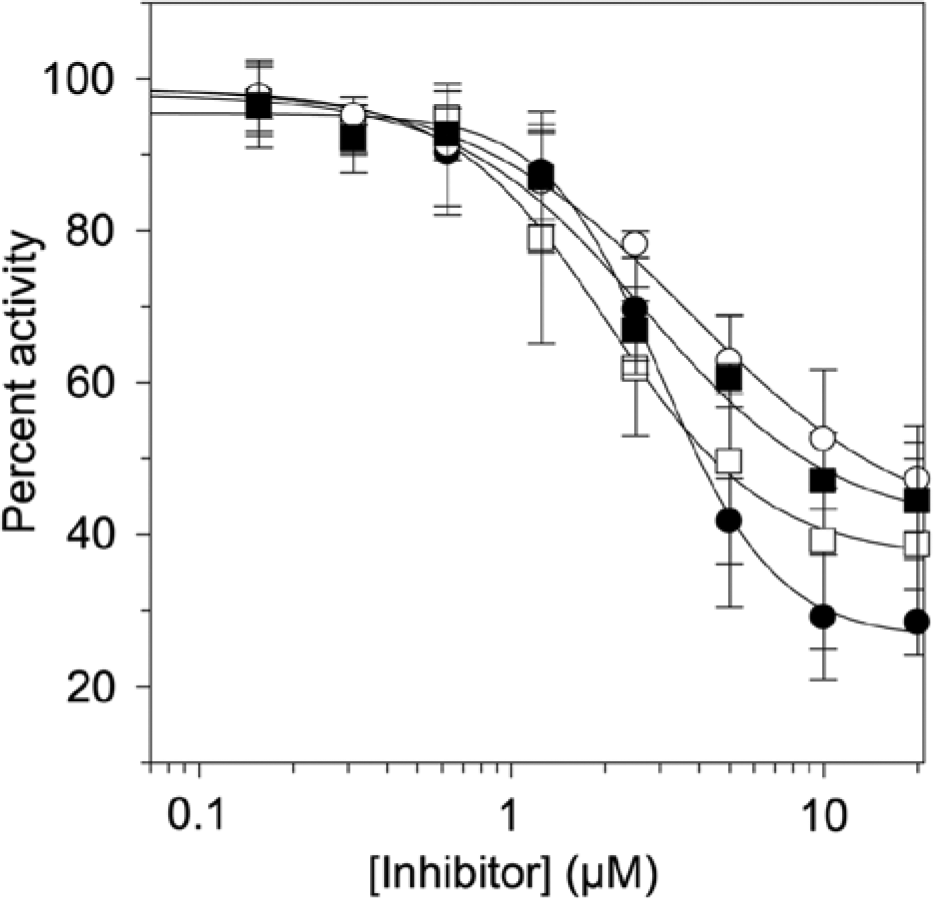
Inhibition of PpAroE by epigallocatechin gallate (EGCG) (•) and epicatechin gallate (ECG) (○) and inhibition of the AroE domain of Arabidopsis thaliana DHQ-SDH by EGCG (■) and ECG (□). Data points represent the average of three trials. Error bars represent the standard deviation.
In addition to EGCG and ECG, a closely related group of polyphenols, including epigallocatechin-3,5-digallate, 2,2-bisepigallocatechin gallate, theaflavin monogallate, epitheaflavin monogallate, theaflavanin, theaflavin-3,3-digallate, taxifolin, quercetin, and baicalein, inhibited the activity of PpAroE by 25% to 50% ( Table 1 ). Other inhibitors discovered in our screen included ellagic acid, dianthrol, purpurogallin, and pyrogallin, all of which reduced the apparent turnover rate of PpAroE by ~50% ( Table 1 ). Additional, lower-potency inhibitors included merbromin, aurintricarboxylic acid, diethylenetriamine pentaacetic acid, ebselen, nordihydroguaiaretic acid, cardiolipin, hydroquinone, pyridoxine, and a novel dibrominated small molecule. The presence of these compounds reduced the turnover rate of PpAroE by ~25% during screening ( Table 1 ).
Inhibitor Sensitivity of A. thaliana DHQ-SDH
While P. putida is not a pathogenic bacterium, PpAroE exhibits significant primary sequence similarity with the AroE enzymes from a number of clinically important species. For instance, PpAroE shares ~50% amino acid sequence identity with the AroE enzyme from enterohemorrhagic E. coli O157:H7 and ~70% sequence identity with the AroE enzyme from the opportunistic human pathogen, Pseudomonas aeruginosa ( Fig. 3 ). In particular, these enzymes share a strictly conserved collection of active site residues, which includes residues involved in substrate and cofactor binding as well as those required for catalysis ( Fig. 3 ).

Amino acid alignment of AroE orthologs. Sequences are from Pseudomonas putida, Aquifex aeolicus, Escherichia coli, Mycobacterium tuberculosis, Pseudomonas aeruginosa, Staphylococcus epidermidis, Thermus thermophilus, Neurospora crassa (as part of the fungal pentafunctional AROM polypeptide), and Arabidopsis thaliana (as part of the plant bifunctional DHQ-SDH complex). Percent identities relative to the P. putida sequence (shown in parentheses) were determined using the ClustalW feature at http://bar.utoronto.ca. Shikimate binding residues are outlined in black; catalytic residues are underlined; key residues in the cofactor binding domain (glycine-rich loop and NADP+-specifying motif) are outlined in gray.
PpAroE exhibits lower overall sequence identity (~25%) with representative AroE enzymes from plant species, such as the AroE domain of A. thaliana DHQ-SDH ( Fig. 3 ). However, comparative structural analysis of PpAroE (PDB ID: 4OMU) and A. thaliana DHQ-SDH (PDB ID: 2O7S) 14 demonstrates that these enzymes nevertheless share a highly similar 3D fold ( Fig. 4 ). We therefore predicted that the A. thaliana enzyme might show sensitivity to the PpAroE inhibitors identified in our screen. Indeed, we found that EGCG and ECG inhibited the A. thaliana AroE domain with IC50 values of 2.1 ± 0.3 µM and 2.0 ± 0.2 µM, respectively ( Fig. 2 ). These values are very close to those calculated for PpAroE, demonstrating that EGCG and ECG are potent inhibitors of both plant and microbial AroE enzymes.
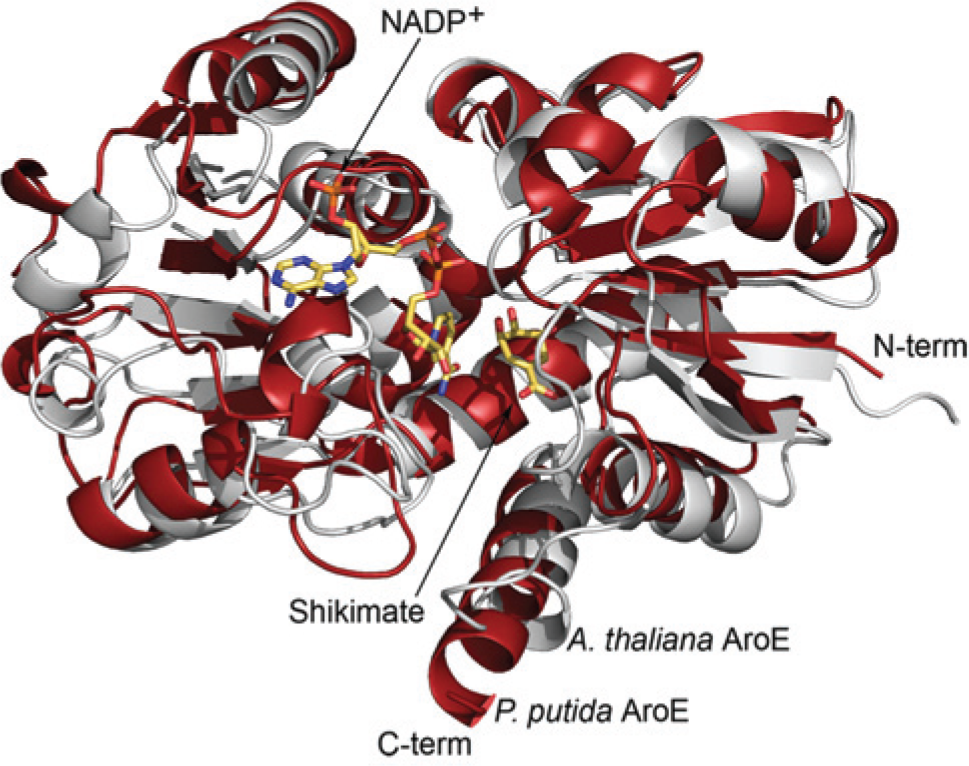
(A) Superimposition of the crystal structures of Pseudomonas putida AroE (red; PDB ID: 4OMU) and the AroE domain of Arabidopsis thaliana DHQ-SDH (gray; PDB ID: 2O7S). Shikimate and NADP+ are present in the active site of the A. thaliana protein.
Antibacterial Activity of EGCG and ECG
Previous studies have reported that EGCG possesses moderate antibacterial properties.18–20 We evaluated the effects of EGCG, as well as ECG, on the growth of P. putida. Using a broth microdilution method, we determined a minimal inhibitory concentration (MIC) for EGCG of 500 µM ( Fig. 5 ). While ECG also appeared to inhibit the growth of P. putida, concentrations up to 1 mM resulted in only ~40% lower growth compared with a solvent-treatedcontrol ( Fig. 5 ).
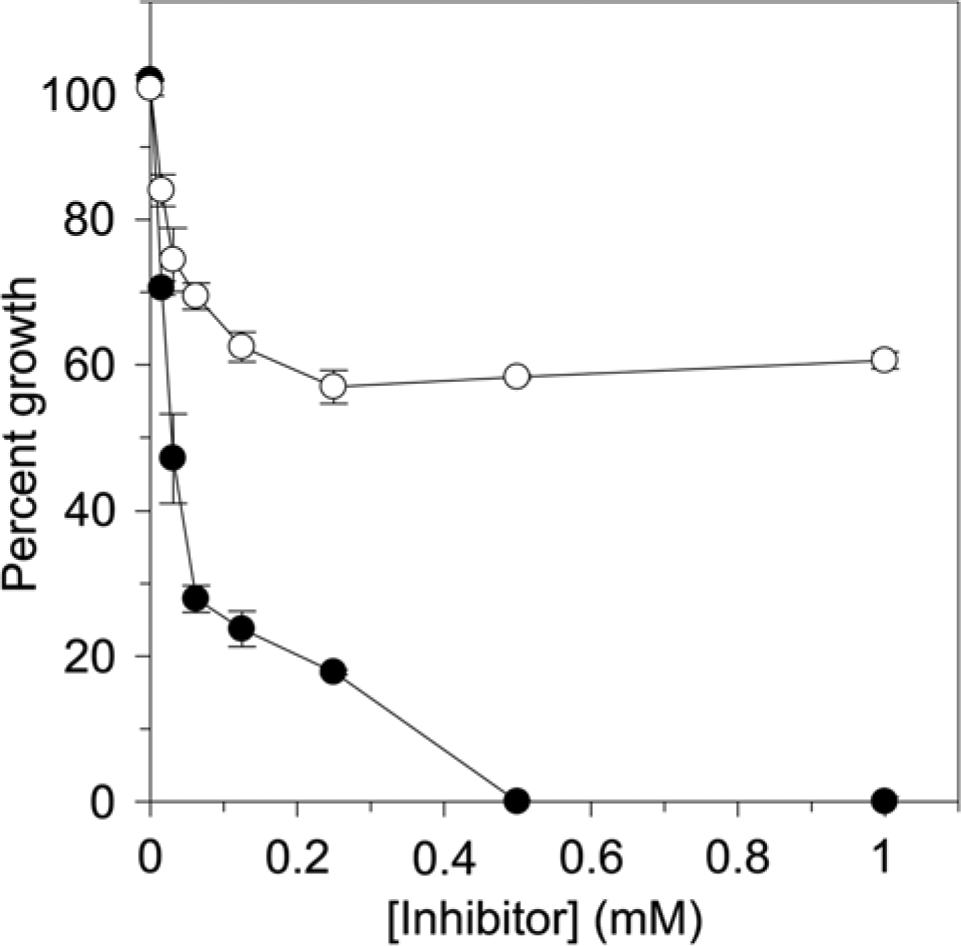
Inhibition of Pseudomonas putida growth by epigallocatechin gallate (EGCG) (•) and epicatechin gallate (ECG) (○). Values were normalized relative to a DMSO-treated control, which was considered to represent 100% growth. Data points represent the average of three trials. Error bars represent the standard deviation.
Discussion
AroE represents an appealing target for herbicides and antimicrobial agents due to the enzyme’s essential metabolic role. Indeed, loss-of-function aroE mutations are typically lethal without supplementation with downstream metabolites. 21 However, only a small number of previous studies have successfully identified inhibitors of the enzyme. Han et al. 10 found five compounds that inhibit Helicobacter pylori AroE with low micromolar IC50 values. Recently, Hsu et al. 11 used a novel pathway-based screening approach to discover a group of inhibitors that exhibited IC50 values <10 µM against the H. pylori enzyme. Our own analysis of more than 5500 compounds identified a group of plant-derived polyphenolic inhibitors, none of which had previously been shown to inhibit AroE ( Table 1 ).
Two compounds identified in our screen, EGCG and ECG, were particularly potent inhibitors of PpAroE. Other related inhibitors we identified included the larger molecules, epigallocatechin-3,5-digallate, 2,2-bisepigallocatechin monogallate, theaflavin monogallate, epitheaflavin monogallate, and theaflavin-3,3-digallate ( Table 1 ). The reduced potency of these chemicals in comparison to EGCG and ECG suggests that the size of the polyphenol backbone and the number of galloyl groups are important determinants of inhibitor strength.
At least one gallate ester appears to be indispensible for strong inhibition by this group of compounds, as a number of polyphenols lacking this moiety, such as theaflavin, epicatechin, and epigallocatechin, were among the chemicals in our libraries that failed to inhibit PpAroE ( Table 1 ). The related molecules, theaflavanin, taxifolin, quercetin, and baicalein, were relatively weak inhibitors ( Table 1 ).
Gallate itself, which has a highly similar chemical structure to the substrate of PpAroE, shikimate, did not exhibit significant inhibitory activity ( Table 1 ). Moreover, a variety of other benzoic acids with alterations in the number and position of attached hydroxyl groups, or those containing halogen or methyl groups, were present in our screening libraries but did not inhibit PpAroE. These findings demonstrate that both a galloyl moiety and a flavanol backbone are necessary for strong inhibitory activity.
Our screen also uncovered inhibitors containing a range of other chemical scaffolds ( Table 1 ). Purpurogallin and pyrogallin, whose structures contain an interesting seven-membered ring, were intermediate-potency inhibitors of PpAroE. Two of the weaker inhibitors identified, pyridoxine and hydroquinone, bear moderate structural resemblance to shikimate. Others, such as merbromin, aurintricarboxylic acid, diethylenetriamine pentaacetic, ebselen, cardiolipin, and nordihydroguaiaretic acid, represent additional unique chemical scaffolds ( Table 1 ). Detailed analysis of the inhibitory activities of these compounds will be the basis of future studies.
We subjected the two strongest inhibitors identified in our screen, EGCG and ECG, to further analysis. We calculated IC50 values of 3.0 ± 0.2 µM and 3.7 ± 0.5 µM for EGCG and ECG, respectively, against PpAroE ( Fig. 2 ). In addition, both compounds showed nearly identical inhibitory activity against the AroE domain of A. thaliana DHQ-SDH (IC50 EGCG = 2.1 ± 0.3 µM; IC50 ECG = 2.0 ± 0.2 µM; Fig. 2 ). The comparable response of PpAroE and A. thaliana DHQ-SDH to these inhibitors can be rationalized based on the highly similar 3D structures of the enzymes. Superimposition of the experimentally determined crystal structure of PpAroE (PDB ID: 4OMU) with that of the AroE domain of A. thaliana DHQ-SDH (PDB ID: 2O7S) 14 reveals virtually complete conservation of secondary structural elements ( Fig. 4 ). Like other AroE enzymes, the structure of PpAroE is characterized by a pair of α/β domains that lie on either side of a cleft that contains the enzyme’s active site. The N-terminal domain contains the majority of residues required for substrate recognition, while the C-terminal domain forms a Rossmann fold responsible for binding the cofactor, NADP+. Mainchain alignment of the PpAroE structure with the structures of AroE orthologs from A. thaliana (PDB ID: 2O7S), Staphylococcus epidermidis (PDB ID: 3DON), and Thermus thermophilus (PDB ID: 1WXD) produces RMSD values of 2.7 Å, 1.9 Å, and 1.7 Å, respectively, demonstrating the high level of structural similarity of the enzymes. We hypothesize that this structural conservation extends to the binding sites for EGCG and ECG, which we are presently attempting to identify by cocrystallization of the compounds with PpAroE.
Previous studies have demonstrated that EGCG exhibits moderate antibiotic activity against a number of bacterial species, including E. coli, 20 H. pylori, 18 and Acinetobacter baumannii. 19 Consistent with these reports, we determined an MIC for EGCG of 500 µM against P. putida ( Fig. 5 ). In comparison, we found that ECG was maximally capable of reducing the growth of P. putida by ~40% at concentrations up to 1 mM. It would be interesting to understand how the subtle structural differences between these two compounds afforded such distinct growth inhibition profiles.
In addition to their antimicrobial properties, a number of other biological activities have been previously ascribed to EGCG and ECG. Studies have investigated the potential benefits of their use in the treatment of a range of diseases, including human immunodeficiency virus infection and various cancers.22–24 Interestingly, both compounds are found in considerable quantities in green tea and therefore have significant appeal as nontoxic therapeutic agents.
While the present study represents the first report describing the inhibition of AroE enzymes by EGCG and ECG, these compounds have been identified as in vitro inhibitors of a number of other enzymes. These enzymes include fatty acid synthase (FAS),20,25 squalene epoxidase, 26 steroid 5α-reductase, 28 and glutamate dehydrogenase (GDH).28,29 In the case of FAS, EGCG and ECG are low micromolar inhibitors of the NAD(P)H-dependent reductase components but not of the elongation-condensing domain. 20 This finding suggests that inhibition by these polyphenols is specific for NAD(P)H-binding proteins.
The specificity of EGCG and ECG for enzymes that use a dinucleotide cofactor has been further shown by their ability to inhibit squalene epoxidase, a monooxygenase that requires both NADPH and FAD, but not other cholesterol biogenesis enzymes such as lanosterol cyclase or lanosterol 14-demethylase. 26 With respect to NAD(P)H-dependent GDH, EGCG and ECG both exhibit sub-micromolar IC50 values. 28 Inhibition of the enzyme by EGCG appears to have significant physiological effects, as the compound is capable of altering levels of insulin secreted by pancreatic β-cells. 28
Our finding that EGCG and ECG are potent inhibitors of AroE enzymes fits the hypothesis that these compounds recognize a conserved feature of dinucleotide-binding proteins. Indeed, the C-terminal Rossmann fold observed in the structures of AroE enzymes ( Fig. 4 ) is a common structural motif among NAD(P)H-dependent enzymes. However, the precise mechanism by which EGCG and ECG affect these proteins is not well established. For the reductase components of FAS, EGCG and ECG have been reported to act as competitive or mixed-type inhibitors with respect to the cofactor, NAD(P)H.20,25 Conversely, for GDH, EGCG was reported to interact with the enzyme in a noncompetitive manner with respect to both its cofactor and substrate. 28 Consistent with this result, structural analysis of GDH in complex with ECG suggests that the compound binds to an allosteric site outside of the enzyme’s active site. 29
Similar to the case of GDH, our own mechanistic analysis of EGCG and ECG using PpAroE did not obey “classical” competitive or mixed-type inhibition models (data not shown). While inhibition of PpAroE by EGCG and ECG is likely to involve recognition of a conserved feature of the C-terminal cofactor binding domain, a detailed description of this interaction awaits the structural characterization of the enzyme in complex with these inhibitors.
Due to the large number of enzymes that use NAD(P)H as a cofactor, the potentially broad specificity of EGCG and ECG must be considered if they are to be applied as enzyme inhibitors in an in vivo context. Nevertheless, these compounds and the others identified in our screen may represent useful chemical scaffolds that can be modified in the future to obtain greater specificity for AroE enzymes.
Footnotes
Acknowledgements
The authors thank the Centre for the Analysis of Genome Evolution and Function (University of Toronto, Toronto, Canada) for providing access to the chemical libraries used in this study. We also thank Dr. Vivian Saridakis (York University, Toronto, Canada) for use of her x-ray generator for preliminary screening of PpAroE crystals.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: D.C. receives funds from a Discovery Grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada.