
Abstract
Protein kinases C (PKC) modulate the activity of the Kv11.1 ion channel current (hERG). However, the differential effects of specific PKC subtypes on the biophysics of the channel are unknown. The pharmaceutical tools to selectively modulate PKC subtypes are not membrane permeable and must be added directly to the intracellular solution in electrophysiology studies. Here, the PatchXpress electrophysiology robot was used to voltage clamp up to 16 cells simultaneously yet asynchronously across individual Sealchip chambers. The precision afforded by repeats of automation procedures minimized the experimental errors typical of these assays. Eight well-known PKC selective peptidomimmetics and general synthetic modulators were used to modulate the protein-protein interactions between hERG and the major PKC subtypes. We identified a specific role for the PKCε inhibitory peptidomimmetics in decreasing PKC-induced hERG τ activation (80%) and half-maximum activation voltage (90%) at steady state; a specific PKCε activator exhibited the opposite effect. Disruption of PKCβ, PKCα, and PKCη interactions also showed significant effects albeit of lower magnitudes. The effect of PKCδ inhibitor was only marginal. A significant correlation was observed between the shifts in τ activation and half-maximum voltage at steady state (R2 = 0.85). Peak current amplitudes and time constant of deactivation remained unaffected in all conditions.
Keywords
Introduction
The protein kinase C (PKC) family comprises more than 10 different isozymes grouped in three general subtypes. Subtypes classification is based on structural similarities and the susceptibility of the PKC regulatory C-terminal to typical modulators such as Ca2+ and diacylglycerol (DAG). 1 Briefly, the current classification system goes as follows. Conventional PKCs (cPKC) are made of PKCα, PKCβ (PKCβ1, PKCβ2), and PKCγ, which are all activated by DAG and Ca2+. The novel PKCs (nPKC) include PKCδ, PKCε, PKCθ, and PKCη, which are also activated by DAG but lack direct modulation by Ca2+. A third subtype comprises the atypical PKCs (aPKC) and includes PKCλ/ι and PKCζ, which are not activated by either Ca2+ or DAG. 1
Typically, kinases exert their signaling activity either directly via a phosphorylation of the final effector protein or indirectly via modulation of intermediate factors. As part of a vast cellular kinome, PKCs are critically involved in the signaling of vital physiological responses, including inflammatory, 2 (auto)immune responses, 3 tumor progression, 4 and cardiovascular functions. 5 PKCs are cytosolic enzymes, although, once activated, they may translocate to the cell membrane using the membrane anchoring receptors for activated C-kinases (RACK). 6 There, they phosphorylate membrane-bound and integral proteins, including several voltage-gated ion channels (VGICs; see below).
A new field of research is dedicated just to the study of ion channel phosphorylopathies and focuses on understanding the detailed modulations of the biophysical properties of ion channels by protein kinases, many of these being directly relevant to pathological states including LQT1 and 2 syndromes. 7 Although the differential roles of the specific PKC isoforms remain largely unresolved, the roles of the PKC family on VGIC regulation have been studied on numerous occasions using recombinant systems expressing neuronal Nav1.8, 8 cardiac Kv4.3, 9 Nav1.5, Cav1.2, 10 and Cav1.3, 11 as well as in recombinant systems expressing the human Kv11.1, which is the focus of the present study. 12
The long QT syndrome, a channelopathy of primarily Kv11.1 and Kv7.1, 13 can lead to the development of “Torsades de Pointes” when the cardiac action potential fails to repolarize and immediately stops the heart in tetanus, eventually causing sudden death if left uncorrected. It has long been known that PKC modulates Kv11.1, 14 although to the best of our knowledge, no data are currently available as to the roles of specific PKC isoforms and their differential implications in regulating the channel biophysics.
In this context, our study is a further attempt to contribute to the understanding of the roles of PKC isoforms on Kv11.1 modulation. Here, we evaluated the effects of selective inhibitory peptides modulating six PKC isoforms (PKCα, PKCβI, PKCβII, PKCδ, PKCη, and PKCε) and those of one nonselective and one subtype-specific PKC activator (for PKCε) in regulating some of the Kv11.1 electrophysiological properties. Since the addition of membrane-impermeable inhibitory peptides must be done via the intracellular solution, this procedure presents a challenge for many automation devices. Automated high-throughput systems gaining whole-cell access via perforated patch methods or achieving seal values in the megohm range would be of limited use in these cases. In turn, the PatchXpress (Molecular Devices, Sunnyvale, CA) instrument delivers an ideal balance of automation benefits with fully compensated gigaseal values and asynchronous independent single-cell recordings, qualitatively similar to manual patch clamping. The automated PatchXpress system has a maximum throughput of 16 parallel recordings that is well adapted to detailed electrophysiological experiments.
Materials and Methods
Peptides and Chemicals
Phorbol-12-myristate-13-acetate (PMA) was obtained from Sigma-Aldrich (St. Louis, MO) and was tested at a final concentration of 10 nM in the extracellular solution. Since our polypeptides would not efficiently cross the cell membranes, all six PKC subtype-specific antagonist peptides (εV1-2, αC2-4, βIV5-3, βVII5-3, δV1-1, and ηV1-2) and the activator peptide (εV1-7) were added at a 100-nM final concentration in the intracellular solution and had greater than 90% purity. Subtype-specific PKC antagonists were custom made at Sheldon Biotechnology, McGill University, Montréal, Quebec, Canada. The sequence and properties of PKC subtype-specific peptides were reported previously.15,16 The peptide εV1-2, like all other PKC isoform selective peptides used here, acts negatively on PKCε by binding to the RACK complex and preventing translocation of the kinases to the cell membrane. The activator εV1-7 instead binds directly to PKCε to increase its intrinsic activity. Practically, the PKCε activator peptide is the only subtype-specific activator peptide reported to date whose activity has been validated by different groups (including ours).
Electrophysiology
Experiments were conducted using the PatchXpress 7000A electrophysiology robot (Molecular Devices) using the Sealchip substrate developed by Aviva Biosciences (AVIVA Biosciences Corp., San Diego, CA). Each well of the Sealchip received between 30,000 and 2,000,000 cells suspended in the 8- to 10-µL extracellular solution maintained in each chamber. Cells were selected for study if they met the following criteria: (1) a gigaseal was achieved (RSeal > 1 GΩ) prior to whole-cell access and within 20 s of cell addition to the Sealchip; (2) whole-cell access was attained using suction, resulting in a membrane resistance (Rm) > 200 MΩ and access resistance (Ra) <10 MΩ; and (3) whole-cell capacitance (Cm) was >4 pF, and peak whole-cell current (IhERG) was >0.1 nA. Throughout an experiment, cells were eliminated if Ra rose above 20 MΩ or Rm dropped below 200 MΩ. Holding potential was set to −80 mV prior to attempting whole-cell access and held throughout each experiment.
The whole-cell currents were digitally sampled at 1 kHz with an analog bandwidth ratio of 5 (200 Hz). The voltage clamp waveforms are reported with their respective data in the figures or in the text. In all time course experiments, Kv11.1 monitoring pulses were delivered once every 10 s. In each trace current, the peak current recorded during deactivation of Kv11.1 (tail current) was corrected for linear leak currents by subtracting the leak currents present in a short 10-ms voltage step to −50 mV (prestep), applied immediately prior to activating the Kv11.1 current (the depolarization step). The electrophysiological parameters of all patched cells used in the study are shown in the supplementary material (the >80 cells retained for this study represented about 35% of all recordings).
The following sequence made up the experimental procedure carried out on each cell asynchronously and with independent feedback control. (1) After whole-cell access, the access was verified for 2 s, and then each cell was monitored with a 12-ms, 16-mV square pulse centered on a −80-mV holding potential for 2 to 120 s until the membrane resistance and access resistance were stable; most cells were stable within 10 s. (2) Upon determination of stable voltage clamp parameters, waveform protocols were applied to determine activation voltage dependence and tail current voltage dependence, and then (3) a monitoring pulse was applied continuously to monitor the stability and amplitude of the whole-cell tail currents. Cells with insufficient tail current amplitudes (as described above) were terminated before any subsequent steps were performed. (4) For cells that met all criteria for inclusion in the study, the first dose of PMA activation was added to the extracellular solution. Each compound addition step comprised two sequential additions of 45 µL of extracellular solution containing the final concentration of PMA, spaced 11 s apart and delivered at 20 µL/s. During each compound addition, the chamber volume in the test chamber was maintained at 10 µL by suction applied at the other side of the test chamber from the point of application. Each compound addition replaced at least 95% of the solution in the extracellular chamber, for an overall solution exchange of 99.8% of the extracellular solution in the test chamber. (5) The monitoring pulse was repeated until the effect of the PMA addition achieved steady state as judged by the last eight data points in the time course having a standard deviation less than 2 pA or a slope presenting with less than 0.1% variance. (6) Once steady state was achieved, the test waveform/protocols for activation voltage dependence and tail current voltage dependence were repeated. In addition, a dual-step waveform protocol with varying activation time was applied to monitor the time constant for onset of activation of the current. (7) Finally, the procedure was marked as completed, but the monitoring pulse was again applied repeatedly for an additional 5 min or until all independent procedures running in parallel on the same Sealchip were also marked as completed.
Buffers and Solutions
The extracellular solution for electrophysiological recordings (ES) contained (in mM) the following: 150 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose, with pH 7.3 (NaOH, 5M) and osmolality 310 ± 5 mOsm. The intracellular solution for electrophysiological recordings (IS) contained (in mM) the following: 140 KCl, 2 MgCl2, 10 HEPES, 6 EGTA, and 5 ATP, with pH 7.2 (KOH, 1M) and osmolality 300 ± 5 mOsm. Test compounds were added to ES or IS immediately before starting an experiment, which was 5 to 10 min before the first recordings for IS compounds and 2 to 3 min before the first recordings for ES compounds. All data were collected at ambient temperature, which was monitored between 22 and 24 °C. We prepared eight different intracellular solutions corresponding to each of the six intracellular PKC inhibitory peptides (in combination with PMA), the selective PKCε activator peptide (in the absence of PMA), and a blank that did not contain either PMA or any peptides (basal). PMA, a nonselective PKC activator that is membrane permeable, was added to the extracellular solution in a compound addition step initiated after the baseline Kv11.1 current had stabilized. All compounds were subsequently allowed to equilibrate for 10 min as indicated elsewhere. 17 The basal condition represents the addition of buffered extracellular solution in the absence of any modulator compounds.
Cell Preparations
HEK-293 Kv11.1 cells (AVIVA Biosciences) were cultured in an incubator maintaining 37 °C and a 5% CO2 environment, in T-75 flasks (VWR, Radnor, PA) containing 15 mL Dulbecco’s modified Eagle’s media (DMEM) with F-12 nutrient supplement and with fetal bovine serum (FBS; 10% v/v), and 5 mL penicillin/streptomycin antibiotics and a “selector antibiotic” intended to kill nonexpressing cells were added to maintain sterility and hERG expression (Life Technologies, Grand Island, NY). Cells were maintained in the presence of the antibiotics, except for the last passage before electrophysiological recordings.
For passaging and for electrophysiological experiments, cells were seeded at 2 million cells per flask for next-day use, 1 million cells per flask for second-day harvest or passaging, or 0.5 million per flask for third-day passaging (after a weekend). Cells were harvested by the following procedure. (1) The culture media was removed with a transfer pipette and quickly rinsed twice with 5 to 10 mL of calcium- and magnesium-free Dulbecco’s phosphate-buffered saline solution (DPBS; VWR). (2) After 1 min of total exposure, the solution was removed and replaced with 5 mL of a 1:3 Accumax enzyme solution (Innovative Cell Technologies, San Diego, CA) in calcium- and magnesium-free DPBS and placed back into the incubator for 2 to 5 minutes. (3) On completion, the digestion enzymes were quenched with three volumes of 10 mL calcium- and magnesium-free DPBS containing 5% FBS and delivered across the flask surface to help dislodge the remaining cells. Each 10-mL addition was removed and collected into a single 50-mL plastic centrifuge tube. (4) The tube was then centrifuged at 600 to 800 g relative centrifugal force (RCF) (~14 cm radius) for 1 min and the supernatant removed. (5) The pellet was immediately resuspended using Dulbecco’s PBS that contained a full complement of calcium and magnesium (DPBSc) to yield approximately 1 million cells per milliliter (approximately 5–8 mL). (6) The tube, containing the cells in DPBSc, was temporarily stored at 37 °C and tilted, to reduce sedimentation pelleting, until the cells were needed for the experiments. It is important to note that exposure of the cells to the calcium- and magnesium-free media (steps 1 and 2) should be limited to a maximum of 5 min to preserve long-term membrane stability in the whole-cell mode. In addition, the small amount of calcium contributed by the 5% FBS was intended to stimulate closure of gap junctions before overloading the cells with a full complement of calcium.
It is also important to note that pellets should be promptly, yet gently, resuspended to preserve gigaseal success rate. At the start of each experiment, a 1.5-mL aliquot of cells was collected in an Eppendorf tube and centrifuged 40 to 45 s at 800 to 1000 g RCF (7 cm radius) and resuspended into 140 µL of fresh DPBS containing calcium and magnesium. This suspension was added to the PatchXpress robot for direct use on the Sealchips.
Statistical Analysis
Data from all experiments were analyzed using DataXpress 2.0 (Molecular Devices) and ClampFit 10.2 (Molecular Devices). Single exponential, Gaussian, and Boltzmann curve fitting, as well as one-way analysis of variance (ANOVA), checked with Bartlett’s homogeneity of variance test and further drilled down with Dunnett’s multiple comparisons post hoc test, were all performed with Prism 5.0 (GraphPad Software, La Jolla, CA). Statistical significance was assumed when a confidence level of at least >95% was achieved (p < 0.05). Results are displayed as mean ± standard error (SE), with counts in parentheses (n).
Results
Effect of PKC Modulators on Kv11.1 Tail Current Amplitudes at Steady State
Figure 1a shows a representative trace current of Kv11.1 in transfected HEK cells. The Kv11.1 current was elicited by a classical two-pulse protocol from a holding potential at −80 mV. As the peak current comprises a balance between the activation time course and the time course of voltage-dependent inactivation, the current from the second pulse (tail current) is used as a more robust indicator of the time course of Kv11.1 amplitude. Aside from the Kv11.1 current, a small transient current from the background was apparent only in the first milliseconds of the initial depolarization to +40 mV, likely attributable to a small endogenous sodium conductance that does not have any measurable deactivating current and therefore would not contribute to the measurements of the tail currents. Aside from noting its occasional presence and negligible amplitude, we made no further attempts to characterize the inward transient.
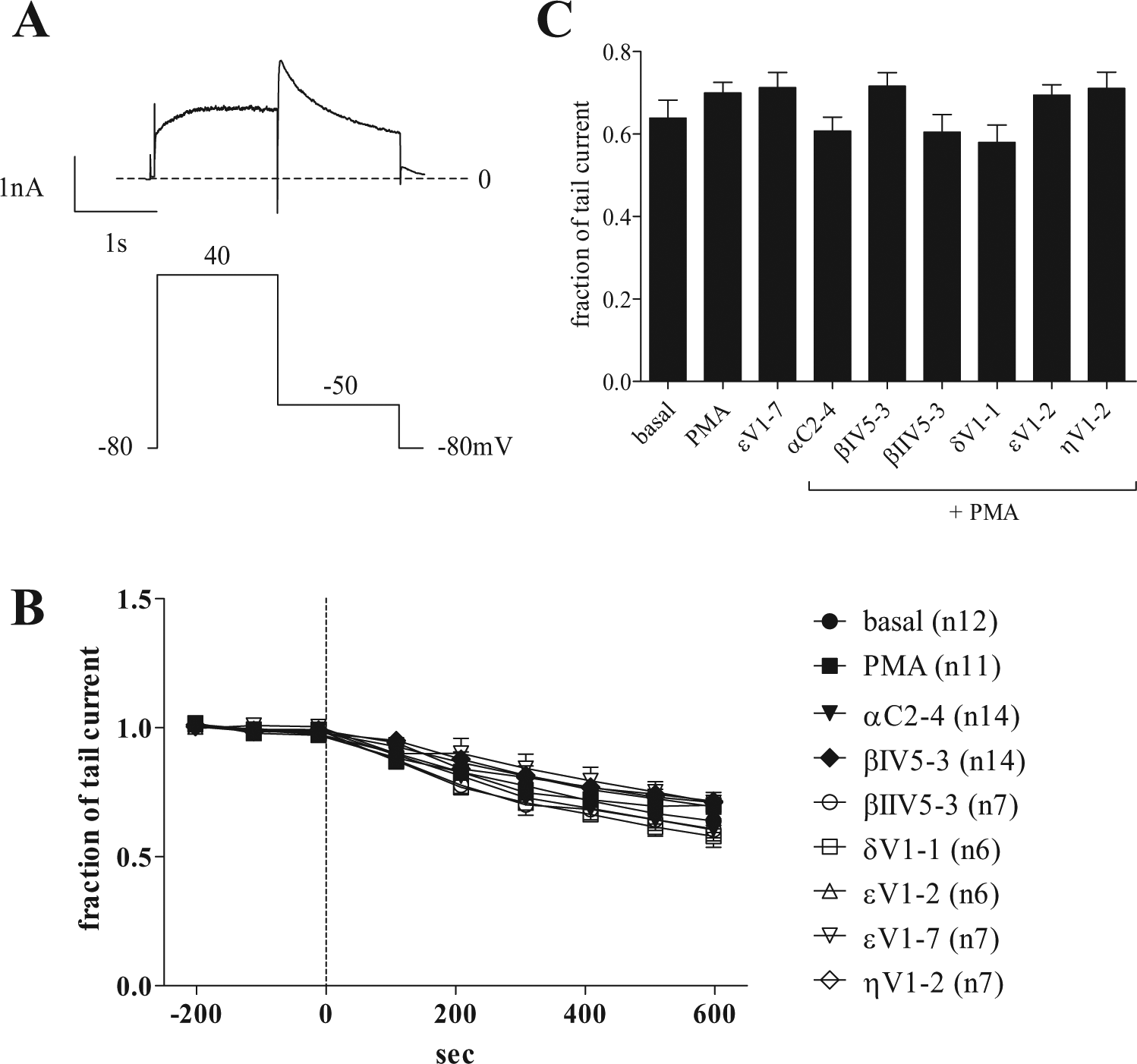
Monitoring of Kv11.1 tail current. (
To begin, we monitored the progression of Kv11.1 tail current amplitude over time. Currents were normalized to the initial current amplitude after baseline stabilization and throughout a 10-min incubation time in the presence of the PKC modulators described below ( Fig. 1b ). At zero time point, after baseline stabilization, the membrane-permeable and general PKC activator, PMA, was included directly in the extracellular solution to produce PKC-dependent activation during prior incubation with each of the six isoform-specific PKC inhibitory peptides present in the intracellular solution, similar to methods reported elsewhere.17–19 Nine different conditions were tested: (1) basal level represents a negative control with the addition of buffer alone without PMA and without modulators in the intracellular solution; (2) PMA represents the PKC-activated state, induced by the addition of buffer with PMA but without modulator peptides in the intracellular solution; (3–8) the same PMA-induced activation but now in the continuous presence of each of six intracellular PKC isoform-specific inhibitors: αC2-4 (PKCα), βIV5-3 (PKCβ1), βIIV5-3 (PKCβ2), δV1-1 (PKCδ), εV1-2 (PKCε), and ηV1-2 (PKCη); and finally (9) εV1-7 represents the addition of buffer without PMA but in the continuous presence of a specific PKCε activator peptide (εV1-7) in the intracellular solution.
Figure 1c compares the tail currents recorded at the end of the 10-min incubation time with PKC modulators or in control conditions. The currents are normalized to the current present at the end of the initial stabilization period. We noted no significant differences in the mean peak current amplitudes using one-way ANOVA analysis across all nine different groups (F = 1.6, R = 0.15). Dunnett’s post hoc analysis was used to run multiple comparisons between groups using either PMA or the basal condition as control groups, yielding no differences with any treatment. This set of data does not support a role for the PKC peptides in the modulation of the peak current amplitudes of Kv11.1. It is worth noting that the typical voltage waveform protocol used here to monitor Kv11.1 tail current sets the channel into a fully activated state (see Zhou et al. 20 ). In the fully activated state, any increase in the maximum amplitude of tails will be detected from a potential shift of Kv11.1 along the time course of the activation curve. Nevertheless, as the data show here, no differences were detected in the tail amplitudes measured at steady state.
Effect of PKC Modulators on the Kv11.1 Tail Current Deactivation Kinetics
The time constant of the tail current deactivation (τ deact.) was determined by simple curve-fitting using a mono-exponential decay fit to each sweep’s tail current over the 1.5 s of the repolarization step. Values for τ deactivation were compared at the end of the 10-min incubation time in the presence of the PKC peptides, in the presence of the PMA control, and in basal conditions. Differences in the means across all groups were identified by one-way ANOVA analysis (F = 3.6, R = 0.28). The multiple comparisons done by Dunnett’s post hoc analysis did not identify significant changes compared with the PMA-activated state. However, differences were found for both inhibitory peptides βIIV5-3 + PMA and ηV1-2 + PMA when compared to the control PMA-activated state (p < 0.05). This seems counterintuitive given that PMA activation showed no differences compared with the basal state or with any other inhibitory peptide given in the PMA-activated state (see supplementary material). A possible explanation is that the PKCη or PKCβ2 isoforms would use an alternative pathway to decrease the kinetics of deactivation that does not rely on the preliminary activation of PKCη or PKCβ2 by PMA. This hypothesis falls outside of the scope of the present study, and separate studies are warranted to verify the roles of these two PKC isoforms with regard to PMA activation. In all cases, these effects were minor compared with those observed on the other biophysical parameters measured in this work (see below).
Effect of PKC Modulators on the Activation Gate of Kv11.1 and Shift in Voltage Dependency of Tail Current Activation
Figure 2a displays a classic voltage-dependency protocol applied to Kv11.1. The parameters of the voltage dependency were calculated from the Boltzmann curve fit to the data, as seen in Figure 2c . Voltages of half-activation (V1/2) are reported in the histogram of Figure 2b . A one-way ANOVA with the Boltzmann half-activation voltage (V1/2) shows significant variations between the nine groups (F = 20.34, R = 0.71). Dunnett’s post hoc analysis highlighted significant differences (p < 0.05) between the basal state and all other PKC conditions, except for the PKCε inhibitory peptide with PMA (εV1-2), which was the only condition that did not significantly differ from the basal condition. When Dunnett’s analysis was conducted against the control PMA-activated state, all other conditions proved statistically different, except that of the inhibitory βIIV5-3 + PMA, δV1-1 + PMA, and ηV1-2 + PMA. Here, in contrast to the low-magnitude changes of the time constants of deactivation, very significant differences were seen upon activation and inhibition of the various PKC isoforms. Upon addition of the basal condition, Kv11.1 exhibited V1/2 = −3 ± 2 mV; treatment with PMA resulted in an increased value to V1/2 = +15 ± 1 mV. The amplitude and direction of the shift observed here are consistent with those independently reported with a general PKC inhibitor in another recombinant system. 21 Among the PKC peptides, εV1-2 blocking PKCε, displayed the largest difference in V1/2 values with a V1/2 more than 90% lower than in the PKC-activated state (from 15 ± 1 mV in PMA to 1 ± 2 mV in PMA + εV1-2). By order of decreasing amplitudes, the second most important reduction was observed with the inhibition of PKCβI isoforms (V1/2 = 4 ± 1 mV), showing a 73% lower V1/2 compared with the PMA-activated state. PKCα inhibition showed a 40% reduction (p < 0.0001), while the two remaining inhibitory peptides showed statistically significant differences but of minor amplitudes: PKCη and PKCβII with, respectively, 27% (p < 0.0001) and 13% (p < 0.05) lower values than that of the PMA-activated state. This data set, along with the others, is summarized in Table 1 . PKCε inhibition showed the largest reduction in V1/2. On the other hand, a specific treatment with PKCε activator peptide (εV1-7) showed just the opposite effect. The V1/2 value from a selective PKCε activation alone was equivalent to almost 50% of the PMA signal, potentially indicating an important effect for the PKCε isoform in the downstream modulation of Kv11.1 voltage dependency. The slope factors, in turn, stayed essentially equal across all conditions tested and were no different from that of PMA control or basal condition (k = 10 ± 1 mV).
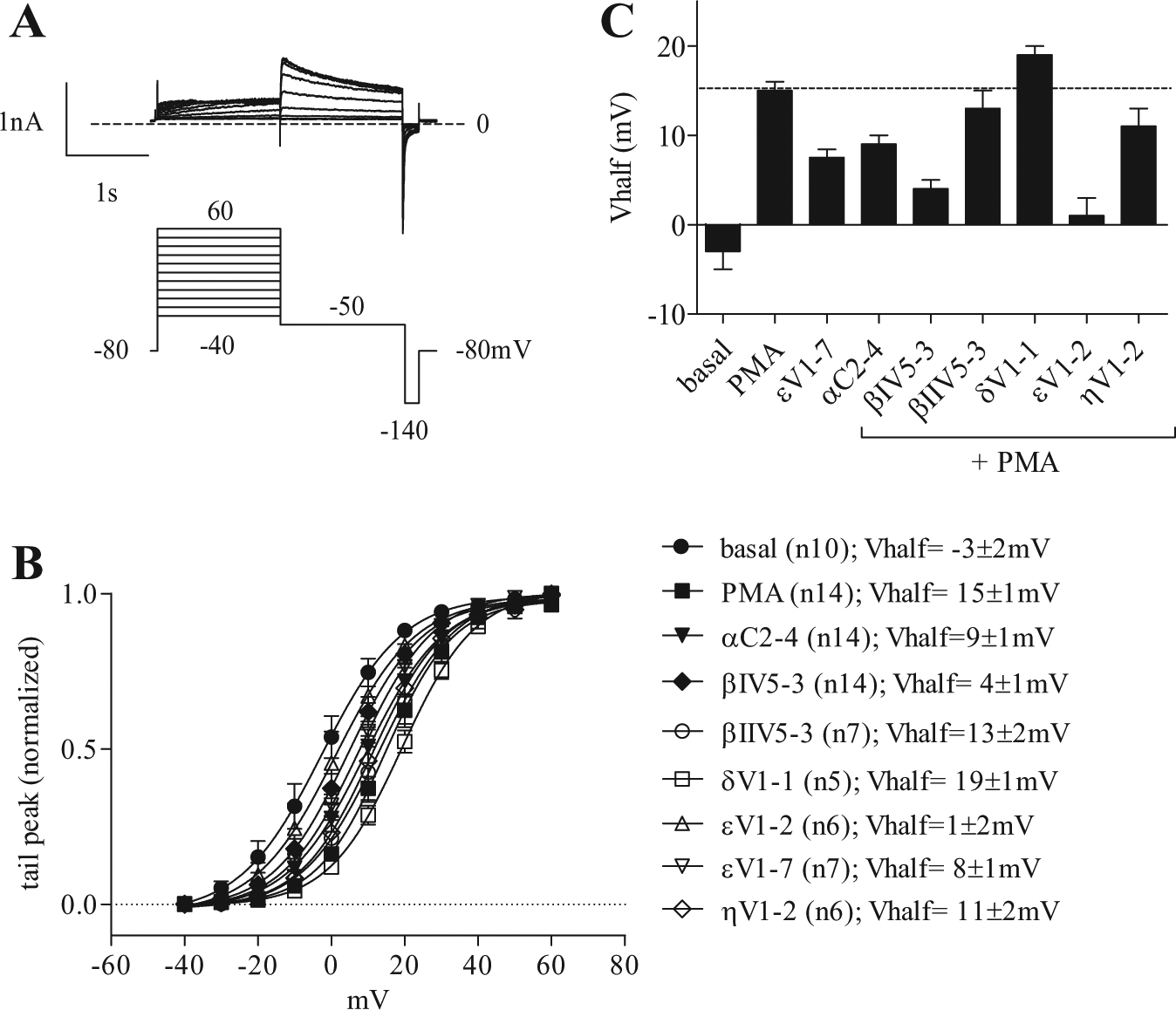
Steady-state activation of hERG tail current. (
Summary Table of the Modulation of Kv11.1 Electrophysiological Properties by PKC Modulator Peptides.
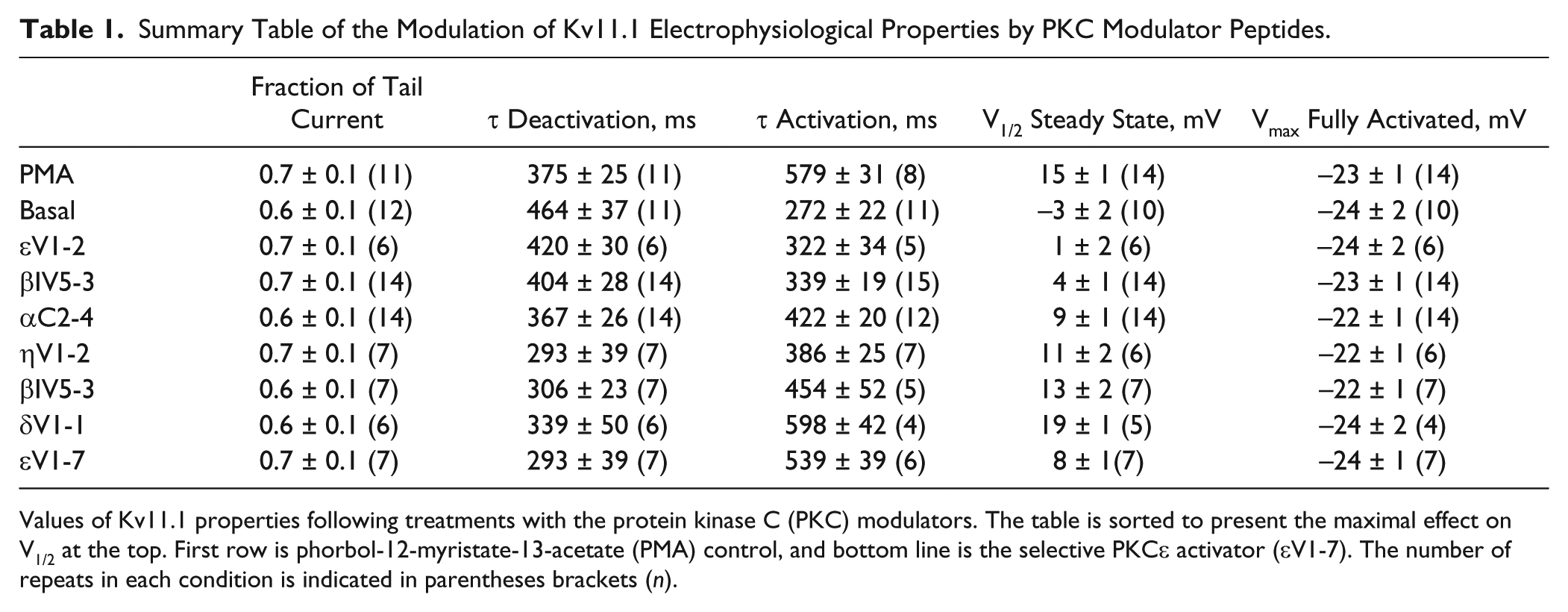
Values of Kv11.1 properties following treatments with the protein kinase C (PKC) modulators. The table is sorted to present the maximal effect on V1/2 at the top. First row is phorbol-12-myristate-13-acetate (PMA) control, and bottom line is the selective PKCε activator (εV1-7). The number of repeats in each condition is indicated in parentheses brackets (n).
Effect of PKC Modulators on the Kinetics of Kv11.1 Tail Current Activation
The kinetic of tail current activation is measured by removing the accumulating voltage-dependent inactivation at various time points throughout the activation step. The peak of the tail current is representative of the amount of activation at each pulse duration ( Fig. 3a ). In the envelope of tails protocol, a two-step voltage pulse is repeated with increasing duration of depolarization time from 6 to 1500 ms. 22 The resulting tail activation kinetic analyses are shown in Figures 3b and 4c by plotting the amplitude of the tail current as a function of the depolarizing pulse duration. The time course of activation was determined by fitting a single exponential curve to the envelope of tails. The time constants of activation were significantly different across treatments with the PKC modulators ( Fig. 3c ). As in the case of the V1/2, a one-way ANOVA indicated that the time constant values also presented significant differences between groups (F = 14.3, R = 0.64). Dunnett’s comparisons indicated that the differences observed between the basal and every other condition were significant (p < 0.05), except for βIV5-3 + PMA and the PKε inhibitory peptide with PMA (εV1-2). When Dunnett’s analysis was conducted using the PMA-activated state as a reference, all other conditions also proved statistically different at p < 0.05, except for δV1-1 + PMA and the selective PKCε activator (εV1-7), which showed no difference versus the nonselective PMA activator. The PKCε inhibitory peptide (εV1-2) displayed the largest reduction in time constant value (84%) compared with the PMA-activated state (from 579 ± 31ms in PMA to 272 ± 22 ms in εV1-2+PMA), almost reaching the value of the basal state. As for the rightward shift in V1/2, PKCβI (78% reversal; τ activation = 339 ± 19 ms) and PKCη inhibitory peptides (63% reversal; τ activation = 386 ± 25 ms) also showed a significantly lower value in the time constant of activation with respect to the PMA-activated control (p < 0.001). Finally, inhibition of PKCα (τ activation = 422 ± 20 ms) resulted in a 51% lower activation time constant (p < 0.001), closely followed by PKCβII inhibitory peptide exhibiting a 41% reduction in τ activation value (τ activation = 454 ± 52 ms; p = 0.001). Table 1 lists all individual values of τ activation along with the V1/2 and the other biophysical parameters examined in our study. Here, the rank order of the shifts induced by PKC modulators was essentially preserved when comparing the time constants of tail current activation and the values of V1/2. A linear correlation could be drawn between V1/2 and τ activation, as shown in Figure 4 . The linear fit to the data indicates that a coupling between these parameters is preserved despite the differential effects caused by specific PKC isoform peptides (R = 0.85; b = 0.05 ± 0.01). This will be commented on further in the Discussion.
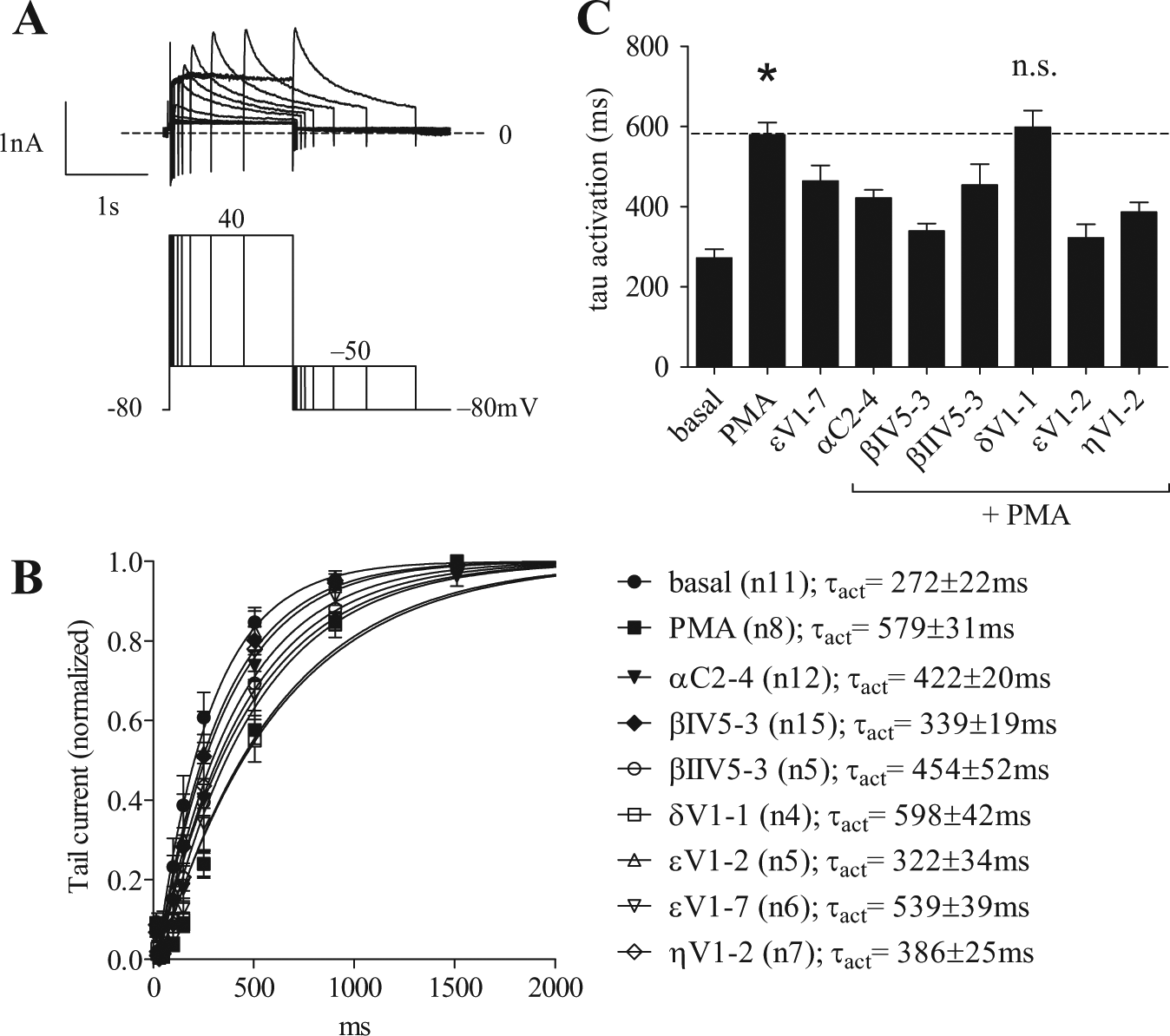
Time course of hERG current activation using the envelope of tails. (
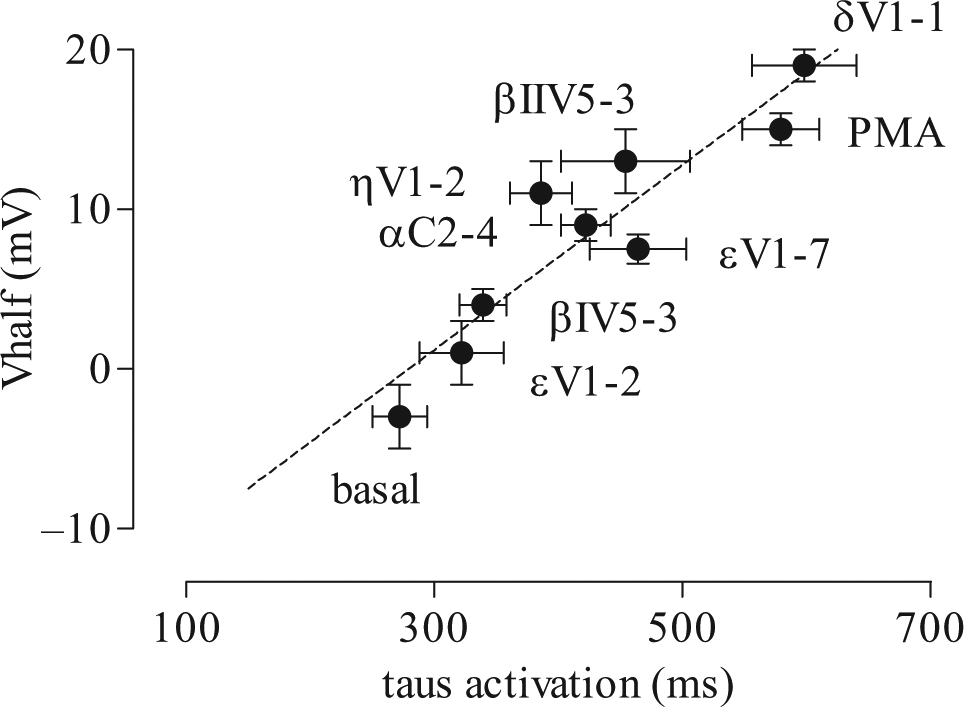
Coupling between time course of Kv11.1 current activation and shift in V1/2 activation at steady state. Coupling between the time constants of Kv11.1 activation and the corresponding shifts in the voltage dependency of activation at steady state. Values of τ activation are plotted as a function of V1/2 for each condition. A linear fit to the data is drawn as a dotted line (R = 0.85, b = 0.05 ± 0.01). Data plotted ± SE. The number of repeats (n) for each observation is the same as in Figures 2b and 3b .
Effect of PKC Modulators on the Fully Activated Voltage Dependence of Kv11.1 Tail Current
Because the PKC modulators exhibited a prominent effect in the gating of Kv11.1 activation, we decided to characterize whether PKC modulators would also affect the gating of Kv11.1 tail currents when the channels are in the fully activated state. To this end, we programmed a classic two-pulse protocol depolarization step that kept the first step constant at +40 mV (>95% of maximal current activation) while the voltage of the second “repolarization” step varied between −140 and +60 mV by increments of +20 mV. The voltage of maximum tail current activation (Vmax), typically reported to characterize Kv11.1 voltage dependency, was determined by a best fit to the normalized tail current amplitudes between the interval of −140 mV and +40 mV. As a result, the calculated Vmax values were −23 ± 2mV for all treatments, indicating no significant differences (one-way ANOVA F = 0.4, R = 0.04). No effects could be identified either on the half-maximum of the fully activated voltage dependency (basal = −86 ± 2 mV, n = 10; PMA = −83 ± 2 mV, n = 14). The voltage protocol and tail currents analysis are shown in the supplementary material.
Discussion
With this work, we aimed to study the contribution of each PKC isoform to the modulation of Kv11.1, as determined by measuring different biophysical modalities of the ion channel. We used a series of specific PKC isoform modulators and a general PKC activator to report on their regulation of biologically relevant channel characteristics. Namely, we studied: (1) maximal tail current amplitude, (2) kinetics of tail current deactivation, and (3) time course of tail current activation, with voltage dependency assessed either at (4) steady state or (5) in the fully activated state. We concluded that the PKCε isoforms played a prominent role in one of these modalities that is the gating of Kv11.1 activation. The blocking of PKCε showed the most robust and significant effect in lowering the V1/2 of steady-state activation compared with the PMA-activated state ( Fig. 2 ). A similar effect was observed on the time constant of tail current activation (τ activation), as seen in Figure 3 . When a specific PKCε activator was used, it showed a direct opposite effect to the inhibitory peptide on both the voltage dependency and the kinetic components of the gating of activation. Although it was not expected a priori, the rank order of the specific PKC inhibitor peptides was quite consistent between the values of τ activation and the V1/2 shifts ( Fig. 4 ). As a matter of fact, both the V1/2 and the τ activation participate in the gating of activation of Kv11.1 and may be equally coupled to PKC-induced conformational changes. From prior data on Kv11.1 mutants, it appeared that a direct coupling was not always observed. Namely, the N-terminus of Kv11.1 and its C-terminus may present with known long QT syndrome mutations that typically affect channel gating via distinct biophysical properties. 23 The N-terminus is known to physically interact with the S4-S5 linker that is further coupled to the charged voltage-sensing machinery of the transmembrane segment 4. 24 In Kv11.1, discrete portions of this domain also seem to mediate differential effects on the gating of activation. 25 As seen below, depending on the exact nature of Kv11.1 mutations, either a direct coupling between activation kinetics and voltage dependency of activation is observed, either these properties are affected independently and thus appear to be uncoupled. A direct effect on voltage dependency may be accompanied by a robust shift in the deactivation kinetics while showing no effects on the activation kinetics. In an early study by Spector et al., 26 conducted with a Kv11.1 mutant with a truncated N-terminus (aa 2–354), a notable shift was observed in the voltage dependency of activation (V1/2 shift of +20 mV), yet the activation time constant remained unchanged (τ activation). Instead, in that same study, the deactivation rate appeared much increased following the truncation. In contrast, using a different Kv11.1 mutation that was missing only the proximal part of the N-terminus (aa 138–373), another group showed that a shorter kinetics of channel activation (τ activation) seemed coupled to a leftward shift of the voltage dependency (V1/2 of activation). 25 Our observations with PKC isoforms appear in line with this last set of data. Finally, a study on two single-point mutations associated with long QT syndromes showed that both N33T and R56Q (N-terminal) displayed a notable shift in the V1/2 of activation, whereas no changes could be seen in the activation time constants (H70R and L86R, in turn, were silent on both parameters). 27 It thus appears that the V1/2 and τ activation of the gating of activation in Kv11.1 can be either coupled or uncoupled depending on the exact nature of the aa modifications and their location. It is perhaps reasonable to think that discrete phosphorylation of amino acids by PKC isoforms could also modulate the voltage dependency of activation in either ways. Nonetheless, the correlation of Figure 4 indicates that a robust coupling is preserved across all conditions tested. A physiological interpretation of the observation falls beyond the scope of this study; it is worth noting, however, that general PKC sites have long been identified in several domains of Kv11.1, where some have been shown to be pathologically relevant. 14
Our results indicated a strongest effect for PKCε modulator peptides, followed closely by the PKCβI and PKCα, whereas PKCη and βII isoforms displayed effects of much lesser amplitudes. It was previously reported that PKCε also played a significant role in the regulation of many other voltage-gated ion channels (VGIC), as in the case of the L-type Ca2+ channels, and the sodium channels Nav1.7 or Nav1.8.8,11,18,19 However, the effects of the PKCε isoforms appeared to be context dependent, with PKCε either increasing channel activity by lowering the threshold for activation, as in the case of NaV1.8 in neurons, or decreasing the activity as for L-type CaVs and NaVs in recombinant systems. In this context, it is perhaps not surprising that the PKCε isoforms also stood out as a significant contributor to the modulation of the gating of activation in our Kv11.1 study. These effects were easy to observe in the PatchXpress, thanks to the tighter time control and highly reproducible protocols available when using programmable robotic systems. These recordings are typically more resistant to variations due to aging of solutions, aging of cell preparations, and minute variations in compound addition that may be observed in other systems. The perfusion system itself and the intelligent feedback of the programming lines allow for adaptive patch clamp procedures to be applied to each cell, which greatly improve the resolution of measurements where comparisons between cells are typically prone to experimental variances.
Despite the high measurement accuracy, no effects were observed on the maximal tail current amplitudes in the fully activated state, the kinetics of deactivation, or the voltage for maximal activation. Without further structural data, we may only hypothesize that in the fully activated state, the activation gate of Kv11.1 may be locked in a conformation where more subtle changes by downstream PKC modulation would simply be masked. In turn, at more physiological potentials (like those at steady state), this may not be quite the case, allowing for the observation of the differential effects of PKC isoform modulation on both the voltage and time constant of activation. It is worth noting that a prominent role for PKCε on cardioprotection has been inferred for various studies conducted on ischemic models.28–30 Differential effects on the prevention of ventricular tachyarrhythmia were also recently reported using the same modulatory peptides as those used in our study, with a predominant effect observed following application of PKCε peptides to the physiological system. 31 In the light of the data presented here, the predominant role of PKCε in the modulation of Kv11.1 may thus participate in the cardioprotective effect that this particular PKC isoform seems to confer in native tissues, although further studies are clearly warranted to further substantiate this hypothesis.
Footnotes
Acknowledgements
We thank Crystal Bantados from AVIVA Biosciences for her helpful contribution in data collection and preparation of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Funding for this study were provided by the Biomedecine Research Center from the Hopital du Sacre-Coeur de Montreal, Canada.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.